

Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Niemann-Pick disease type C is an autosomal recessive neurovisceral and neurodegenerative lysosomal storage disorder caused by biallelic variants in either the NPC1 (most cases) or NPC2 gene. It is characterized by impaired cellular trafficking of cholesterol, which leads to cholesterol and sphingolipid storage in various tissues, and a complex pathogenic cascade that is still incompletely understood. The clinical spectrum is particularly broad: most commonly, the initial presentation is visceral (including a rapidly fatal neonatal form), followed by onset of neurologic symptoms at a later period ranging from early childhood to late adulthood. Currently, laboratory diagnosis combines measurement of biomarkers in plasma or dried blood spots with genetic testing. Progress in the management and treatment of patients includes the recent approval of two pharmacological agents in the United States and ongoing clinical trials.
• Niemann-Pick disease type C has a very broad spectrum of clinical phenotypes and is most likely underdiagnosed in adults. | |
• For laboratory diagnosis, first-line orientation tests include the measurement of biomarkers (in plasma or for some of them in dried blood spots): oxysterols [cholestane-3beta-5alpha−6beta-triol (C-triol)], a bile acid derivative [3beta,5alpha,6beta-trihydroxy-cholanoyl-glycine (TCG)], and palmitoyl-phosphocholine-serine (PPCS). These biomarkers are sensitive but not specific to Niemann-Pick disease type C. | |
• Confirmation of the clinical diagnosis of Niemann-Pick disease type C is primarily based on molecular study of the NPC1 and NPC2 genes. Whenever two pathogenic variant alleles cannot be identified, demonstration of impaired trafficking of endocytosed cholesterol in living cells (filipin test) may still be needed as the most specific functional test. | |
• Miglustat was the first disease-modifying pharmacological agent aiming to stabilize or slow the progression of neurologic manifestations in Niemann-Pick disease type C; it is currently approved in 42 countries but not for this indication by the U.S. Food and Drug Administration. In September 2024, two other drugs, arimoclomol and levacetylleucine, were approved by the U.S. Food and Drug Administration. | |
• Among experimental therapies, two compounds are currently under study in clinical trials: intravenous administration of 2-hydroxypropyl-beta-cyclodextrin and oral administration of nizubaglustat. |
Historically, the eponym "Niemann-Pick disease" encompassed autosomal recessive lysosomal lipid storage disorders with common features of hepatosplenomegaly and sphingomyelin storage. In 1958, Crocker and Farber showed in 18 patients a wide variability in age of onset, clinical expression, and level of sphingomyelin storage (27). This led Crocker to propose a classification of Niemann-Pick disease into three main groups, A, B and C (26). Type C was characterized by a subacute nervous system involvement with a moderate and slower course than type A, and a milder visceral sphingomyelin storage than types A and B. Patients that were similar but all of Nova Scotia Acadian origin were individualized as type D (later shown to belong to type C). From 1966 to 1968, Brady and associates demonstrated a severe deficiency of acid sphingomyelinase activity in tissues from patients with type A (corresponding to the original cases of A. Niemann and L. Pick) and with type B, but not in those from patients with types C and D.
The defect underlying types C and D remained an enigma, but patients with a retrospective diagnosis of Niemann-Pick disease type C were published in the 1960s and 1970s under various names: juvenile Niemann-Pick disease, juvenile dystonic lipidosis, atypical cerebral lipidosis, neurovisceral storage disease with vertical supranuclear ophthalmoplegia, maladie de Neville, DAF syndrome, adult dystonic lipidosis, adult neurovisceral lipidosis, giant cell hepatitis, and lactosyl ceramidosis (119). In 1982, a consensus was reached in a meeting in Prague (35) that Niemann-Pick disease type C was a distinct and specific entity. Soon after, following seminal observations by Peter Pentchev and collaborators, the concept of Niemann-Pick disease type C evolved to that of a cholesterol--rather than sphingomyelin--storage disorder (120; 121; 122; 154; 82; 175; 119). Today, “Niemann-Pick disease type C " designates disorders characterized by unique abnormalities of intracellular transport of endocytosed cholesterol and ancillary sphingolipid storage (170).
Major later advances have been the description of two genetic complementation groups and the isolation of the underlying genes. NPC1, located at chromosomal segment 18q11, is involved in 95% or more of the families (including those with type D). NPC2, located at chromosomal segment 14q24.3, is involved in rare families (161; 171; 19; 48; 108). Although the global functions of the NPC1 and NPC2 proteins are not fully understood, their sequential role in egress of endocytosed cholesterol from the cellular late endosomal/lysosomal compartment is now well established (75; 124; 134).
In the strict sense, one should distinguish between Niemann-Pick disease type C1 (www.ncbi.nlm.nih.gov/omim/257220) and Niemann-Pick disease type C2 (www.ncbi.nlm.nih.gov/omim/607625). The distinction is made from the genotype, but because the clinical manifestations and biochemical abnormalities of types C1 and C2 are similar, the generic terms of "Niemann-Pick disease type C," “Niemann-Pick C disease,” or “NPC” remain widely used in practice and in literature.
Finally, it is essential to remember that any mention of a diagnosis as "Niemann-Pick disease" without specification of a subgroup--either acid sphingomyelinase deficiency (ASMD) (SMPD1 mutations), or Niemann-Pick C (NPC1 or NPC2 mutations)--is ambiguous and a potential source of error in genetic counseling or even management of patients.
|
• The broad clinical spectrum of Niemann-Pick disease type C, already evident in Crocker and Farber's series, is now well recognized (179; 176; 39; 66; 148; 177; 58; 59; 42; 144; 158; 192; 112; 117; 159; 194; 60; 57). Several helpful overviews have been published (169; 43; 08). | |
|
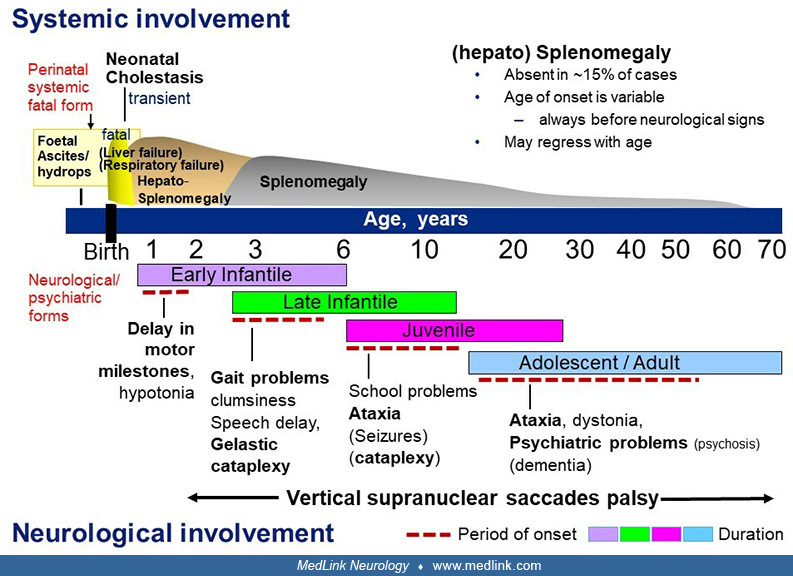
• The age of presentation is highly variable, ranging from the perinatal period to late adulthood. The initial manifestations may also vary from systemic (involving liver, spleen, and lungs) to neurologic or psychiatric in nature. Note that systemic and neurologic manifestations follow independent courses and that the systemic disease, if present, always precedes the onset of neurologic symptoms. | |
|
• Based on a widely used classification scheme of the disease, patients can be divided into a rare perinatal systemic rapidly fatal form (fetal hydrops, death in the first months of life from liver, respiratory, or multivisceral failure) and four main neurologic forms defined by age of onset of neurologic symptoms: early infantile (younger than 2 years), late infantile (2 to 6 years), juvenile (6 to 15 years) and adolescent/adult (15 years and older); a “waiting” category should be added to include diagnosed patients of any age who show only systemic symptoms (eg, prolonged neonatal cholestatic jaundice, splenomegaly, hepatosplenomegaly) and, therefore, are still nonclassifiable within a neurologic form. The rationale for proposing such a classification was the early observation that life expectancy (except for the rare neonatal rapidly fatal cases) was in large part related to the age of onset of the neurologic or psychiatric disease and not to that of the first symptom, which often--especially in children--is of a systemic nature and can occur very early in life for any of the neurologic forms. | |
|
• Liver involvement of varying severity is often present in the first months of life or even prenatally. During the course of the disease, a moderate, sometimes transient (hepato)splenomegaly is a common finding, but this sign can be absent, and it is generally of no clinical consequence. | |
|
• In typical patients, the neurologic disorder consists mainly of cerebellar ataxia, dysarthria, dysphagia, and progressive dementia. Importantly, the vast majority of cases show a characteristic vertical supranuclear gaze palsy (better named saccades palsy). Gelastic cataplexy, seizures, and dystonia are other quite common features. Psychiatric disturbances are frequent in late-onset patients (179; 39; 185; 42; 144; 192; 169; 97; 101; 117; 136; 106). Mild to moderate hearing loss (67) and sleep problems (135) may occur. In the rapidly progressive neurologic variant with early infantile onset (179; 143; 40), delayed development of motor milestones and hypotonia, followed by pyramidal signs, are the main features. |
Schematic representation of the clinical aspects of Niemann-Pick disease type C, with emphasis on period of onset and type of the main first neurologic manifestations. (Modified from: Vanier MT. Niemann-Pick disease type C. Orp...
