







Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Pleomorphic xanthoastrocytoma is a rare subtype of low-grade glioma that predominantly affects a young adult population. These tumors frequently arise in the temporal lobes and often present with seizures. This review covers the latest developments in our understanding of pleomorphic xanthoastrocytoma, including its more aggressive anaplastic variant, its reclassification into the category of circumscribed astrocytic gliomas (51), and the ongoing efforts to classify unique molecular alterations identified in this type of primary brain tumor, as well as the recent trends in the treatment of this unique CNS primary tumor.
|
• Pleomorphic xanthoastrocytoma is generally regarded as a WHO grade 2 neoplasm most frequently diagnosed in children and young adults. | |
|
• Seizures are the most common presentation of pleomorphic xanthoastrocytoma. | |
|
• WHO 2021 classifies the tumor within the category of circumscribed astrocytic gliomas, differentiating it from the diffuse gliomas. | |
|
• An anaplastic variant of pleomorphic xanthoastrocytoma has a poorer overall prognosis; furthermore, classic pleomorphic xanthoastrocytoma (WHO grade 2) has the potential for transformation into a high-grade tumor (WHO grade 3). | |
|
• Histopathologic features include nuclear and cytoplasmic pleomorphism, xanthomatous changes, multinucleated cells, presence of pericellular reticulin, and eosinophilic granular bodies. It is histologically characterized by a mixture of pleomorphic (often multinucleated and sometimes lipidized), epithelioid, or spindle cells, frequent eosinophilic granular bodies and lymphocytic infiltration, and genetically by recurrent BRAF p. V600E mutation (76.1%) and CDKN2A homozygous deletion (94%). Based on the mitotic count, it is classified as grade 2 or grade 3, where grading remains a strong predictor of survival (93). | |
|
• Owing to its histological features, pleomorphic xanthoastrocytoma should be distinguished from several other tumors, including giant‐cell glioblastoma. This is a subtype of giant‐cell glioblastoma that features a gross circumscription and is histologically characterized by multinucleated giant cells in a background of small, often fusiform cells, and lymphocytic infiltrate. The molecular landscape of giant‐cell glioblastoma is dominated by impairment of the TP53/MDM2 and PTEN/PI3K pathways (03). Therefore, strong and diffuse p53 immunostaining favors giant‐cell glioblastoma in the differential diagnosis of pleomorphic xanthoastrocytoma. | |
|
• Well-circumscribed gliomas include low-grade lesions such as pilocytic astrocytoma and pleomorphic xanthoastrocytoma, which may be cured surgically if resected in their entirety. | |
|
• Pleomorphic xanthoastrocytoma has the potential to spread via the cerebrospinal fluid. | |
|
• Extent of resection, WHO grade designation, and BRAF V600E mutation are main prognostic factors. | |
|
• The majority of cases have been shown to harbor CDKN2A/B homozygous deletions and MAPK pathway alterations, the most common of which is the BRAF V600E mutation. |
Pleomorphic xanthoastrocytoma is a rare, circumscribed astrocytic tumor. The first cases were reported in 1973 as meningocerebral fibrous xanthomas, which were presumed to be of mesenchymal origin (34). In 1978, the discovery of the astrocytic marker GFAP allowed Kepes and his colleagues to reevaluate these previously reported cases, along with several new ones, and to confirm their astrocytic lineage. Their subsequent report of 12 patients with pleomorphic xanthoastrocytoma was the first to describe its features systematically and to name this new entity--one that reflects its most salient pathologic characteristics (35). An evaluation of a pleomorphic xanthoastrocytoma excised in 1930 from a patient who survived for 40 years has yielded what is apparently the earliest known example of this tumor (18). The recognition of this tumor is critical for patient care because its natural history markedly differs from most astrocytic tumors, as do the implications for its management. Moreover, identification of the targetable molecular alterations can help define subtypes of this tumor, achieve more accurate prognostication, and offer new potential treatment avenues.
Pleomorphic xanthoastrocytomas are tumors most often found in supratentorial cortical regions (69; 28), often with a cystic component on imaging, which can make it difficult to distinguish them from pilocytic astrocytomas (28) or higher-grade infiltrating astrocytomas. Most frequently, the temporal lobe is affected in up to 65% of cases, the parietal lobe being next in frequency, and the occipital and frontal lobes are less commonly involved (67; 91; 20). They can rarely occur within deeper structures, such as the thalamus, brainstem, and cerebellum (44; 97; 69; 79; 02; 23; 01) and on occasion can be intraventricular (100). Rare primary occurrence in the spinal cord has been reported (11; 105; 25; 12), and even an extramedullary intradural tumor has been observed (39). There have also been occasional reports of multicentric tumor occurrence (59; 80; 105). Pleomorphic xanthoastrocytoma primarily arises in children and young adults, with a median age in the early 20s, but it can also arise in children younger than 1 year of age and adults reaching their ninth decade of life (69). There is no significant gender predilection.
