




Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Rasmussen syndrome is a rare disease defined by refractory epilepsy, progressive hemiparesis, and deterioration of mental capacity with usual onset in childhood. The epilepsy often manifests as epilepsia partialis continua. Focal motor seizures are most commonly refractory to antiseizure medications. A definite etiology has not been delineated, but mounting evidence indicates that it is an autoimmune disorder. Numerous immunosuppressive therapies have been attempted due to the presumed inflammatory etiology based on histopathology and immunological studies. The only curative measure has been hemispherotomy or hemispherectomy.
|
• Rasmussen syndrome is a progressive disorder, predominantly with childhood onset and characterized by refractory epilepsy, hemiparesis, and neurologic decline. | |
|
• Rasmussen syndrome is a rare immune-mediated reaction for which an etiology has yet to be determined. | |
|
• Various treatments, such as antiseizure medications, steroids, plasma exchange, immunoglobulins, and other immunomodulatory drugs, have been attempted to slow the clinical progression of Rasmussen syndrome. | |
|
• Complete hemispheric disconnection, which may be either a hemispherectomy or a hemispherotomy, is the only effective treatment to control seizures and prevent cognitive deterioration. | |
|
• Controversy exists as to whether hemispherotomy should be performed early in the disease course due to the anticipated disability or after deficits become fixed. |
The first cases of what is now called Rasmussen encephalitis or Rasmussen syndrome were referred to as chronic focal encephalitis in 1958 by Dr. Theodore Rasmussen and his colleagues at the Montreal Neurological Institute (54). Their report describes three children with focal seizures and progressive hemiparesis refractory to antiseizure medications. Hemispherectomy in two cases resulted in the improvement of hemiparesis and resolution of seizure activity. Pathology of brain tissue in all three cases demonstrated inflammatory changes, leading to the theory that the disease process was secondary to focal encephalitis. An etiology was not determined, but the disease was thought to be virally mediated. Many years later, the etiology remains elusive, but studies indicate an autoimmune disease with T cell-mediated neuronal death (60; 49; 35; 66; 74).
However, there are reports of cases described by Kozhevnikov, a Russian neurologist in the 1880s, suggesting that this disease process has existed for centuries. He described patients with unilateral focal motor seizures, using the term epilepsia corticalis sive partialis continua to depict a continuous epilepsy syndrome. His account describes men of varying ages suffering from persistent focal motor seizures that progressed to generalized convulsions in some. These men then developed unilateral weakness that was observed to be progressive on the side affected by seizure activity. He did not find an etiology to explain the syndrome but did suggest a chronic inflammatory process (37). It is fascinating that Kozhevnikov thought of operating on these patients, as he could find no cure. Unfortunately, he was refused the opportunity to perform autopsies on this group of patients. This syndrome has become known as Kozhevnikov epilepsy, thought to be common in Siberia.
In the 1989 International League Against Epilepsy (ILAE) classification, Rasmussen syndrome was introduced as a synonym of Kozhevnikov type 2 syndrome; eventually, the name Kozhevnikov was removed (51). The revision of seizure terminology by the ILAE Commission on Classification and Terminology lists Rasmussen syndrome under the category “distinctive constellations” (08).
A study of 48 cases with pathology consistent with Rasmussen syndrome has been critical in delineating features of this disease. Mainly a childhood illness, the diagnosis may not be so obvious at first. The age of onset is between 1 and 14 years, with epilepsy beginning before 10 years of age (47). Onset in adulthood is less common, comprising about 10% of cases (11). There is no gender or ethnic preference.
Rasmussen syndrome is manifested by the onset of seizures, with the semiology varying, but most commonly focal motor seizures. The Montreal Neurological Institute’s series of 48 patients found that generalized tonic-clonic seizures were the initial symptom in 30% of patients. Complex partial and simple partial seizures, classified as focal seizures with and without impaired awareness, respectively, were seen in 26% of patients (22). Status epilepticus, whether generalized or epilepsia partialis continua, occurred in 20% of patients (47). Long-term follow-up of patients has found varying manifestations of seizure activity, with simple partial motor seizures occurring most commonly. Epilepsia partialis continua, a form of focal status epilepticus, has an incidence of 56% to 92% in Rasmussen syndrome (11). However, the first symptom may be motor dysfunction, with seizures being observed only months or years later (28).