Systemic manifestations.
Fetal and neonatal presentations. The earliest (rare) presentation of Niemann-Pick disease type C is with fetal hydrops or, more frequently, fetal ascites (158; 24); many of these newborns develop liver or respiratory failure and die rapidly.
In the neonatal period, prolonged cholestatic icterus associated with progressive hepatosplenomegaly is present in at least one third of patients (179; 66; 196; 117; 10; 41). In most cases, the jaundice resolves spontaneously by 2 to 4 months of age, eventual portal hypertension also resolves, and only hepatosplenomegaly remains, preceding onset of neurologic symptoms at a variable age. Liver and spleen size tend to diminish with time.
In a small subset (fewer than 10%) of patients with prolonged neonatal jaundice, the cholestasis quickly worsens and leads to liver failure and death, in most cases before the age of 6 months (41). Some other infants, especially those having mutations in NPC2, present with a severe respiratory insufficiency (with hepatosplenomegaly or more severe liver disease) that may also be rapidly fatal. Infants with this dramatic "acute" perinatal rapidly fatal systemic form die before onset of neurologic symptoms (other than a non-specific general hypotonia). Many examples in multiplex families have shown that this presentation can be associated with a neurologic form in the same sibship. Only infants who have died early from their systemic disease should, therefore, be included in this category.
Systemic involvement in infants, children, or adults with Niemann-Pick disease type C. In infants and children without a history of prolonged neonatal cholestatic jaundice, isolated hepatosplenomegaly or splenomegaly is often the presenting symptom. This may stay the only sign for many years or decades, until onset of neurologic symptoms (58; 192; 169; 41). Splenomegaly is usually mild to moderate and tends to diminish with time. Importantly, it may not be observed in 10% to 15% of patients with a childhood neurologic form and in a larger proportion of patients with adolescent or adult onset of the neurologic symptoms. On the other hand, the finding of three patients aged 53 to 63 years with isolated splenomegaly and a biochemical and molecular diagnosis of Niemann-Pick C disease suggests the existence of a rare non-neuronopathic form of the disease (34). Such cases may remain undiagnosed (188). Nevertheless, apart from these exceptional cases and from infants with early death, more than 90% of patients with Niemann-Pick disease type C develop neurologic symptoms.
Neurologic and psychiatric manifestations.
Patients with Niemann-Pick disease type C are best classified according to the age of onset of their neurologic symptoms because age correlates with the subsequent course and lifespan (179; 39; 177; 119; 192; 169; 117; 159; 60; 57; 10).
Severe early infantile neurologic form. In most affected infants, prolonged neonatal jaundice has been observed, and hepatosplenomegaly has been present since birth or the first months of life (117; 143; 40). Delay of developmental motor milestones and central hypotonia become evident between the ages of 6 to 9 months and 2 years. Subsequent clinical course includes a loss of acquired motor skills, proportionally less marked mental regression, followed by pronounced spasticity with pyramidal tract involvement. Many affected children never learn to walk. Intention tremor has frequently been noted; when closely monitored, supranuclear gaze palsy is present in this form, but not as an early sign (40). Seizures can occur. Brain imaging shows signs of myelination delay and, later, of leukodystrophy and cerebral atrophy. Survival rarely exceeds 5 to 7 years (179; 59; 192; 143; 40).
Late infantile neurologic form. A history of prolonged neonatal jaundice is frequent; hepatosplenomegaly or splenomegaly has usually, but not constantly, been present for a varying period. The child often presents with gait problems, frequent falls, and clumsiness between 3 and 5 years of age, due to ataxia. Language delay is frequent. The motor problems worsen, and cognitive dysfunction appears. Gelastic cataplexy is frequent and sometimes can be the initial symptom. More than half of patients develop epileptic seizures of all types (117; 10). Vertical supranuclear gaze palsy, which should more appropriately be renamed vertical supranuclear saccade palsy is a nearly constant sign.
Juvenile neurologic form. A preexisting moderate splenomegaly is common, although absence of a detectable organomegaly (even by ultrasonography) has been reported to occur in at least 15% of cases. Clear neurologic symptoms appear between 6 and 15 years of age, but onset is more variable. Vertical supranuclear gaze/saccade palsy also constitutes a typical sign. In younger children (some of them overlapping with the late infantile category), school problems with difficulty in writing, impaired attention, or apraxia are often the first signs and may lead to misdiagnosis. The child then develops clumsiness and shows more learning disabilities, and ataxia becomes obvious (falls often, cannot run properly). Seizures or gelastic cataplexy may occasionally be the presenting symptom and can become a major problem in more than half of these patients. In pre-adolescents, the onset is often more insidious, with slower progression of ataxia and other symptoms. Movement disorders more often include dystonia and, sometimes, myoclonic jerks. This “late juvenile” form shows overlaps with the adult-onset form, as shown from data in several large cohorts where siblings were classified in either category (60; 57).
The late infantile and juvenile forms, considered the “classic” Niemann-Pick type C, constitute in most countries about half of the patients. The disease progression is globally similar. As ataxia progresses, dysphagia, dysarthria, and dementia develop. Action dystonia is also frequent, mostly in the (late) juvenile form. Cataplexy, without or with narcolepsy (110; 86), typically laughter-induced, is another common and specific sign, observed in about one third of patients (64). Motor impairment is major, and intellectual decline may be variable. The neurodevelopmental and neurocognitive phenotypes in children and adolescents have been studied (165; 164). About half of the patients with the classic form develop seizures, which may be partial, generalized, or both. They generally respond to standard treatment, but refractory cases may occur, with some patients dying from status or complications of seizures. Severe epilepsy is of bad prognosis and significantly shortens the lifespan of the patients. At a later stage, dysarthria worsens, and patients ultimately stop talking. The patients develop pyramidal signs and spasticity and pronounced swallowing problems. Most will require gastrostomy. Death usually occurs between 7 and 12 years of age in late infantile-onset patients. The lifespan is quite variable in the juvenile form; some patients are still alive at the age of 30 years or later (192).