Patients most commonly present with seizures, often experiencing epilepsy for some time before diagnosis, given the relatively indolent nature of pleomorphic xanthoastrocytoma. Patients may also present with signs of increased intracranial pressure (nausea, vomiting, papilledema), focal neurologic deficits, or both (50; 28; 79). Rarely, the initial presentation is an intracranial hemorrhage (100).
The prognosis for pleomorphic xanthoastrocytoma is generally more favorable than what is observed in patients with high-grade infiltrating gliomas. Investigations into the differences between grade 3 pleomorphic xanthoastrocytoma and conventional grade 2 pleomorphic xanthoastrocytoma revealed that the anaplastic variant more closely resembles higher-grade gliomas with less favorable outcomes, even following gross total resection (28; 86; 09; 79). Conventional grade 2 pleomorphic xanthoastrocytoma is also known to undergo late recurrences. Kepes’s original report of 12 patients included one patient who suffered her first recurrence 18 years after initial resection and subsequently died of a second recurrence 25 years after her original diagnosis. Other studies have reported primary recurrences and survival greater than 25 years following initial diagnosis (69; 28).
It has been consistently demonstrated that 5-year and overall survival rates are significantly different between grade 2 and grade 3 tumors (96; 93). In a reported series of 408 pleomorphic xanthoastrocytomas, a substantially inferior survival was observed for patients with grade 3 histology compared to patients with grade 2 histology (median survival: 51 months vs. not reached) (76). In another series of 67 pleomorphic xanthoastrocytomas reported in the prior year, grade 2 tumors had a 60% 5-year progression-free survival and 81% overall survival rates, whereas their grade 3 counterparts had a 40% 5-year progression-free survival and 48% overall survival rates, corroborating the data from prior studies (21; 69; 17; 62; 28; 93). Tumor grade (grade 2 vs. grade 3 pleomorphic xanthoastrocytoma) has been implicated in the prediction of overall survival, with extent of resection and age also proving significant in a number of studies (69; 17; 28; 79; 93). In one study, strong T1 enhancement, infiltrative tumor margins, peritumoral edema, WHO grade, and gross total resection predicted the progression-free survival in the univariate analysis, whereas multivariate analysis revealed that the WHO grade and infiltrative tumor margins (p = 0.008) influenced both the progression-free survival and overall survival (47).
More recent work has shown that most pleomorphic xanthoastrocytomas harbor alterations in the MAPK pathway, the majority of which involve BRAF. Further investigation of BRAF alterations shows that they comprise three classes: class I includes V600E mutations resulting in ligand-independent activation of the MAPK pathway; class II consists of BRAF fusions and non-V600E mutations, which also lead to increased MAPK pathway activation; and class III, which have impaired or absent kinase activity (81). The most common alteration is the activating BRAF V600E mutation, generally estimated to occur in up to 80% of pleomorphic xanthoastrocytomas and associated with a favorable overall prognosis (14; 40; 28; 86; 79; 92; 93). This represents an exciting avenue for further research given the development of targeted BRAF and MEK inhibitor combinations, including those with histology-agnostic regulatory approval (US FDA 2022). Additionally, the approval of the pan-RAF inhibitor tovarafenib for pediatric patients can be a viable consideration in this disease. It may be warranted to subclassify pleomorphic xanthoastrocytomas into two groups, those with MAPK pathway aberrancies and those without, as this may underlie differences in clinical response to treatments.
An 8-year-old girl presented to the clinic with a 3.5-year history of medically refractory partial complex seizures. She had a febrile convulsion at the age of 2, but she developed spontaneous seizures at 4.5 years of age, consisting of 20 to 40 seconds of staring with bilateral arm twitching, sometimes accompanied by chewing movements or picking at her clothes. These occurred daily despite concurrent treatment with four anticonvulsants. Neurologic examination was normal. MRI scan demonstrated a 1.5 cm lesion centered in the right superior temporal gyrus, which was felt to be consistent with a cavernous hemangioma. In retrospect, this lesion was thought to have been present on a scan done at 2 years of age, albeit without the dense calcification seen subsequently.
On video-EEG monitoring, her seizures were not well localized. Right temporal resection was performed 6 months after the MRI diagnosis, with pathology demonstrating a pleomorphic xanthoastrocytoma. She was tapered to a single anticonvulsant and remained seizure-free. Eighteen months after her initial resection, a routine follow-up MRI revealed a recurrent tumor, which was grossly totally resected and pathologically confirmed. The recurrent mass was partially cystic. One year after her second resection, she was well and without recurrent seizures.