Epilepsia partialis continua is characterized by continuous clonic activity of a group of muscles, usually the face, arm, or leg. It can persist for a period of hours to months as a focal motor status epilepticus. Epilepsia partialis continua is very frequent in Rasmussen syndrome, and its development should raise concern; however, it is also found in structural lesions such as tumors, developmental abnormalities, and neurodegenerative conditions. Kozhevnikov and later Omorokov described this phenomenon in Russia. Omorokov, a Russian neurologist in the early 1900s, kept account of 52 cases from his clinic in Siberia and noted the onset of epilepsia partialis continua during the spring and summer, which was thought to be Russian spring-summer encephalitis (48).
As the seizures become difficult to manage, the development of hemiparesis ensues. One study has divided Rasmussen syndrome into three stages based on clinical symptoms and MRI findings akin to the traditional three stages mentioned in the Montreal Neurological Institute (MNI) series. A prodromal phase, acute disease, and residual stage have been described that correspond with MRI findings of hemispheric volume loss (13). The prodromal phase consists of infrequent seizures with minimal hemiparesis. This is similar to MNI Stage 1, which spans from the onset of seizure activity to the development of hemiparesis. The next phase, acute disease, is characterized by increased seizure frequency and hemiparesis. MRI imaging during this time reveals hyperintense T2 and FLAIR signals suggesting inflammation. It was found that most hemispheric volume loss contralateral to the side of weakness occurred at this time. Again, the definition of this acute phase is similar to MNI stage 2, which is characterized by increased seizure frequency with different semiology and clinical deterioration. In the last stage, the residual stage, seizure frequency decreases while a fixed hemiplegia develops, which is consistent with MNI stage 3 definition of fixed neurologic findings of hemiplegia, cognitive impairment, and deficits such as visual field cuts. However, after a period of time, the seizure frequency may increase. This study implies that treating Rasmussen syndrome during the acute phase may have effects on future brain atrophy.
|
Stage |
Clinical symptoms |
MRI findings |
MNI Stages |
|
Prodromal phase |
Low seizure frequency, rare hemiparesis |
Normal to mild changes |
Stage 1 |
|
Acute disease |
Frequent polymorphic seizures, developing hemiparesis to fixed deficit |
Hyperintense signals, greater degree of volume loss |
Stage 2 |
|
Residual disease |
Fixed hemiparesis, decreased seizure frequency |
Stable volume loss |
Stage 3 |
Variants of Rasmussen syndrome have been described. As stated previously, about 10% of cases occur in adults (29; 11). The disease course is prolonged and milder with less severe hemiparesis in adult-onset Rasmussen encephalitis (29). Compared to the classical childhood-onset, patients with late-onset Rasmussen encephalitis, including adolescent and adult-onset, have more frequent focal seizures with impaired awareness, less frequent epilepsia partialis continua, a slower evolution with less cognitive deterioration, and a better outcome (21).
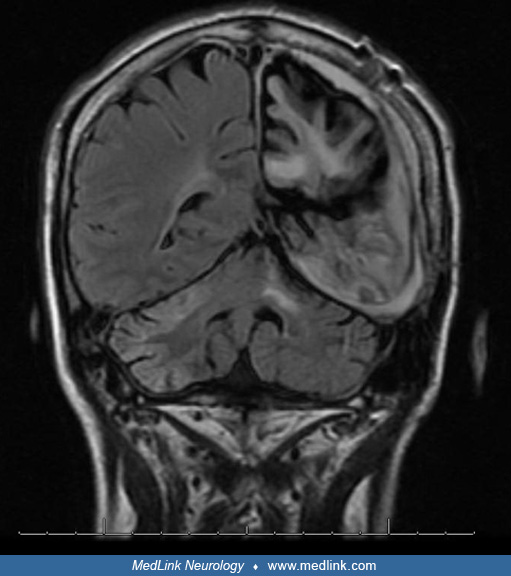
MRI discloses progressive atrophy and signal changes through the affected hemisphere, but rarely gadolinium enhancement, which, when observed, requires a brain biopsy to distinguish Rasmussen encephalitis from an alternative diagnosis as unihemispheric vasculitis (19; 17).