Adolescent and adult form. With increasing awareness, more patients with a late neurologic onset (often in the second or third decade, but as late as 50 years or older) have been identified in the past 10 years, although this diagnosis likely remains underestimated (148; 185; 69; 144; 107). The global symptomatology is that of an attenuated juvenile form with an insidious onset, but patients are likely to present with fragmentary phenotypes. About one third of cases may have a psychiatric presentation that can be isolated for several years before the onset of motor and cognitive signs. Psychiatric signs are most often consistent with psychosis, including paranoid delusions, auditory or visual hallucinations, and interpretative thoughts (112; 11; 106). Onset may be acute or progressive, eventually with relapses. At this stage, the neurologic examination may be normal. Other types of psychiatric disturbances are depressive syndrome, behavioral problems with aggressiveness, or social isolation. Cases have also been reported with bipolar disorders, obsessive-compulsive disorders, or transient visual hallucinations. Catatonia can also be seen. Some patients may also enter the disease with cognitive decline and mainly frontal syndrome. More in continuum with the juvenile form, many patients present with a gait disorder and cerebellar dysfunction, often associated with dystonia and, more rarely, with myoclonus. Movement disorders (dystonia, parkinsonism, chorea) are more frequent than in the juvenile form. A literature review of 72 adult-onset patients indicated that the most common initial movement disorder was cerebellar ataxia (75%), followed by dystonia (15%), tremor (5%), and myoclonus (5%), whereas generalized chorea was rare (79). The cognitive dysfunction can be very variable. Epilepsy is rare in adult-onset Niemann-Pick disease type C. Absence of clinically detectable splenomegaly has been reported in a significant proportion of patients, but abdominal sonography may reveal a slightly enlarged spleen. Vertical supranuclear gaze palsy is nearly invariably present but not always detected because slow pursuit is maintained for a long period (70; 112; 15). The later disease progression is essentially similar to that in the juvenile form.
In all forms of Niemann-Pick disease type C, vertical supranuclear gaze palsy will develop. Parents may notice compensatory head thrust when the child wants to look downward or upward. The initial sign, which may require careful examination, is an increased latency and slowing of voluntary vertical saccadic movements, slowly evolving towards vertical supranuclear gaze palsy; horizontal supranuclear saccades palsy occurs later (138; 01). A prospective study of 72 subjects described ocular motor manifestations of Niemann-Pick disease type C in detail (15).
Several clinical (neurologic) disability scales have been developed for clinical follow-up of patients and evaluation of therapies. A first scale was proposed by Pineda's group, and assessed four domains: ambulation, manipulation, language, and swallowing (59). It was later modified to add seizures and ocular movements (128). The 17-domain Niemann-Pick type C clinical severity scale (NPCCSS) proposed by Porter’s group at the National Institutes of Health provides the most complete panorama of the disease course, but it is time-consuming in regular clinical practice. It evaluates nine major domains (further including cognition, memory, and hearing) and eight modifiers (195). An abbreviated five-domain version (ambulation, manipulation, speech, swallowing, and cognition), or its four-domain version without cognition and a revised swallowing scoring preferred by the Food and Drug Administration, appears to be a valid alternative in regular clinical practice (95; 36; 115). To evaluate and compare individual rates of progression, an annual severity increment score (ASIS) has been proposed, in which the total score is divided by the age of the patient (25).
The disease is considered to invariably lead to death. However, the rate of progression and lifespan show considerable variation. Estimates of age at death in patients have been compiled, either globally (09), or categorized from the age of onset of neurologic disease (192; 169). Some (5% to 10%) patients with severe neonatal liver disease or respiratory distress may die before 3 to 6 months of age. Patients with the severe neurologic early infantile form rarely live longer than 6 to 7 years of age. Patients with a late-infantile and juvenile neurologic onset usually survive until their teenage years or later. A sizable proportion of patients reach the age of 30 years or more. Of note, because symptomatic management of patients (eg, gastrostomy) has considerably improved since 1990, current survival of patients might be longer than found in very early historical cohorts. In a large study, no significant change in survival was found over the past 20 years (09). Owing to improved diagnosis of patients with the adult-onset form in recent years, as of 2021, slightly more than half of the living patients are aged between 20 and 60 years or older in the United Kingdom, France, or Germany. Three proven NPC1 patients aged 53 years or more with splenomegaly, but no neurologic symptoms, have been documented (104; 34). In neurologic patients, two natural history surveys using different disease-specific disability scales found a linear progression over time (193; 195); however, the slopes varied when a broader range of clinical phenotypes was studied (193; 159).
The severity of the disease is primarily determined by the degree of nervous system involvement; however, liver failure may cause rapid death in 5% to 10% of neonates presenting with a severe cholestatic icterus, and a few patients (most of them with a severe NPC2 mutation) have died early from severe pulmonary insufficiency. Neonatal cholestatic jaundice is otherwise transient and usually resolves spontaneously by 4 months of age. In a study of 34 patients with early liver disease, hepatocarcinoma (four patients with Niemann-Pick disease type C reported in the literature) was not observed (41). Motor involvement is often more severe and more rapidly progressive than intellectual disability. Progressive and severe dysphagia requiring gastrostomy is a common complication. Whenever epilepsy becomes very severe or intractable, the downhill course of the disease accelerates. Splenomegaly very rarely leads to severe hypersplenism. Recurrent pulmonary infections occur, and patients are recognized with severe pulmonary involvement. Psychiatric disturbances, in rare cases, may be prominent or even dramatic. Progressing deterioration in swallowing is an important risk factor for Niemann-Pick disease type C morbidity and likely mortality (155; 156). The commonest cause of death is aspiration pneumonia, related to a combination of brainstem failure and pulmonary infection.
Niemann-Pick disease type C is also a rare cause of fetal hydrops or fetal ascites. Mild to moderate hearing loss involving the frequencies most important to speech understanding has been described in 75% of a cohort of 50 patients, and a recommendation was made to perform regular audiological monitoring in patients with Niemann-Pick disease type C, beginning at the time of diagnosis (67). Patients with NPC1 mutations seem to have an increased susceptibility to early-onset fistulizing colitis with granuloma formation reminiscent of Crohn disease, which has been linked to defective autophagosome function (141). The limited available data advocate the use of treatment with anti-TNF agents in children with Niemann-Pick disease type C and inflammatory bowel disease (190).
Importantly, patients with a purely systemic perinatal fatal form may have siblings with a neurologic form. Otherwise, affected siblings are usually categorized within the same global neurologic subtype of the disease. Fairly similar age at neurologic onset is generally observed within the early- or late-infantile forms, but more discordant courses between siblings have been described (76) and are more frequent in later onset forms.
Case 1. A female with an unremarkable neonatal period was noted to have hepatomegaly without splenomegaly at 3 years of age. From the age of 10 years, she showed clumsiness and learning problems at school. At the age of 13 years, moderate hepatosplenomegaly, cerebellar tract involvement, and vertical (upward) supranuclear gaze palsy were present. Slow waves were noted on EEG. At 15.5 years, the diagnosis of Niemann-Pick disease type C was established; then status epilepticus and repeated generalized seizures developed. At 17 years of age, enlargement of liver or spleen was no longer present, but the patient lost the ability to walk. Pyramidal tract involvement developed later. At 22.5 years of age, she was bedridden, dystonic, but still understood simple sentences. She died at 28.5 years of age. In retrospect, this patient was found homoallelic for the p.I1061T NPC1 mutation.
Case 2. A male infant developed neonatal cholestatic icterus that resolved spontaneously at 5 months of age. At 3 years of age, he was noticed to have hepatosplenomegaly. He had neurologic problems beginning at 5 years of age, with unsteady gait and loss of language. At 7 years, onset of laughter-induced cataplectic attacks occurred, and a diagnosis of Niemann-Pick disease type C was established. At 8 years of age, ataxic gait, symptoms of autism, and vertical supranuclear gaze palsy were present. Expressive language was impaired. At 10 years of age, epilepsy, dystonic features, and pyramidal tract involvement developed; cataplexy (three to four attacks per day) and narcolepsy worsened but improved with clomipramine and modafinil. At 16 years of age, he could no longer walk and swallowing problems were such that a gastrostomy became necessary, and he died at the age of 18. He was a compound heterozygote for the p.I1061T and p.S940L NPC1 mutations.
Case 3. A female, with a twin sister who died at 3 months of age from the cholestatic rapidly fatal form of Niemann-Pick disease type C (diagnosed from autopsy material), had neonatal cholestatic icterus that was resolving by 3 months of age. Hepatosplenomegaly and delayed motor milestones (she sat independently at 10 months of age and could not stand independently at 12 months of age) were present in infancy. Motor regression and a progressive intention tremor began at 15 months of age. At the age of 28 months, she could no longer stay in sitting position or maintain head control. She also developed swallowing problems. Severe pyramidal tract involvement occurred at the age of 33 months. The child died at 38 months. This patient was found homoallelic for a p.Q775P NPC1 mutation, located in the putative sterol-sensing domain of the protein.