Comment. This case illustrates the typical age, location, and presentation of this tumor in a patient. If her initial febrile seizure is considered symptomatic, the interval between initial symptom and diagnosis was 6 years. It also illustrates the potential for recurrence even with low-grade histology and no symptoms.
The etiology of this tumor is unknown.
Pleomorphic xanthoastrocytoma is pathologically distinct from other astrocytomas and is now classified in the category of circumscribed astrocytic gliomas (51).
Macroscopically, pleomorphic xanthoastrocytomas usually appear as well-circumscribed, firm to partially cystic, superficial cortical masses, commonly involving the overlying leptomeninges. However, the superficial circumscription is deceptive because the deeper portions often exhibit underlying parenchymal infiltration. The solid parts of the tumor generally appear as ivory-colored or gray-white firm avascular masses without evidence of necrosis. Some tumors may appear yellowish in color due to extensive lipidization of tumor cells. The cystic part is either uni- or multiloculated and may contain clear, yellow, or xanthochromic fluid; it is usually located deep to the solid part. Invasion of the dura, exophytic growth, and leptomeningeal dissemination has been observed in some cases (20).
Histologically, a key hallmark of these neoplasms is the superficial location associated with leptomeningeal involvement. This can be identified under the microscope by the identification of (a) leptomeningeal large caliber muscularized arteries embedded within the tumor and (b) tumor cells surrounding cortical vessels in the perivascular Virchow-Robin spaces. In addition to the discrete solid superficial located component, many tumors show an element of invasion into the underlying brain parenchyma (70).
In general, pleomorphic xanthoastrocytomas are moderately cellular, consisting of astrocytes with pleomorphic nuclei, bizarre multinucleated giant cells, and fascicles of elongated or polygonal cells. Lipid droplets are present in many cells (hence, the prefix "xantho-") and are sometimes large enough to force the cytoplasm into a thin rim around the droplet. The nonlipidized portion of the cytoplasm stains positive for GFAP, a glial immunohistochemical marker (35; 107). In addition to nuclear pleomorphism, GFAP positivity, and lipidization, the fourth cardinal histologic feature is the presence of reticulin fibers surrounding many of the cells.
Pleomorphic xanthoastrocytomas are cellular tumors with pleomorphic cells in fascicular or storiform patterns, resembling mesenchymal tissue. Cells may be spindle-shaped with elongated nuclei or large polygonal epithelioid forms, sometimes multinucleated. Their cytoplasm can appear eosinophilic or vacuolated due to lipid accumulation.
These xanthomatous cells with foamy cytoplasm are diagnostically helpful, but were seen in approximately 25% to 83% of cases in different series. One of the most characteristic features of pleomorphic xanthoastrocytomas is deposition of patchy to widespread intercellular reticulin networks, either individually around tumor cells, or more commonly surrounding small groups of tumor cells (as cell clusters).
Focal perivascular or intratumoral collections of mature lymphocytes with occasional plasma cells are a frequent finding. Unique histopathology with abundant clear cells and focal papillary appearance or rare presence of pigment-containing cells has also been reported (101).
This feature led to its initial misclassification as a mesenchymal tumor, as pure gliomas are reticulin-negative. Notably, lipidization and reticulin staining may each be present focally. A less-appreciated finding is that of eosinophilic granular bodies, which are often present (20). A variable amount of lymphoplasmacytic infiltrate may also be present.
Necrosis is usually absent, and mitoses are low or absent (fewer than 5 mitoses/10 HPF or 2.5 mitoses/mm2, equating 1 HPF to 0.23 mm2 in area or 0.54 mm in diameter). MIB-1 labeling index is generally less than 1%. The relative lack of mitosis in the setting of marked pleomorphism is a characteristic feature in the diagnosis of pleomorphic xanthoastrocytoma. However, it must be emphasized that the histological features of pleomorphic xanthoastrocytoma are variable; not all of the typical characteristics are present simultaneously or in all cases, which may well cause a diagnostic challenge, particularly in distinguishing pleomorphic xanthoastrocytoma from the mimics (54). Also, it should be noted that the presence of mitotic index ≥ 5/10 HPF and necrosis is associated with decreased overall survival (28). Electron microscopic studies demonstrate basal laminae, an ultrastructural correlate of positive immunohistochemical staining for reticulin, between the cells (54). Importantly, these tumors do not demonstrate the extensive parenchymal brain infiltration observed with diffuse gliomas, which is the reason they are considered circumscribed.