MRI atrophy of the contralateral hemisphere has been described occasionally (40), but a study using voxel‐based morphometry demonstrated that hemispheric gray matter volumes declined over time in the affected and unaffected hemispheres (59). Another MRI voxel‐based morphometry study in 21 patients with Rasmussen encephalitis showed that compared to controls, the contralateral hemisphere of the patients revealed an increased cortical volume but a reduced subcortical gray matter volume (18). The authors hypothesize that the comparatively increased cortical volume in the contralateral hemisphere could be a result of compensatory structural remodeling in response to atrophy of the ipsilateral hemisphere (18). Another MRI morphometric study found that cortical atrophy was accompanied by ipsilateral atrophy of the nucleus accumbens, caudate nucleus, putamen, thalamus, and contralateral atrophy of the nucleus accumbens and caudate nucleus, but in patients with disease onset greater than 6 years, there was a contralateral volume increase of the amygdala, hippocampus, pallidum, and thalamus (06). The same group of investigators, using MRI diffusion-tensor images, showed that white matter microstructural changes occurred in both hemispheres and correlated with disease duration (31). Contralesional epileptiform activity was persistent postoperatively in 6% to 50% of cases in another study, reducing the probability of postoperative seizure freedom and, thus, questioning the concept of strict unilaterality of Rasmussen encephalitis (07).
Bilateral Rasmussen syndrome is extremely uncommon. Different theories have been proposed, like Wallerian degeneration or early treatment with immunotherapy, to explain spread to the other hemisphere (11). Movement disorders such as dystonias or chorea have been reported in cases where atrophy of the basal ganglia is seen on imaging. Two cases have been described where atrophy and hemiparesis preceded the onset of seizure activity by several months. Bihemispheric disease, basal ganglia, and brainstem variants also exist, though rare (20).
Atrophy in Rasmussen syndrome also involves the cerebellum and brainstem. An MRI morphometric study in 57 patients with Rasmussen syndrome showed bilateral cerebellar and mesencephalic atrophy predominantly ipsilateral to the affected hemisphere (55). The interpretation was that the contralateral cerebellar damage was due to crossed cerebellar atrophy, whereas the ipsilateral atrophy was due to the inflammatory changes (55).
Rasmussen syndrome is a progressive disease that results in fixed hemiparesis and intellectual decline. Immunotherapies may play a role in the rate of progression, leading to preserved motor function for a period (39). However, inevitably, surgical treatment is needed to manage refractory seizures as well as to improve motor deficits. The timing of surgery has been a challenge with the advent of medical treatment. However, a shorter preoperative seizure duration is a significant independent predictor of long-term seizure freedom after hemispherectomy surgery (42; 44).
Hemispherectomy has been the only method of treatment to improve seizure frequency and stabilize motor deficits, though the risk of producing severe deficits exists. Long-term follow-up of patients treated with hemispherectomy, for the most part, have shown positive outcomes with 79% of patients achieving seizure freedom (39; 42; 44).
In addition, before developing complete hemiparesis, some patients present frequent episodes of convulsive status epilepticus with high morbidity and mortality risk (11; 20), which is a potential indication for functional hemispherectomy before the development of hemiparesis.
A 31-year-old woman was left-handed, with normal birth and development. She had her first unprovoked generalized tonic-clonic seizure at 8 years of age. CT, MRI, and lumbar puncture done at that time were normal. An EEG revealed sharp transients in the left central region.