Case 4. This female patient had no early medical history. She attended school without problems but was noticed to be mildly clumsy since the age of 14 years. Neurologic problems were first noticed at the age of 17 years, including dysarthric speech, gait and limb cerebellar ataxia, and mild dysphagia. At the age of 19 years, vertical ophthalmoplegia was also observed, together with brisk deep tendon reflexes and dystonia of right lower and upper limbs. She also experienced several sudden falls while standing, without loss of consciousness, which were described as a loss of tonus and were suggestive of cataplexy. General examination did not show splenomegaly, which was found with ultrasonography. At 20 years, her mental status was still well preserved. Her motor status continued to deteriorate, but she was still able to walk. She was treated with miglustat from the age of 23 years and died at 31 years. One of her NPC1 alleles carried a p.G992E mutation, the second one a p.G993 frameshift mutation.
|
• Biallelic pathogenic variants in either of two separate genes, NPC1 and NPC2, can cause the disease. Approximately 95% of patients have mutations in NPC1 (18q11-q12), which encodes a large multipass membrane protein. The remainder have mutations in NPC2 (14q24.3), which encodes a small soluble protein. Both proteins localize to the late endosomal/lysosomal compartment. | |
|
• Pathology includes variable enlargement of the liver and spleen with foam cell infiltrates and sea-blue histiocytes. In the brain, the main features are neuronal storage and progressive neuronal death, particularly of Purkinje cells, as well as neuroaxonal dystrophy, neurofibrillary tangles, ectopic dendritogenesis, meganeurite formation, and signs of neuroinflammation. | |
|
• A defect of either NPC1 or NPC2 results in a complex lipid storage profile, which differs in systemic organs where cholesterol and sphingomyelin predominate, and in the brain where a marked increase of gangliosides GM2 and GM3 is the most evident anomaly. Other important biological abnormalities include impairment of Ca++ release from acidic compartments, abnormal autophagic flux, endoplasmic reticulum stress, and neuroinflammation. | |
|
• It is now established that the NPC2 and NPC1 proteins work in a sequential fashion for egress of endocytosed cholesterol from the late endosomal/lysosomal compartment and that a loss of function of either protein results in accumulation and sequestration of this and other compounds in this cellular compartment. The fine mechanism for cholesterol transport is in large part elucidated. The NPC1 protein seems to be also involved in other regulatory processes, which further complexifies our understanding of the pathophysiology. | |
|
• About 700 NPC1 variants are known, among which at least 450 cause disease. There are few common variants but many recurrent ones, some of which may be related to a geographical or ethnic origin. Approximately 60 families have been documented with mutations in NPC2. |
Years after the breakthrough discovery by P Pentchev of abnormal cholesterol trafficking as the cellular hallmark of Niemann-Pick disease type C, the existence of two genes responsible for the disease was established by complementation studies using cell hybridization and linkage analysis (161; 171). This also showed, before identification of the NPC1 gene, that at least 95% of families belonged to the main NP-C1 group, which still holds true. Early studies in cells and tissues from NP-C1 and NP-C2 patients could not disclose any biochemical marker that was specific to any of the groups. The NPC1 gene was identified in 1997 by positional cloning (19), the NPC2 gene in 2000 by proteomics (108). Clinical and biochemical comparison between double mutant mice deficient in both NPC1 and NPC2 and the single mutants demonstrated a nonredundant functional cooperativity of the two proteins (153) and established that mutations in either of the genes result in a similar cellular lesion.
Neuronal storage with meganeurite formation and extensive growth of ectopic dendrites, as well as formation of neurofibrillary tangles, are important neuropathological features together with neuroinflammation and neuroaxonal dystrophy. The paired helical filament tau is indistinguishable from that in Alzheimer disease (119). Alterations of the amyloid metabolism have also been described (91). As the disease progresses, neuronal death becomes prominent, affecting more specifically certain regions, particularly Purkinje cells of the cerebellum; the basis of this selective neuronal vulnerability is unclear (119; 181). The primordial role of NPC1 expression in neurons was underscored by studies in conditional mutant mice, showing that neuron-specific expression of NPC1 in null mutant Npc1 mice rescued disease, whereas neuron-specific NPC1 deficiency was sufficient to cause disease). NPC1 expression in both neurons and oligodendrocytes appeared important for CNS myelin formation and maintenance (197; 74), but astrocyte-specific deletion of NPC1 did not cause a phenotype.
The pattern of accumulating lipids is different in brain and in non-neural organs, but similar profiles have been observed in NPC1 and NPC2 disease (patients and mouse models) (153; 169; 170). In liver and spleen, a complex lipid storage pattern, with no predominating compound, is observed. Accumulated lipids include unesterified cholesterol and sphingomyelin, bis (monoacylglycerol) phosphate (also named LBPA or BMP), glycolipids (essentially glucosylceramide, lactosylceramide, globotriaosylceramide, and ganglioside GM3), free sphingosine and sphinganine. In human patients (at variance with the mouse and cat models), accumulation is more pronounced in spleen than in liver, where changes may be subtle. In brain tissue, neither cholesterol nor sphingomyelin overtly accumulate, and free sphingosine is only mildly elevated. But histochemical staining (by filipin or BC-theta) reveals a significant storage of unesterified cholesterol in the late endosomal/lysosomal compartment of neurons. By biochemical analysis of cerebral gray matter, a striking increase of GM2 and GM3 gangliosides is the most conspicuous lipid abnormality. Myelin lipids are markedly affected in the severe infantile neurologic form only (170).
Indication of oxidative stress participation in pathogenesis led to reappraisal on the status of cholesterol oxidation products in the Npc1 mouse model. In liver, elevated levels of multiple nonenzymatically formed species were observed early in life, whereas in brain, only cholestane-3ß,5a,6ß-triol was significantly elevated. Increased levels of these metabolites also occurred in plasma. Conversely, both in brain and plasma, there was a decrease of the enzymatically (CYP46A1) formed 24(S)-hydroxycholesterol, which plays a major role in brain cholesterol homeostasis. Findings in murine plasma were recapitulated in plasma of patients, suggesting that oxysterols could serve as biomarkers of disease (132; 166).
The mature native NPC1 is a large transmembrane glycoprotein (1252 amino acid for the mature protein) that resides primarily in late endosomes and interacts transiently with lysosomes. Its 2-D structure (32) was confirmed by later cryo-electron microscopic or cristallographic studies (81). By contrast, the NPC2 protein is small (132 amino acids for the mature protein), soluble, and transported to the lysosome via the mannose-6-phosphate receptor; at variance with NPC1, it is secreted and recaptured. The cholesterol-binding domain of NPC2 has been well identified (162).
Mechanism of lysosomal cholesterol transfer. There is ample evidence that both NPC1 and NPC2 are required for cholesterol egress from the endolysosomal system. The early "hand-off" model proposed for the sequential role of the two proteins (75) was confirmed from structural observations (124). In cells such as hepatocytes or fibroblasts, after endocytosis of LDL via LDL-receptor, unesterified cholesterol, released from cholesteryl esters by lysosomal acid lipase, first binds to NPC2, which then hands it off to NPC1. A defect in either of the two proteins will, thus, lead to a progressive accumulation of unesterified cholesterol in the endolysosomal compartment. Further knowledge regarding the intimate mechanism of cholesterol transfer can be summarized as follows: facilitated by bis(monoacylglycero)phosphate, NPC2 binds cholesterol from intraluminal vesicles, docks into the NPC1 middle luminal domain, and transfers the sterol to the N-terminal loop of NPC1. The latter rotates to form a tunnel between the middle luminal loop and the C-terminal loop, allowing cholesterol to pass through the glycocalyx and reach the sterol-sensing domain (191; 124; 134).
Due to the prevention of lipoprotein uptake from circulation by the blood-brain barrier, cholesterol essentially originates from local synthesis in the brain (mostly by oligodendroglial cells and, to a lesser extent, by astrocytes and neurons). However, neurons also acquire cholesterol by glial delivery through endocytosis of an apo-E-cholesterol-phospholipid lipoprotein (06). Although lysosomal trafficking of this cholesterol source won’t need hydrolysis by the acid lipase, it will still require functional NPC2 and NPC1 proteins (05). This pathway is quantitatively small, which could explain why biochemical measurement does not show a significant increase in cholesterol concentrations in cerebral gray matter (170); however, excess of unesterified cholesterol is clearly detectable in neuronal bodies using filipin staining (153). Studies in cultured sympathetic neurons from Npc1 mutant mice gave further indication that cholesterol did accumulate in cell bodies but was decreased in distal axons, leading to a distribution imbalance (167).