A number of cases that have all the cardinal features of classic pleomorphic xanthoastrocytoma, and additionally exhibit anaplastic features such as frequent mitoses, necrosis, nuclear pseudopalisading, microvascular proliferation, and hypercellularity, have been described (53; 04; 28; 09; 86; 79).
Although the original description of this tumor expressly excludes such features, an apparent consensus stipulates that the remarkable pathologic and clinical similarity to more typical xanthoastrocytomas justifies the labeling of these tumors as grade 3 pleomorphic xanthoastrocytoma (28; 86; 09; 79).
These reports have fueled speculation about the cellular origin of this tumor. The cells of a xanthoastrocytoma are GFAP-positive and have basal laminae between them. Thus, the prevailing hypothesis has been that this tumor derives from the subpial astrocyte, as these are the only astrocytes that possess a basal lamina (33). The fact that these tumors are nearly always superficial and frequently involve the leptomeninges has supported this hypothesis. The observed association with gangliogliomas and dysplasias, as well as the finding of tumor cells that stain with both glial and neuronal markers, has led some to hypothesize that the pleomorphic xanthoastrocytoma derives from a primitive neuroectodermal precursor that can differentiate along either neuronal or glial lines with a marked tendency toward the latter. The formation of the tumor may then be associated with abnormal neuronal migration and a tendency for the dysplastic cells to become neoplastic. Such a hypothesis places pleomorphic xanthoastrocytoma in close relationship to other indolent, mixed glioneuronal tumors associated with epilepsy, such as ganglioglioma, dysembryoplastic neuroepithelial tumor, and the subependymal giant-cell astrocytoma of tuberous sclerosis (SEGA) (46; 73; 94).
In the last decade, more has become known about the molecular biology of this neoplasm. Earlier studies failed to consistently reveal aberrations commonly found in other glioma types (32), and in the following years, some of those were elucidated. Mutations of the oncogene p53 were found in two of eight (25%) patients in one study (68), but a different group describing 55 tumors demonstrated that only 2% had a high degree of p53 immunostaining (19). Only a single grade 3 pleomorphic xanthoastrocytoma case with a p53 mutation was reported in a cohort of low- and high-grade pleomorphic xanthoastrocytomas (93). A group of 50 xanthoastrocytomas was studied using comparative genomic hybridization, revealing loss of material from chromosome 9 in 50% of cases; this genetic abnormality was not seen in any of a control group of more typical astrocytomas (98). Finally, a unifying mutation in the BRAF gene, a component of the MAPK pathway, was found and confirmed in the majority (up to 80%) of xanthoastrocytomas (14; 28; 86; 79; 92), including 96% of tumors arising in the temporal lobe (14; 40). Both low- and high-grade pleomorphic xanthoastrocytomas were noted to harbor it. However, its presence was much less frequent in grade 3 pleomorphic xanthoastrocytoma compared to classic grade 2 pleomorphic xanthoastrocytoma. Significantly longer overall survival has been reported in the V600E-mutant tumors (28). The observation that the BRAF V600E mutations occur frequently in several other glial and glioneuronal tumors, including gangliogliomas, pilocytic astrocytomas, chordoid gliomas, and astroblastomas, further supports the hypothesis that pleomorphic xanthoastrocytoma shares a common cell of origin with them (10; 49). Pleomorphic xanthoastrocytomas without the canonical V600E mutation have been shown to harbor other MAPK/ERK signaling pathway alterations, such as mutations and fusions in the RAF family of kinases (12; 93) and an NTRK gene fusion (99). Another recurrent alteration found in pleomorphic xanthoastrocytomas is homozygous deletion of CDKN2A/B, which is reported in 60% to 94% of cases (98; 92; 71). It has been demonstrated that combined p16 and MTAP immunostains correctly detect CDKN2A homozygous deletion in pleomorphic xanthoastrocytoma and may serve as surrogate markers in the absence of molecular studies (95). Interestingly, concurrent MAPK alterations and CDKN2A/B deletions are frequent in epithelioid glioblastoma, pediatric secondary high-grade glioma types, and a high-grade astrocytoma with piloid features (75).
Of note, pleomorphic xanthoastrocytomas with TERT promoter mutations are associated with shorter overall survival (15). Some studies also reported TERT promoter mutations and amplifications, which are more prevalent in recurrent grade 3 pleomorphic xanthoastrocytoma, suggesting that it may be a late genetic event associated with anaplastic transformation and recurrence and less favorable overall prognosis (71; 93; 15; 104). The definitive prognostic significance of some of these alterations in pleomorphic xanthoastrocytoma still remains to be elucidated in larger studies.