She continued to have residual twitching of her right foot that she described as a sensation of her foot falling asleep. This progressed to involve the entire right side of her body except her face. Despite multiple medication trials, her seizures became continuous and progressed to epilepsia partialis continua. Her language and understanding were intact throughout the initial years of her disease. Sometime after the onset of epilepsia partialis continua, she began to have intellectual decline, requiring additional help in school. Multiple EEGs revealed theta activity with continuous high-voltage spike-wave discharges over the left hemisphere. Repeat MRI scans demonstrated progressive atrophy of the left hemisphere.
Four years after the onset of epilepsia partialis continua, mild right hemiparesis developed and continued to worsen with time. She developed a typical hemiparetic gait and progressive weakness of her right arm but had relatively intact language function. Roughly 16 years after her initial presentation, epilepsia partialis continua progressed to involve facial twitching, resulting in deviation of her mouth to the right and a gaze deviation. She lost the ability to verbalize at this time and remained mute for a few months. Her speech gradually returned, but she was not able to speak in complete sentences.
Throughout the course of her illness, she was admitted multiple times to a tertiary care center. She was treated with steroids, plasmapheresis, and IVIg. High doses of intravenous steroids over weeks seemed to provide temporary relief of symptoms. Plasmapheresis was of questionable benefit, and IVIg was of no benefit.
She and her parents were resistant to hemispherectomy despite counseling for years until she was 24 years old. At the age of 26, she finally underwent a functional hemispherectomy. A preoperative left-sided intracarotid Amytal test showed that most of her language had shifted to the right. She underwent hemispherectomy without any deterioration of her motor or language function. Pathology revealed evidence of chronic inflammation, gliosis, and perivascular lymphocytic cuffs, typical of Rasmussen syndrome.
She has remained seizure-free for 7 years. She has been weaned from five antiseizure medications to just one. In addition, her speech output and ability, as well as her mobility, have increased. She now volunteers at a local library and for Meals on Wheels.
The etiology of Rasmussen syndrome remains a mystery (38). Pathological findings of an inflammatory reaction have led to studies focusing on virology. Early studies to detect a viral etiology as the pathogenesis of Rasmussen syndrome have been unsuccessful (05). Other studies have implicated cytomegalovirus and herpes simplex virus, as some patients demonstrated positive cytomegalovirus polymerase chain reaction, but results remain unsatisfying (34). In some patients with Rasmussen syndrome, polymerase chain reaction for cytomegalovirus has been positive but not distributed in a particular way in the brain, and the virus could not be cultured from affected tissue (46).
Studies have hypothesized a T-cell mediated response as a mechanism for the inflammatory response seen in Rasmussen syndrome (62). CD8+ lymphocytes were found in tissue samples of patients with encephalitis close to neurons (60; 49; 35; 66; 74). They release granzyme B, a protease that plays a role in cell apoptosis. However, an etiology that sets off this cascade has not been identified (09). One study using spectratyping of brain tissue and peripheral blood mononuclear cells from patients with Rasmussen syndrome gave support for antigen-driven, MHC class-I restricted, CD8+ T-cell mediated neuronal/astrocytic damage as one of the main pathogenic mechanisms in this form of encephalitis (60). Results for one study support the hypothesis of an antigen-specific attack of peripherally expanded CD8+ lymphocytes against brain structures in Rasmussen encephalitis (62). However, the antigen that triggers the CD8+ T cell response remains unknown.
The role of autoantibodies to the glutamate receptor 3 subunit (GluR3) in the pathogenesis of Rasmussen syndrome was proposed when it was found that rabbits immunized with GluR3 protein developed symptoms similar to Rasmussen syndrome and high titers of antibodies to GluR3 (57). These antibodies were also found in the sera of some patients with Rasmussen syndrome, but not all. One patient treated with plasmapheresis to remove GluR3 antibodies showed brief improvement in symptoms that correlated with decreasing titers during treatment. Subsequent studies have failed to detect antibodies in a larger sample of Rasmussen syndrome patients on a consistent basis and have found the presence of antibodies in other neurologic diseases and noninflammatory epilepsies (73; 72). Therefore, the detection of GluR3 antibodies is not specific for Rasmussen syndrome. It is likely that pathological mechanisms in Rasmussen encephalitis involve a combination of antibody-mediated degeneration, T-cell cytotoxicity, and microglia-induced degeneration (60; 50; 69).