Wider role of the NPC1 protein and some prominent features of the pathophysiological cascade. A key mechanism by which the lysosome communicates with other organelles involves membrane contact sites and fusion events. At the lysosome-endoplasmic reticulum membrane contact sites, the efflux of cholesterol appears mediated by NPC1 and the transfer protein Gramd1b/Aster (94). At the limiting lysosomal membrane, NPC1 also interferes with mTORC1 kinase. In Niemann-Pick C, hyperactivation of mTORC1 due to defective NPC1-SLC38A9 signaling (33) and decreased endoplasmic reticulum cholesterol will result in a cascade of events influencing virtually every organelle, including secondary mitochondrial dysfunction through membrane contact sites or other mechanisms (20). NPC1 also appears to be directly implicated in lysosomal export of sphingosine (04), a process that is defective in Niemann-Pick C (54). Sphingosine storage can exert various cellular deleterious effects (182) and likely participates in the dysregulation of lysosomal Ca++ stores observed in the disease (84; 53). Of note, a reduced TRPML1-mediated lysosomal Ca++ release in Niemann-Pick C cells was also described (147). On the other hand, the increase in ganglioside GM2 and other glycolipids appears due to the cholesterol-induced inhibition of their lysosomal degradative pathway (14). In Niemann-Pick C, autophagy is another important mechanism connecting NPC1 dysfunction to neuronal loss. An impairment of the autophagic flux with defective amphisone formation leading to retarded autophagic cargo clearance has been reported (139).
Because 60% to 70% of the NPC1 mutations (including the common p.Ile1061Thr and many others) are missense variants, which often induce reduced protein levels through increased degradation via ubiquitin-proteasome, endoplasmic reticulum quality control pathways are also important to study. Work is in progress to investigate the rescuing effect of proteasome inhibitors, pharmacological chaperones, or molecular chaperones (44; 140; 187).
The NPC1 gene maps to chromosome 18q11-q12, spans 56 kb, and contains 25 exons (19). About 700 NPC1 variants have already been reported, among which at least 450 are considered pathogenic, with only a few that are common or recurrent, often showing geographical or ethnic distribution (29; 49). All types of mutations have been described, including large deletions (137) and deep intronic mutations, but approximately two thirds are missense variants. Missense mutations can be located in all domains of the protein, but a larger proportion affects the third cysteine-rich luminal loop, also called the C-terminal domain, including the two well-studied frequent alleles p.Ile1061Thr and p.Pro1007Ala. The p.Ile1061Thr variant constitutes about 25% of alleles in the United Kingdom, 10% in France, and is also frequent in the United States, but it is less frequent in Germany, the Czech Republic, and Italy (4%) (103; 44). In the homoallelic state, it correlates with a juvenile or adolescent neurologic onset form. In the heteroallelic state, it has been found to be associated with all clinical forms except the most severe infantile neurologic-onset form. The Pro1007Ala variant, frequent in Europe but also in Brazil and the United States, appears to be more commonly associated with an adult or juvenile form than with a late infantile one. The mutational profile of representative national cohorts has been reported for Spain (38; 85), the United Kingdom (57), the Czech Republic (60), and Italy (29), among others.
Genotype-phenotype studies in homoallelic patients have generally shown good correlation between nonsense or frameshift mutations and the most severe neurologic course. The frequency of null alleles is highest (about 60%) for patients with the early infantile neurologic form, compared with 10% in the juvenile or adult forms, in which missense variants (about 85%) largely predominate. At the other end of the clinical spectrum, some NPC1 variants (particularly p. Gly992Arg and p.Asp874Val, but also some others) appear to be associated with an adult-onset form, even in combination with a null allele. From observations in multiplex families, it can be concluded that mutations show correlations with the global subtype of neurologic disease but are not predictive of the systemic course: one sibling may have died from fetal hydrops and another one suffered from a juvenile neurologic onset form (43). The complexity and limitations of genotype/phenotype correlations in NPC1 have been discussed in a review (76). Some missense mutations have underscored the functional significance of specific domains of the NPC1 protein: p.Arg518Gln, and to a lesser extent p.Arg404Trp, were shown to alter NPC2 binding. Homozygous mutations in the sterol-sensing domain were often found very deleterious (104). The cysteine-rich luminal loop contains mutations with a variable cellular and clinical impact; interestingly, variants leading to a minor impairment of cellular cholesterol trafficking (eg, p.Pro1007Ala, p.Gly992Arg); “biochemical variant” phenotype) are typically located on this loop (174).
NPC2 was shown to correspond to a previously identified gene, HE1, mapped to chromosome 14q24.3 (108). The NPC2 mutational pattern (26 pathogenic mutations described to date) shows one more common nonsense mutation (p.Glu20*) and many mutations leading to a truncated protein, associated with very severe phenotypes, including a large genomic deletion. Described missense mutations correspond to various phenotypes, including juvenile- and adult-onset patients; p.C99R appears frequent in Moroccan patients; p.Ser120Pro, which has been instrumental to confirm the functional significance of the cholesterol-binding site of the NPC2 protein, has been described in patients with the juvenile or adult form from Turkey and Iran.
Several spontaneous animal mutants involving Npc1 are known (two in the mouse and one in the cat). An Npc2 mouse mutant has been generated (153), one mouse harboring a p.Asp1005Gly Npc1 missense mutation has been characterized (92), and an Npc1 p.Ile1061Thr mouse has also been made (133). These models are particularly useful to study brain dysfunction and facilitate various types of experiments, including preclinical therapeutic trials.
|
• The disease shows autosomal recessive inheritance and is panethnic. | |
|
• Its minimal incidence at birth, calculated from diagnosed patients, is estimated to be about 1 in 100,000. |
In Western Europe, the minimal incidence, estimated earlier to be 1 in 120,000 live births, appears closer to at least 1 in 100,000 (188; 43). The population prevalence is estimated to be approximately 1 to 1.5 in 1.106. The most common NPC1 variants differ according to the geographical or ethnic origin of the patients. One main genetic NPC1 isolate has been described in French Acadians of Nova Scotia (initially described as type D Niemann-Pick disease) (p.Gly992Trp variant). The p.Gly992Trp variant appears also highly prevalent in Palestinian patients. The frequency of families with NPC2 mutations varies greatly between countries. A significant number have been described from North Africa, Turkey, and Italy.
|
• Niemann-Pick disease type C is inherited following an autosomal recessive mode; genetic counseling is, therefore, primordial. | |
|
• Identification of the pathogenic mutations in the index case and his or her parents is a prerequisite for prenatal or preimplantation diagnosis and detection of heterozygotes in blood relatives. |
Prenatal diagnosis is best achieved using chorionic villus sampling at 10 to 12 weeks, as studies on amniotic fluid cells will lead to a later diagnosis (169; 112). It is performed by molecular genetic analysis, which can be applied to uncultured chorionic villus sampling. As a prerequisite, both mutated alleles must have been identified in the index case, and parents must also be studied. Preimplantation genetic testing is, in principle, possible. Prenatal diagnosis by cellular biology testing has been abandoned because the procedure is difficult, less reliable, and not applicable to “biochemically variant” families.
Detection of heterozygotes is not reliable using biochemical or cellular biology methods and must be done by genetic testing. Identification of mutations in an index case, followed by parental study, allows reliable prenatal diagnosis for the couple as well as precise genotyping in their blood relatives.
Differential diagnosis has been discussed by Patterson and colleagues (112).
In the neonate and young infant, Niemann-Pick disease type C must be differentiated from idiopathic neonatal hepatitis and other causes of cholestatic jaundice. Onset of cholestasis usually occurs in the early neonatal period. Associated splenomegaly is a useful orienting sign. Niemann-Pick type A or Gaucher type 2 are other differential diagnoses.