An epigenetic study examined methylation in a group of 10 both grade 2 (n=8) and grade 3 pleomorphic xanthoastrocytomas (n=2) and found that the two anaplastic cases had increased methylation of five different genes (CD81, HCK, HOXA5, ASCL2, and TES) that have been described in glioblastomas (56). A genome-wide methylation profiling study of 46 cases performed against a comprehensive reference data set assigned 40 of them to the pleomorphic xanthoastrocytoma methylation class, with the remaining six cases grouped with the methylation classes of ganglioglioma, pilocytic astrocytoma, anaplastic pilocytic astrocytoma, or control tissue (93). Overall survival was significantly different between aleomorphic xanthoastrocytoma and anaplastic pleomorphic xanthoastrocytoma (5-year overall survival 80.8% vs. 47.6%; P = 0.0009) but not progression-free survival (5-year progression-free survival 59.9% vs. 39.8%; P = 0.05). These results confirmed the importance of WHO grading in histologically and epigenetically defined pleomorphic xanthoastrocytoma (93). No prognostic difference was reported in association with the methylation class based on DNA methylation; systemic metastases of pleomorphic xanthoastrocytoma, although very rare, can now be diagnosed using methylation profiling (30).
Alterations in many other genes in the genetic landscape of anaplastic pleomorphic xanthoastrocytomas (including SMARCB1, BCOR, BCORL1, ARID1A, ATRX, PTEN, FANCA, FANCD2, FANCI, FANCM, PRKDC, NOTCH2, NOTCH3, NOTCH4, and BCL6) have been described with unclear pathogenetic significance in pleomorphic xanthoastrocytomas (71).
Due to the extreme rarity of this tumor, no precise information regarding its incidence or prevalence is available. Pleomorphic xanthoastrocytoma accounts for less than 0.3% of primary CNS tumors, with an annual incidence of fewer than 0.7 cases per 100,000 population (65). Pleomorphic xanthoastrocytoma is estimated to account for less than 1% of astrocytic tumors (82), and it represented 1.5% of all pediatric low-grade gliomas in one Canadian-based population study (43). In the setting of epilepsy referral centers, chronic epilepsy is found to be due to pleomorphic xanthoastrocytoma in roughly one out of every 650 cases (61).
Presence of BRAF mutation, earlier age of seizure onset, and smaller tumor size are associated with seizure freedom after initial resection prior to progressive disease. It also appears that BRAF-mutated and BRAF-wildtype pleomorphic xanthoastrocytomas have distinct epilepsy phenotypes, yet similar survival rates are observed between patients with and without tumor-related epilepsy (106).
Pleomorphic xanthoastrocytoma is classified under the umbrella term “unique astrocytoma variants.” In SEER Cancer Statistics Review, only 214 cases were reported between 1981 and 2007 in the United States. This is approximately one-tenth the incidence of pilocytic astrocytomas, another rare CNS tumor (64).
There are no means of preventing these tumors. Pleomorphic xanthoastrocytoma is not associated with any other disorders, nor is any particular group (besides the young) known to be at risk. Several case reports of xanthoastrocytomas occurring in patients with Sturge-Weber syndrome or neurofibromatosis type 1 are available, but whether these are chance occurrences or harbingers of a population at risk has yet to be determined (66; 45; 78; 38; 80; 87). Hypothetically, the presence of Sturge-Weber syndrome might create a unique environment that could predispose an individual to the development of other brain tumors, such as pleomorphic xanthoastrocytoma.
The differential diagnosis for this lesion consists of all the other glioma subtypes, including noninfiltrative gliomas and glioneuronal tumors such as pilocytic astrocytoma and ganglioglioma, as well as infiltrative gliomas such as grade 2 and 3 astrocytoma and oligodendroglioma, and glioblastoma (22). High-grade glioma with piloid features, a tumor type defined by it’s DNA methylation profile, may also have both histopathologic and molecular similarities. Leptomeningeal involvement is uncommon in all of these except pilocytic astrocytoma. Dysembryoplastic neuroepithelial tumor may also arise in this location. Meningioma is also an important diagnostic consideration because of the superficial location of these lesions, which may even produce a "dural tail" on imaging (72). Cavernous hemangioma is another important consideration in an enhancing temporal lobe lesion that fails to change over time. Metastases may produce a similar appearance but are usually not a consideration in the younger age group in which these tumors most frequently arise. Two cases of pleomorphic xanthoastrocytoma mimicking an inflammatory granuloma were reported (13). As there is no pathognomonic feature of pleomorphic xanthoastrocytomas, the diagnosis can often be difficult in clinical practice.
Pleomorphic xanthoastrocytomas are frequently associated with seizures. Epilepsy is the most frequent presenting sign, likely due to the tumor’s predilection for the temporal lobes, particularly the cortical (as opposed to subcortical) involvement. It is also possible that the tumor biology and the tumor cell-microenvironment interactions facilitate the epileptogenicity of the tumor.