One study showed that the cellular immune response in Rasmussen encephalitis involves both classical αβ T cells and non-classical γδ T cells (49). In addition, the authors found identical δ1 subtype clones in brain samples with focal cortical dysplasia, implying a common inflammatory pathway in both Rasmussen encephalitis and focal cortical dysplasia. In line with this finding, a retrospective review of resection specimens from 31 patients with Rasmussen encephalitis found that 74% had focal cortical dysplasia type Ib (25). Interestingly, in another study using tissue samples from controls, Rasmussen encephalitis and focal cortical dysplasia showed aberrant activation of mTORC1 and mTORC2 pathways in patients with Rasmussen encephalitis (53).
A study using whole exome sequencing, RNAseq, and proteomics in brain tissue of patients and autopsy controls identified activated immune signaling pathways and immune cell type annotation enrichment, suggesting innate and adaptive immune responses, as well as HLA variants that may increase vulnerability to Rasmussen syndrome (41).
Genetic studies suggest an abnormal adaptive immunity, probably associated with polygenic single-nucleotide polymorphisms, which predisposes to Rasmussen syndrome (02; 64). However, these findings need further confirmation. Experimental mouse models of Rasmussen syndrome may help to shed some light on the pathogenesis of this still obscure disease (35).
Studies and signs in isolation will not lead to the diagnosis of Rasmussen syndrome; however, when analyzed together, a diagnosis can be made. As seizure activity is the initial manifestation of the disease, certain findings are typical. EEG recording reveals abnormal background activity displayed by polymorphic delta activity. Usually, findings are asymmetrical and unilateral with low voltage. Interictal discharges are either localized over one focus, lateralized over one hemisphere with multiple foci, or bilateral with multiple foci. Even with bilateral interictal discharges, a background asymmetry will still be present to indicate which lobe is diseased (63).
Imaging studies should be used in conjunction with other testing, as findings alone are nonspecific. Early MRI may be normal, or FLAIR/T2 hyperintensities may be seen in grey or white matter representing gliosis (20). Serial imaging reveals cortical atrophy corresponding to disease progression. Atrophy of the frontal lobe and insula correlated significantly with epilepsy duration in a series of 19 patients with Rasmussen encephalitis (71). A study analyzing 13 patients with histologically proven Rasmussen syndrome measured hemispheric ratio on MRI and found that the period of maximal brain atrophy corresponded to the acute phase of the disease (20). Early brain SPECT or PET may prove beneficial by demonstrating hypoperfusion or hypometabolism of the diseased hemisphere when imaging modalities such as MRI and CT are normal (20; 68). Bilateral cerebellar and mesencephalic atrophy predominantly ipsilateral to the affected hemisphere is present in later stages of the disease (55).
Specific histopathologic changes are seen on brain biopsy, confirming the diagnosis of Rasmussen syndrome. Early in the disease process, microglial nodules, perivascular round cells, and glial scarring are seen. There may be associated neuronophagia. As the disease progresses, nodules decrease, but there is increasing evidence of cuffs of perivascular lymphocytic round cells, neuronal loss, and gliosis (11). Perivascular round-cell cuffs are seen with subarachnoid inflammation as well. Spongiosis is less extensive than in spongiform encephalopathies. Cortical destruction with few areas of active inflammation suggests the disease has reached a “burnt-out” stage (56).
Interestingly, a study used MRI diffusion tensor imaging analysis of the perivascular spaces as a surrogate marker of the glymphatic system function in vivo in patients with Rasmussen encephalitis (23). The authors concluded that their findings indicate that impaired perivascular clearance is correlated with altered cortical excitability and focal atrophy in the affected hemisphere of patients with Rasmussen encephalitis. Whether these findings are directly related to the classical perivascular round cell cuffs in histopathology remains to be determined.
There are no epidemiological studies about Rasmussen encephalitis.
No preventive measures have been identified.