In a young child with or without a history of transient neonatal cholestatic jaundice, the differential diagnosis of various causes of splenomegaly or hepatosplenomegaly should include, among lysosomal storage disorders, not only Gaucher disease and acid sphingomyelinase deficiency (Niemann-Pick disease type B or AB) but also Niemann-Pick disease type C because the systemic manifestations in the latter may remain isolated for a long period.
In children and adults, the diagnosis may be difficult in patients without overt visceromegaly (at least 15% of the cases). Depending on the symptoms, other conditions with cerebellar ataxia, dystonia, psychosis, and vertical supranuclear gaze palsy should be considered (70; 112; 71). A few patients mimicking Huntington disease or progressive supranuclear palsy have been described (79; 90). Targeted next-generation sequencing gene panels (eg, for ataxias) are useful and are increasingly leading to the diagnosis of Niemann-Pick disease type C. The most difficult differential diagnosis occurs in patients with psychiatric manifestations and no systemic symptoms.
Demonstration of foam cells in bone marrow or, better, typical inclusions on electron-microscopic examination of a skin biopsy were useful discriminating signs in the past, but these examinations are no longer common practice. Among lipidoses, differential diagnosis before onset of the neurologic disease involves mostly acid sphingomyelinase deficiency (ASMD) type B but also ASMD types A or AB, or possibly Gaucher disease or acid lipase deficiency. It is important to note that in leukocytes or dried blood spots, acid sphingomyelinase activity is not abnormally decreased in Niemann-Pick disease type C.
|
• Complementary tests, such as routine biological tests, cerebral MRI, abdominal echography, chest x-ray or scan, ophthalmologic examination, or EEG, may provide useful orientation information and can be important for differential diagnosis, but abnormalities in Niemann-Pick disease type C are inconstant and not specific. | ||
|
• Specific laboratory testing: | ||
|
- Whenever the diagnosis of Niemann-Pick disease type C is considered clinically, the best first-line testing is currently the study in plasma (or dried blood spots for TCG and PPCS) of the new biomarkers discussed below. A combination of several biomarkers is optimal to increase sensitivity and specificity. Measuring these metabolites is also indicated as complementary testing after a phenotype-specific gene panel or whole exome sequencing has given an indication of a Niemann-Pick disease type C diagnosis. | ||
|
- In all cases, confirmation of the diagnosis requires genetic testing, with the study of the NPC1 and NPC2 genes, and the identification of biallelic pathogenic variants in either gene. This may require more refined studies than gene sequencing. | ||
|
- In case of uncertain pathogenicity of the observed variants, a complementary study by the filipin test in cultured skin fibroblasts (formerly the gold standard assay) can be useful. Currently, it is rarely necessary. | ||
General complementary tests (suspicion index tools). Routine laboratory tests give normal results, except in patients with cholestatic jaundice or hypersplenism. Low HDL cholesterol is a common but not universal finding.
Brain imaging is not particularly useful for diagnosis of the patients (except for differential diagnosis) as findings are unspecific, but it has proven useful for follow-up of patients and therapies. MRI and CT scans may be normal or show cerebellar or cortical atrophy or, in the severe infantile form, white matter changes. Specific studies, including size and volume of the corpus callosum, have been associated with disease severity and used for follow-up therapy (184; 183; 186; 78; 12; 77; 89). Longitudinal MRS studies have also been proposed for follow-up of therapy (163; 142), as well as [(18)F]FDG PET-scan (152). Serial acoustic reflexes, brainstem auditory evoked responses, EEGs, and somatosensory evoked potentials studied in 36 patients showed early absence of the acoustic reflexes, later changes of the brainstem auditory evoked responses, EEG abnormalities in 57%, and abnormalities in median or peroneal nerve somatosensory evoked potentials in all patients (52).
Foam cells and sea-blue histiocytes are often present in bone marrow aspirates, but these findings are not specific for Niemann-Pick disease type C (they are also observed, among others, in acid sphingomyelinase deficiency (Niemann-Pick disease types A and B); importantly, storage cells may be missing (41).
Ultrastructural studies on conjunctival, skin, liver, or rectal biopsies can provide strong support to the diagnosis, but false-negative results often occur on liver biopsy studied by light microscopy only (66).
The clinical diagnosis of Niemann-Pick disease type C is relatively easy in patients with the most typical symptoms, such as combined splenomegaly, ataxia, and vertical gaze palsy. However, strikingly different clinical presentations exist, especially in infants and neonates. Also, neurologic onset may be delayed until adolescence or adulthood, and isolated spleno- or hepatosplenomegaly can be the presenting symptom. The disease may not be suspected in cases lacking overt organomegaly. Finally, psychiatric illness may precede motor problems (70). To help clinicians, a suspicion index tool for patients older than 4 years of age has been elaborated (189), and there is also a similar tool for young children (127). A post hoc evaluation was published (125). Clinical patient groups with an increased probability of suffering from Niemann-Pick disease type C have also been discussed (111).
Specific laboratory testing. Of note, in cases with isolated (hepato)splenomegaly, it is recommended to first exclude Gaucher disease and acid sphingomyelinase deficiency. In Niemann-Pick disease type C, chitotriosidase activity is often (but not constantly) moderately elevated, and this finding is very aspecific. Acid sphingomyelinase activity is normal in leucocytes or dried blood spots, but it is often partially deficient in cultured skin fibroblasts.
Several plasma metabolites have emerged as sensitive Niemann-Pick disease type C biomarkers, and their study, completed by sequencing of the NPC1 and NPC2 genes, constitutes the recommended first-line testing once Niemann-Pick disease type C is suspected clinically. Because identification or interpretation of certain genetic variants can be difficult, the historical filipin test, although requiring a skin fibroblast culture and an experienced laboratory, keeps its usefulness in uncertain cases as the best functional test (172; 111; 43). An algorithm outlines a proposed strategy for laboratory diagnosis of Niemann-Pick disease type C.
Orientation tests in plasma (or dried blood spots): biomarker studies.
Oxysterols (cholestane-3ß-5α−6ß-triol and/or 7-ketocholesterol). Following the initial publication, an increasing number of laboratories have implemented these assays (132; 172). Elevated values of cholestane triol (C-triol) or 7 ketocholesterol (7-KC) in plasma are observed in more than 90% of patients with Niemann-Pick disease type C (albeit close to cut-off for some of them), but a mild increase also occurs in 25% of heterozygotes. Importantly, an increase of both metabolites is not specific to Niemann-Pick disease type C but is also found in acid sphingomyelinase deficiency, acid lipase deficiency, cerebrotendinous xanthomathosis, and other causes of neonatal cholestasis, including some congenital disorders of glycosylation (28). An increase of the less specific 7-KC further occurs in Smith-Lemli-Opitz syndrome and some peroxisomal diseases.
Bile acid metabolites. The groups of DS Ory in the United States and of P Clayton in the United Kingdom proposed an alternative bile acid-based approach (62; 93; 150). Measurement of 3β,5α,6β-trihydroxy-cholanoyl-glycine (TCG) appears to offer many technical advantages over the cholestane-3ß-5α−6ß-triol (C-triol) assay (no in vitro "self-generation," no need for derivatization). Discrimination between patients and heterozygotes is also much improved. At variance with C-triol, it can be applied to dried blood spots and, therefore, to neonatal screening (63); TCG is incorporated in a 14-disease pilot study that has started. Of note, an increase of TCG has also been reported in acid sphingomyelinase deficiency (62). It appears to be the most Niemann-Pick disease-specific blood biomarker to date, but few diagnostic laboratories worldwide currently offer measurement of this metabolite.
Palmitoyl-phosphocholine-serine (PPCS) ("LysoSM509") and lysosphingomyelin measurements. The simultaneous measurement of N-palmitoyl-O-phosphocholineserine (PPCS) [termed “lysosphingomyelin-509”(LysoSM509)] (45) before elucidation of its structure (87; 149), and of lysosphingomyelin is a useful tool for the initial screening of all types of Niemann-Pick disease, types A or B (ASMD) as well as C. Striking elevation of PPCS/lysoSM509 occurs in type C but also in types A and B (ASMD), whereas a large increase of lysosphingomyelin only occurs in ASMD, with marginal or no elevation in type C (45; 73; 149; 151). In most cases, their combined assay provides a discrimination between Niemann-Pick disease type C and ASMD (73; 123; 130). Multiplex lysosphingolipid methods have been developed (123; 130; 131), allowing simultaneous measurement of glucosylsphingosine (lyso-Gb1), a biomarker for Gaucher disease (another potential differential diagnosis of Niemann-Pick disease type C) in the same assay. Of note, an increase in PPCS has been reported for some cases with a congenital disorder of glycosylation (28). The measurement of PPCS/lysoSM509 in dried blood spots, initially said to be less reliable (73; 131), has been successfully used in recent practice (72).