Localizable symptoms correlate directly with the neuroanatomy involved with the tumor. Nonlocalizable symptoms are related to the elevation of intracranial pressure and include headaches, nausea, vomiting, diplopia, and somnolence. The mean duration of symptoms before diagnosis is a few months; rarely, they are diagnosed incidentally via imaging performed for other reasons (82).
As with all brain neoplasms, the diagnosis of pleomorphic xanthoastrocytoma generally requires cerebral imaging for identification and localization, followed by surgical resection (or biopsy) for pathologic examination. MRI is the imaging modality of choice.
In general, pleomorphic xanthoastrocytoma is a hemispheric tumor often located peripherally with cortical with or without leptomeningeal involvement; in decreasing incidence, they are found in the temporal, frontoparietal, and occipital lobes. Pleomorphic xanthoastrocytoma is classically cystic with a solid mural nodule; the nodule often abuts the pial surface. It can have an enhancing dural tail of leptomeningeal attachment. There is often with infiltration of adjacent parenchyma beyond the margin of the visible tumor. There might be accompanying adjacent cortical dysplasia.
MRI, the imaging modality of choice, often demonstrates a cyst with a mural nodule. The nodule is usually hyperintense on T2-weighted images and hypo- or isointense on T1-weighted images, with gadolinium enhancement in almost all cases (28; 60).
CT scan reveals a superficial cortical tumor with a cyst visible in 50% to 60% of cases (67; 91; 20). The tumor itself frequently appears as a solid mural nodule and may be hypodense or isodense to the surrounding brain. Calcification is rare, as is bony erosion. Contrast enhancement is an almost invariable feature, but the pattern of enhancement is variable (50). Edema is not prominent. Angiography is usually normal, though occasional tumors are hypovascular (67).
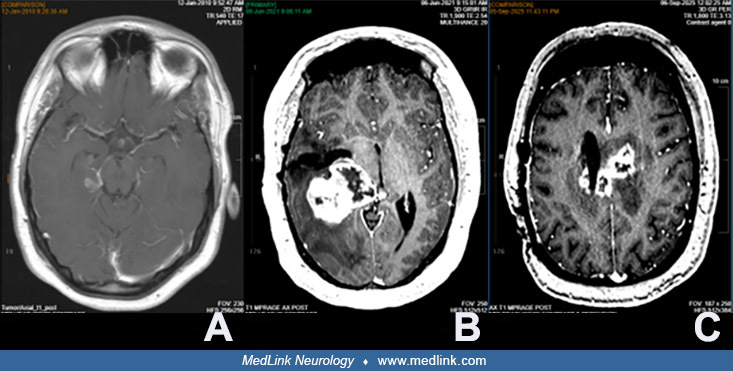
(A) Jan 2010. Enhancement of mass in the right parahippocampal gyrus, currently measuring 1.1 x 2.9 cm. (B) June 2021. Previous right transtemporal mesial temporal pleomorphic xanthoastrocytoma resection with redemonstration of...
On MRI, the cystic component is hypointense on T1- and hyperintense on T2-weighted images, whereas the solid component, including mural nodules or thick cyst walls of tumors, is hypointense or isointense on T1- and slightly hyperintense on T2-weighted images. Marked enhancement of the solid component and peripheral rim enhancement of the cyst is seen on T1-weighted imaging with gadolinium enhancement (103).
Leptomeningeal enhancement is reported in approximately 14% of cases, which contrasts with the fact that leptomeningeal contact is evident in 92% of the lesions.
On angiography, pleomorphic xanthoastrocytomas are hypovascular (102).
Fluorodeoxyglucose PET shows high glucose metabolism, and SPECT perfusion scans show hypoperfusion (06). Compared to pleomorphic xanthoastrocytomas, anaplastic pleomorphic xanthoastrocytomas typically are larger lesions with more heterogeneous-appearing masses. These tumors are more likely to have leptomeningeal spread and have increased perfusion parameters (63).
Analysis of imaging characteristics has shown that the BRAF V600E-wildtype grade 2 pleomorphic xanthoastrocytoma presented with more aggressive conventional and advanced MR imaging features than the V600E-mutant grade 2 pleomorphic xanthoastrocytoma (26). However, there were no significant differences in both ADC min and ADC mean between the BRAF p. V600E-mutant type and wild-type anaplastic pleomorphic xanthoastrocytoma groups. A study of FDG-PET, though it included only grade 3 tumors, showed a strong correlation between the degree of metabolism and the tumor grade (16). PET with FDG correlates with histopathology, and increased uptake may be a marker for more malignant or aggressive tumors. The clinical utility of this, however, is unclear.