Due to the implications that come with the diagnosis of Rasmussen syndrome, accurate identification is necessary. Lesions causing focal seizures or even epilepsia partialis continua (eg, cortical dysplasia, tuberous sclerosis, or tumors) can be evaluated for by imaging. Though epilepsia partialis continua favors the diagnosis of Rasmussen syndrome, other syndromes can be diagnosed by certain distinguishing features.
Tuberous sclerosis presents as early-onset seizure activity, often accompanied by intellectual disability. The cutaneous manifestations, such as shagreen patches and adenoma sebaceum, may appear later, confusing the picture. Sturge-Weber syndrome consists of a vascular nevus in the distribution of the ophthalmic division of the trigeminal nerve, with the onset of unilateral seizure activity with spastic hemiparesis.
Metabolic disorders such as MELAS can be verified with genetic testing, and the imaging changes are distinct from those in Rasmussen syndrome (11).
Autoimmune encephalitis may present with clinical features resembling Rasmussen encephalitis (04; 01). Therefore, evaluations for autoimmune etiologies should be considered in patients with initial manifestations of Rasmussen encephalitis.
The association of Rasmussen encephalitis with other comorbid autoimmune diseases raises the possibility of shared mechanisms of susceptibility, including common immunogenetic or environmental risk factors, or both (03). Studies have suggested an association with Parry-Romberg syndrome, in which hemiatrophy could result from genetic mosaicism (61; 16; 43; 74). One hypothesis is that genetic mosaicism in patients with Rasmussen encephalitis could lead to differential expression of an antigen in only one hemisphere, thus explaining the unilateral inflammation and progressive atrophy (74).
The diagnosis of Rasmussen syndrome is based on clinical presentation, EEG, MRI, and histopathology. The challenge is recognizing early cases where motor deficits may not be present yet. Clinically, increasing seizure activity with progressive cortical deficits is characteristic. The absence of a structural lesion on MRI will help rule out a secondary cause of focal seizures or weakness. EEG, especially early in the disease process, will show unilateral focal slowing in the delta range. MRI within at least 4 months of onset of symptoms will reveal focal cortical atrophy, white matter hyperintensity, and caudate head atrophy. A hyperintense signal in T2-weighted and FLAIR images in cortical and subcortical regions is present in most patients in the early stages of the disease and in practically all patients in more advanced stages. The hyperintense signal and atrophy may be heterogeneous, but they most frequently affect the perisylvian region. Atrophy of the head of the caudate is very frequent, even in early stages. Serial imaging reveals progressive atrophy. FDG-PET demonstrates hemispheric or diffuse hypometabolism even in early stages of Rasmussen encephalitis, even when MRI shows mild changes or is considered normal (69). Brain biopsy is indicated if suspicion for the diagnosis persists after review of the clinical presentation, EEG, and MRI. As mentioned earlier, biopsy will reveal perivascular cuffing, microglial nodules, and gliosis as evidence of chronic inflammation. One study reviewed 12 patients with Rasmussen syndrome to identify early manifestations of this disease in efforts to help solidify early diagnosis and treatment and reduce the need for brain biopsy. Three main features were noted 4 to 6 months after symptom onset: refractory focal motor seizures with rapidly increasing seizure frequency; slow focal EEG activity with progressive unihemispheric background flattening; and focal cortical atrophy, white matter hyperintensity, and caudate head atrophy on MRI (26).
The medical therapies studied in Rasmussen syndrome are based on the presumed immune-mediated pathogenesis of the disease. Antiepileptic medications are prescribed based on semiology with the onset of seizure activity.
Immunosuppressive treatments, such as intravenous gammaglobulin (IVIg), high-dose steroids, tacrolimus, and alpha interferon, are some of the agents that have been used for treatment once Rasmussen syndrome is suspected or confirmed (45; 30; 10; 69).
Experience over the years indicates that immunotherapy may be recommended in the early stages of the disease, in patients with slow disease progression, and for patients not eligible or who refuse surgery (39; 33; 58). No single agent or combination of agents aborts disease progression, although seizures may be controlled temporarily by immunotherapy, in particular with intravenous gammaglobulin (24).