In short, measurements of C-triol or TCG, or of PPCS/lysoSM509 (optimally coupled to that of lysosphingomyelin), preferably in combination, constitute today a powerful primary screening test for Niemann-Pick disease type C (61). But in all cases, the diagnosis must be confirmed by genetic testing.
Definitive diagnosis by molecular analysis of the NPC1 and NPC2 genes. Mutation analysis should be considered mandatory to confirm the diagnosis of Niemann-Pick disease type C. More than 95% of patients have variants in the NPC1 gene (25 exons); the remaining have variants in the NPC2 gene (five exons). Next-generation sequencing technologies and Sanger sequencing provide accurate and sensitive methods for genetic analysis. Some variants, however, can be overlooked, especially by whole exome or even more whole genome sequencing, as an increasing number of deep intronic variants and the deleterious effect of synonymous variants have been described. Copy number assessment is, of course, mandatory, but further complementary testing may be needed. Whenever sequencing or a more refined study identifies a pathogenic variant in the two alleles for either the NPC1 or the NPC2 gene, the diagnosis is certain. To ensure segregation of alleles, it is important that at least one of the parents (or both, in case of an apparent homozygous mutation) has also been studied (112; 111; 172). In practice, interpretation of the data remains uncertain in a few cases, mostly due to the finding of variants of uncertain significance or because one presumably mutant allele has remained unidentified.
The "filipin test" and its current use. Historically and up to the past 8 to 10 years, the "filipin test" (ie, demonstration in cultured cells of an accumulation of unesterified cholesterol in perinuclear vesicles, visualized by fluorescence microscopy after staining with filipin) was considered the gold standard diagnostic assay for Niemann-Pick disease type C (170). It remains a useful functional test but is very rarely used today due to its lack of practicality and difficult interpretation in about 10% of cases, even in expert hands. To begin with, it requires a skin fibroblast culture prior to actual testing, resulting in a lengthy turn-around time. Second, if cells from 85% of cases show intense fluorescent vesicles in a perinuclear distribution (classical profile), approximately 15% display a milder accumulation. This "biochemical variant" profile (defined by specific mutations, eg, p.Pro1007Ala, p.Gly992Trp/Arg) may overlap with that seen in some heterozygotes or in other diseases (especially ASMD). It is also more frequently observed in patients with a late-onset neurologic form (170), who often have less typical symptoms.
• At this time, Niemann-Pick disease type C is not curable, but multimodality treatment and management can greatly improve the quality of life of patients and to some extent, the course of the disease. | |
• Miglustat has long been approved for neurologic manifestations of Niemann-Pick disease type C in the European Union and in many other countries, but it is not approved for this indication by the U.S. Food and Drug Administration. However, the agency approved two other pharmacological agents in September 2024: arimoclomol and N-acetyl-L-leucine (levacetylleucine). Additional drugs are currently at various stages of clinical trials (hydroxy-propyl-beta-cyclodextrin, nizubaglustat) or may be used as new investigational drugs. | |
• Symptomatic multimodality treatment of patients with Niemann-Pick disease type C is of utmost importance. It should aim to manage the symptoms and signs of the disease, including epilepsy, gelastic cataplexy, dystonia, dysarthria and dysphagia, spasticity, and psychiatric troubles. Intercurrent infections must be treated early. Patients should also benefit from appropriate physical, occupational, and speech therapy. |
Historically, treatment based on the hypothesis that cholesterol was the offending metabolite was proposed in the early 1990s. The combination of hypocholesterolemic drugs and a low-cholesterol diet partially reduced the cholesterol load in the liver, but no amelioration of the neurologic disease was seen in patients after 2 years of treatment. It is important to note that at variance with Niemann-Pick disease type B, type C patients usually do not have elevated cholesterol values in serum.
Therapeutic approaches by supplementation of the NPC1 protein are challenging considering that NPC1 is a membrane protein and, thus, does not allow cross-correction. Rare attempts of bone marrow transplantation in animals and humans with NPC1 pathogenic variants had no effect on the neurologic disease. The few successful results of liver transplantation (105; 80) are still lacking a sufficient long-term follow-up in view of the mutational profile of these patients. There has been active research related to gene therapy in the Npc1 murine model, with results clearly better than expected (22; 55; 31; 56). More experimental work is still needed, including viral vectors and combinatorial approaches with pharmacological agents. No gene therapy trial is planned for the near future. Conversely, the NPC2 protein is mannose-6-phosphorylated and undergoes secretion-recapture; bone marrow transplantation has, therefore, been tried in a handful of children with NPC2 mutations, but the results have so far been discouraging (13; 143).
An extensive review covering therapeutic approaches has been published (18).
All clinical trials in humans have been focused on downstream intervention strategies using different pharmacological molecules. Only two of them are currently approved by the United States Food and Drug Administration.
Miglustat. Miglustat has been registered for neurologic manifestations of Niemann-Pick disease type C in the European Union since 2009 and in many other countries since then. Although not FDA-approved for this indication, miglustat is used off-label for treatment of many patients with Niemann-Pick disease type C in the United States. Miglustat is a small iminosugar partially able to cross the blood-brain barrier, administered orally, whose action includes inhibition of glucosylceramide synthase but appears more complex in Niemann-Pick disease type C. Studies in the cat model indicate that miglustat ameliorates several aspects of the brain pathology and may do so in large part through mechanisms different from substrate reduction (160). Its inhibitory effect on the nonlysosomal glucosylceramidase GBA2 may be involved (88). Indications of miglustat as well as its clinical utility and monitoring have been discussed (192; 112; 43). The numerous studies in patients published before 2018 have been comprehensively reviewed (Pineda Walterfang 2018). Outcomes of the treatment are discussed below.
Arimoclomol. This hydroxylamine derivative was primarily considered as a heat shock protein 70/40 inducer in cells under stress (68), but its mode of action is likely more complex (145). A randomized, blinded, placebo-controlled phase 2/3 trial using oral administration of arimoclomol followed by an open-label extension phase was conducted in 50 patients aged 2 to 18 years old, a majority of whom also received miglustat (clinicaltrials.gov/NCT02612129). Results of the 12-month trial have been published (99). The primary endpoint (comparison of disease progression between the treated and placebo groups) was reached, and the product appeared to be well tolerated. However, complementary results from the open-label extension phase and those of an early access program were needed before FDA approval, as well as a reassessment of data using a rescored 4-domain NPC Clinical Severity Scale (4D-NPCCSS). Compared to the original 5D-NPCCSS (115), this 4D-NPCCSS omits the cognition domain and includes a revised scoring of the swallowing domain (100). Arimoclomol was approved for use in combination with miglustat for the treatment of neurologic symptoms of Niemann-Pick disease type C in adults and in pediatric patients 2 years of age and older. It was, thus, the first drug approved by the FDA to treat Niemann-Pick disease type C (September 20, 2024). Arimoclomol, in combination with miglustat, halted disease progression through 12 months of treatment, as shown by a 0.2-point decrease from baseline using the 4D-NPCSS compared to 1.9 points of progression for miglustat alone. Further confirmative evidence was obtained from a 48-month open-label extension when compared to a matched natural history cohort studied at the National Institutes of Health (96).