The mainstay of treatment for this tumor is surgical resection (93). In one reported study, all grades underwent surgical resection at high rates: 95.1% (n = 329) of patients with grade 2 pleomorphic xanthoastrocytoma and 98.4% (n=61) of patients with anaplastic pleomorphic xanthoastrocytoma (p=0.332) (76). Most evidence supports more favorable outcomes with a greater extent of resection (69; 17; 62; 28; 79). There appears to be a correlation between gross total resection with longer progression-free survival; therefore, gross total resection has often been considered the mainstay of treatment (85). Several large case series and reviews have found 5-year progression-free survival and overall survival rates of 28% to 71% and 48% to 81%, respectively, for pleomorphic xanthoastrocytoma of all grades (20; 69; 17; 62; 28), and extent of resection proved significant in a number of studies (69; 17; 28; 93).
However, a study from Northwestern in 2023 looked at 17 patients with pathologically confirmed pleomorphic xanthoastrocytoma. Two patients were excluded due to incomplete treatment information or less than 6 months of follow-up; 15 patients were analyzed (median follow-up 4.4 years). Six patients had grade 2 pleomorphic xanthoastrocytoma, and nine had grade 3 anaplastic pleomorphic xanthoastrocytoma. The 2-year and 5-year progression-free survival for the cohort was 57% and 33%, respectively; 2-year and 5-year overall survival was 93% and 75%, respectively. Overall, the poor survival of cohorts, especially with grade 3 tumors, suggests the need for more aggressive treatment, including maximal resection followed by intensive adjuvant therapy (85).
The role for adjuvant radiation and chemotherapy remains less clear, with multiple studies unable to reconcile the risks and benefits (53; 67; 88; 08; 57; 41; 63; 74; 58; 69).
Data regarding the routine use of radiotherapy in pleomorphic xanthoastrocytoma are insufficient, and although some reports have noted an association with the use of postoperative radiotherapy and improved progression-free survival, no definitive improvement in overall survival has been demonstrated. A meta-analysis of 167 patients revealed use of adjuvant therapy was not found to be a significant factor affecting progression-free survival or overall survival. Radiotherapy was used in salvage treatment in 76.1% of the patients. There is inadequate evidence to recommend routine adjuvant radiation or chemotherapy in all patients with grade 2 pleomorphic xanthoastrocytoma. Earlier incorporation of radiation for subtotal resection or anaplastic pleomorphic xanthoastrocytoma may be considered, given the poorer outcomes associated with these factors. Due to paucity of data, doses ranging between 45 and 54 Gy have been employed in the adjuvant or salvage setting (82). There are also newer modalities being tested, including IMPT for salvage purposes (24). For Grade 3 tumors, aggressive treatment, including maximal safe resection followed by intense adjuvant treatment, is warranted (85). Conflicting data regarding adults with pleomorphic xanthoastrocytoma pointed out that treatment with radiotherapy is independently associated with a significantly higher risk of mortality (36). In this paper, a total of 546 patients were identified, and overall, those who received radiotherapy had a shorter median overall survival (33.3 months) compared to those who did not (more than 128.6 months, p < 0.001). It is unclear whether needing radiotherapy for salvage purposes or having anaplastic features in some cases were inherently associated with worse overall survival (this author’s interpretation). A possible explanation is that radiotherapy may induce the malignant transformation of pleomorphic xanthoastrocytoma into a more aggressive tumor type with associated worse clinical outcomes. At any rate, routine use of this modality in treating pleomorphic xanthoastrocytoma warrants further study.
Chemotherapy traditionally did not have a definitive role in treatment (20; 69; 96), though some early reports noted success with carboplatin and vincristine (52; 63). In one study of 408 pleomorphic xanthoastrocytomas, chemotherapy (regimen not specified) was administered to about 52% of patients with anaplastic pleomorphic xanthoastrocytoma and only about 11% of patients with grade 2 pleomorphic xanthoastrocytoma (76). In pediatric patients, where radiation therapy is of limited use, the use of adjuvant chemoradiotherapy in the treatment of grade 3 pleomorphic xanthoastrocytoma has been supported empirically, consisting of fractionated radiotherapy and concurrent temozolomide in only three patients (77). Currently, the use of systemic therapy continues to be an active area of investigation in the treatment of anaplastic pleomorphic xanthoastrocytoma. The same study reported that about 70% of grade 3 tumors were irradiated; however, only about 18% of grade 2 tumors received radiation. It appears that pleomorphic xanthoastrocytoma does not frequently possess MGMT methylation, raising doubts about the benefits of temozolomide, despite the scattered attempts to use it reported in the literature. The use of traditional alkylating chemotherapy like temozolomide is considered to have limited efficacy against pleomorphic xanthoastrocytoma (84).