Levacetylleucine (N-acetyl-L-leucine). This orally administered modified amino acid is the L-enantiomer of N-acetyl-DL leucine. When given as a short-term treatment to 12 patients with Niemann-Pick disease type C, the latter compound (registered in France since 1957 for acute vertigo) improved ataxic symptoms. Further work showed that the L-enantiomer was more active and exerted a neuroprotective effect. Levacetylleucine is taken up by monocarboxylate transporters, delivered to many tissues, including the brain, and converted to L-leucine, which is proposed to enhance the mitochondria ATP production level, hence, improving neuronal membrane potential but also lysosomal function and autophagy (23; 168). A short international phase 2 open-label, rater-blinded 6-week study (NCT03759639) was first conducted in 32 patients with Niemann-Pick disease type C aged 7 to 64 years, who served as their own control. Improvement in cognitive and motor symptoms was observed (16). The safety and efficacy of the product was then evaluated in 60 patients over 4 years of age in a phase 3 randomized double-blind, placebo-controlled, 24-month crossover trial (successive 12-week periods with or without treatment) (NCT05163288). Patients could receive miglustat. At primary completion, the primary outcome was met (reduction of a functional, modified version of the SARA score compared to placebo), as well as secondary endpoints. The most common side effects were abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting (17). Levacetylleucine was approved by the FDA in September 2024 for the treatment of neurologic symptoms in patients with Niemann-Pick disease type C weighing more than 15 kg. An open-label extension phase evaluating the long-term effect after 12 and 18 months has been conducted (118). Of note, levacetylleucine is not conceived as a drug specific to Niemann-Pick disease type C. Parallel trials have also been conducted in GM2 gangliosidosis (NCT03759665) and ataxia-telangiectasia (NCT03759678).
Hydroxypropyl-beta-cyclodextrin. Promising results had been obtained in preclinical studies after intracerebroventricular administration of 2-hydroxypropyl-beta-cyclodextrin (HPBCD) in Npc1 mouse and cat models (05; 180). This led to a phase 1/2a clinical trial, using intrathecal administration of a VTS-270 (Kleptose-HPB) preparation now called adrabetadex, for which encouraging results were published (109). A blinded international, multicentric, sham-controlled phase 2b/3 trial (NCT02534844) was conducted in 54 patients (who also could receive miglustat) for 1 year. The results, however, failed to show a significant difference between the treated and sham groups in co-primary outcomes, with the puzzling observation that no significant progression occurred either in the treatment group (expected positive effect) or in the sham control one (very unexpected). The open-label extension study was prematurely closed by regulatory agencies due to potentially poor benefit/risk balance (08). Over 100 patients with Niemann-Pick disease type C, however, have been or still are being treated for various lengths of time with intrathecal adrabetadex through various modalities (08). The risk profile of repeated lumbar punctures has been evaluated (03). The future of intrathecal adrabetadex therapy is still under discussion with the United States Food and Drug Administration.
Because intraperitoneal or subcutaneous administration of high doses of HPßCD showed a therapeutic effect in murine, or to a lesser extent feline, models, respectively (30; 83; 180), clinical trials of another HPßCD preparation (Trappsol® Cyclo™) were also conducted using IV long, rate-controlled infusion every 2 weeks. Following a phase 1 trial investigating two doses (1500 and 2500 mg/kg) in 10 adult patients (50), a multicentric, randomized, double-blinded phase 1/2 clinical trial (NCT02912793) was conducted in 12 patients (2 to 39 years old) who received either 1500, 2000, or 2500 mg/kg doses (146). The 48-week results indicated that the treatment was generally well-tolerated and safe, with some improvement in six of nine patients. A randomized placebo-controlled phase 3 study (94 patients aged 3 and older, receiving a dose of 2000 mg/kg) is underway (NCT04860960). Patients may receive miglustat or levacetylleucine. Primary completion (48 weeks) was estimated to be June 2025.
AZ-3102 (nizubaglustat). This agent is a novel orally dispensable, brain penetrant azasugar that inhibits two key enzymes in glycosphingolipid metabolism (glucosylceramide synthase and GBA2) but, at variance with miglustat, not disaccharidases. Due to similarities in mode of action, miglustat treatment is an exclusion criterium during trials with nizubaglustat. Following administration in healthy adults, a phase 2 trial was initiated in 13 patients with Niemann-Pick disease type C or GM2 gangliosidosis to evaluate the safety and pharmacokinetic profile of the drug (NCT05758922). The double-blind study of 12-week duration included six patients with Niemann-Pick disease type C who received two different doses or placebo. Patients then moved to an extension phase (including four patients with Niemann-Pick disease type C) where those on placebo received one of the two nizubaglustat doses. The primary endpoint was reached (46), and a master protocol for a phase 3 study has been announced, which will include patients with Niemann-Pick disease type C who are at least 4 years old with neurologic onset between 2 and 15 years of age.
In human patients with Niemann-Pick disease type C, the lack of good noninvasive biomarkers to monitor CNS response to therapy as well as the broad clinical spectrum, make follow-up of trials particularly difficult. Some approaches, eg, calbindin D (109), neurofilament light chain (02), or total tau (21) may still require CSF studies.
The burden of living with Niemann-Pick disease type C has been evaluated from a patient and caregiver perspective, highlighting the outstanding challenges and the many unmet needs (98; 47). To date, management of patients remains largely symptomatic. The various aspects have been discussed in detail in several reviews and guidelines (112; 111; 43; 08). Seizures generally respond at least partially to antiepileptic drugs until a fairly advanced stage of the disease. Cataplexy can usually be controlled by clomipramine, protriptyline, or modafinil. Anticholinergic agents have been reported to improve dystonia and tremor in some patients. In a pilot short-time study, N-acetyl-DL-leucine showed some improvement of ataxia, and its L-enantiomer N-acetyl-L-leucine is under a phase 3 evaluation (18). Besides pharmacological treatment, physiotherapy is useful in the management of spasticity and the prevention of contractures. Melatonin can be used to treat sleep inversion. Patients with significant hearing impairment benefit from hearing aids. Patients with a slow course of the disease may benefit from special schooling for handicapped children. Prominent splenomegaly and hypersplenism are rare. Spleen size tends to diminish during the course of the disease. Proper management of infections and of swallowing or feeding difficulties (gastrostomy) is essential at an advanced stage of the disease.
Information and support to families can be obtained through organizations devoted specifically to Niemann-Pick diseases (in the United States, Canada, United Kingdom, Germany, Spain, Italy, Argentina, The Netherlands, etc.) or to lysosomal (France) or inherited metabolic diseases (see International Niemann–Pick Disease Alliance). Genetic counseling should be made available for family members.
Treatment with miglustat. Many case series and single reports have been published (128; 37; 51; 65; 116; 107; 126). A comprehensive review by Pineda and colleagues summarizes data published before 2018 (129). Miglustat has no effect on the visceral manifestations of Niemann-Pick disease type C. Initial data indicating an effect on swallowing impairment have been confirmed (157), as has stabilization of patients for 1 year or more and a slower rate of progression of the disease after treatment. Patients with later onset forms, especially juvenile and adult forms, are the best responders (116; 113; 114; 126). But patients with the most severe early infantile neurologic form respond poorly in the experience of one group (40), and some patients, even with the adult form, do not respond (107). A large study combining retrospective and prospective data supported global longer survival in patients treated with miglustat (113). The most common adverse effects of miglustat are gastrointestinal (diarrhea), for which management guidelines are available (07; 43), as well as weight loss and tremor. For optimal benefit, treating early in the course of the disease is important. However, patients diagnosed with Niemann-Pick disease type C early in life on isolated visceral manifestations may not show neurologic symptoms until years or several decades later. Thus, it has been recommended that such patients undergo regular evaluation by a neurologist or metabolic physician and do not start miglustat treatment until some sign of neurologic involvement appears. Starting treatment earlier may, however, be considered in the case of an elder affected sibling, giving a better prediction of the neurologic form (112; 43).
No full reports have yet been published regarding treatment in real life using the recently approved compounds arimoclomol or levacetylleucine.
Several women known to the author have undergone uneventful pregnancies prior to neurologic onset of their disease.
Perianesthetic morbidity in one retrospective study on 32 patients aged 1.8 to 33 years (median age 6.9 years) having undergone 64 anesthesias for diagnostic procedures disclosed need for tracheal reintubation as well as pneumonitis, hypothermia, and seizures (102).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Marie T Vanier MD PhD
Dr. Vanier, at Institut National de la Santé et de la Recherche Médicale received honorariums Sanofi Genzyme as a member of a scientific advisory board.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Neurogenetic Disorders
May. 08, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026
General Child Neurology
Apr. 29, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026