The use of targeted therapies has been an active area of investigation, particularly in the BRAF V600E-mutant pleomorphic xanthoastrocytoma. The overall survival of patients with V600E-mutant tumors was significantly higher than that of those with wild-type tumors, which included 74 pleomorphic xanthoastrocytoma of all grades, 35 of which were treated with adjuvant therapies after recurrence or progression (28). Mitotic index ≥ 5/10 HPF and necrosis were associated with decreased overall survival. This could be confounded by the increased prevalence of the mutation in the grade 2 pleomorphic xanthoastrocytoma group compared to grade 3 cases. Case reports and case series have demonstrated radiographic stability of disease and partial responses in some patients treated with BRAF inhibitors vemurafenib and dabrafenib, which appear to be tolerable in this patient population (07; 27; 90; 48; 05). The VE-BASKET study reported the highest response rate to vemurafenib in low-grade tumors, particularly pleomorphic xanthoastrocytoma (n=7), with a promising clinical benefit rate of 57% (29; 31). One significant challenge that emerged during the medical management of the affected patients is resistance to BRAF inhibitors via acquired reactivation of the MAPK pathway. This led to consideration of the impeding MEK pathway, downstream of MAPK, to increase deep and durable responses as previously shown in melanoma. Therefore, an increasing number of reports support the concurrent use of BRAF and MEK inhibitors in treating pleomorphic xanthoastrocytoma. BRAF/MEK dual-drug inhibitor therapy has been reported to delay tumor progression with good tolerance in the entire cohort with grade 1 to 2 adverse effects, including transient skin rash, fatigue, abdominal discomfort, neutropenia, and diarrhea (31). Although the mechanism is not entirely elucidated, this may be attributed, in part, to a synergistic potential of combining both inhibitors and their specific effects, depending on the specific class of the BRAF alteration harbored by each tumor. In June 2022, dual BRAF and MEK inhibition with dabrafenib and trametinib received histology-agnostic regulatory approval from the U.S. FDA for patients with tumors harboring the V600E BRAF mutation.
In the ROAR trial, which looked at agnostic tumor response rates to dabrafenib and trametinib in various groups, the overall response rate was 54% in the low-grade glioma cohort (n=13); overall survival could not be estimated owing to the low numbers of deaths in the low-grade glioma cohort (n = 4) (83). Although not specific to pleomorphic xanthoastrocytoma, the LGG group performance was somewhat encouraging, with a 47% overall response rate in the entire low-grade glioma cohort. Dual BRAF and MEK inhibition with dabrafenib and trametinib has received histology-agnostic regulatory approval for patients with tumors harboring the V600E BRAF mutation.
Newer generation drugs like pan-RAF Type 2 inhibitor tovorafenib have been recently approved by the U.S. FDA for relapsed pediatric low-grade gliomas. Results are encouraging as the FDA granted approval based on the phase 2 FIREFLY-1 trial (37).
The most common adverse event associated with BRAF or MEK inhibitor treatment is grade 1 to 2 fatigue and skin rash. Dual BRAF and MEK inhibitors were reported to be well tolerated, with reversible grade 1 to 2 adverse events, including transient skin rash, fatigue, abdominal discomfort, neutropenia, and diarrhea. No grade 3 to 4 adverse events were detected (31).
Seizures in patients with pleomorphic xanthoastrocytoma are managed with standard anticonvulsant medications and resection of the lesion. Most case reports indicate that these patients’ epilepsy responds well to tumor resection. It also appears that BRAF-mutated and BRAF-wildtype pleomorphic xanthoastrocytomas have distinct epilepsy phenotypes (106).
Although pregnancy does not appear to increase the risk of developing a pleomorphic xanthoastrocytoma, it can significantly complicate its management. The decision-making process is highly individualized.
Malignant transformation within a 6-month period of a grade 3 pleomorphic xanthoastrocytoma initially diagnosed in a pregnant patient to a glioblastoma postpartum was reported (55). Another patient was reported as having delivered a normal child 4 years after gross total resection and whole-brain radiation for xanthoastrocytoma (42).
No information is available regarding the use of various types of anesthesia in patients with this tumor.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Tolga Tuncer MD
Dr. Tuncer of The University of Kansas Medical Center has no relevant financial relationships to disclose.
See Profile
Rimas V Lukas MD
Dr. Lukas of Northwestern University Feinberg School of Medicine received honorariums from Jazz Therapeutics, Novocure, and Servier for speaking engagements, honorariums from Cardinal Health, Catalyx, Merck, and Novocure for advisory board membership, research support from BMS as principal investigator, and an honorarium from GT Medical Technologies for DSMB membership.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Stroke & Vascular Disorders
May. 03, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026