A study of 19 patients using IVIg, steroids, or both recommended using IVIg as first-line treatment due to fewer side effects (30). Patients treated with IVIg had differing results, ranging from ineffectiveness to a short-lived response to stabilization of the disease. Caraballo and colleagues used a combination of corticosteroids and IVIg in 29 patients, with 14 patients (48%) improving seizure control with a transient cessation of neurologic deterioration (15).
High-dose steroids have also been shown to have a positive effect on seizure control, though, similar to IVIg, results vary among patients (39). Corticosteroids were effective in reducing seizures in 70% of patients (33 of 47) in one study (52). The major limiting factors for long-term therapy are the side effects, such as the development of Cushing syndrome, osteoporosis, infections, and psychosis.
One retrospective study compared outcomes in 30 patients with Rasmussen syndrome who received immunomodulation with azathioprine with 23 patients who were not treated with this drug (52). They showed that azathioprine was a safe alternative to long-term corticosteroid therapy, resulting in better seizure control and delaying the onset of epilepsia partialis continua and hemiparesis, but it did not avoid the progression of hemispheric atrophy and cognitive decline (52).
Tacrolimus has been studied as a treatment modality as well. It is an immunosuppressant traditionally used in transplant patients to prevent rejection. Interestingly, the rate of cerebral atrophy and neurologic decline was slower in treated patients, but no change in seizure frequency was observed (10).
A randomized trial showed that both tacrolimus and IVIg were protective against functional and structural brain damage compared to historical controls. However, this study was underpowered for defining the superiority of each of these two treatments (12).
Overall, plasmapheresis has low responder rates in the long term (39). However, plasmapheresis may be used as an adjunct to other treatment modalities or when other first-line treatments (corticosteroids, IgIV) fail (39).
There have also been a few historical reports on the use of intraventricular interferon alpha in Rasmussen syndrome. Improvements in neurologic decline and epilepsy during the course of treatment with high-dose interferon alpha followed by a regimen of increasing doses over a period of weeks were seen (45). However, epilepsia partialis continua returned, and a second round of treatment was needed.
In conclusion, these immunomodulatory therapies help temporarily reduce the seizure burden of patients with Rasmussen encephalitis and delay, but not stop, the progression of hemiparesis and cognitive decline.
Despite multiple medical trials, only hemispherectomy has been shown to have a definite effect in controlling the disease process of Rasmussen syndrome (39; 65). There is controversy over when to perform surgery. If a fixed hemiparesis is present, the risk of further postoperative motor deficits is minimal, whereas there is a chance of acute worsening due to the operation itself if paresis is minimal (70). Some degree of compensation to the other hemisphere may occur by the time deficits are maximal if surgery is delayed; however, early surgical intervention may allow for better seizure control and neurologic development. Functional hemispherectomy is the preferred surgical approach. It removes a portion of the hemisphere and divides the corpus callosum to “disconnect” the right and left hemispheres.
Long-term outcomes of hemispherectomy or hemispherotomy for intractable epilepsy are particularly good, with 65% to 81% of patients remaining seizure-free (36; 27). Of the 105 children reviewed, 46 had Rasmussen syndrome, and 30 were seizure-free after surgery. A portion of children who remained with seizures felt their quality of life was not affected by the seizure semiology. Five patients who had functional hemispherectomy as treatment of Rasmussen syndrome were still seizure-free at a mean follow-up period of 15.6 years and able to carry out activities of daily living (67).
In a series of 13 consecutive cases with a mean age of 10.6 years (range 5 to 18 years), based on the preoperative duration of seizures, the authors found no difference in outcome, with 63% of patients seizure-free (36% off medication), 100% with improved seizure control, and 90% with improved cognitive function (32). In addition, there was an 83% improvement in language in patients with disease affecting the dominant hemisphere. They suggested that hemispherotomy, even in the dominant hemisphere, can be offered early or late, with good seizure control and functional outcome (32).
Rasmussen syndrome is a progressive disease that results in fixed hemiparesis and intellectual decline. Immunotherapies may affect the rate of progression, leading to preserved motor function for a period. However, inevitably, surgical treatment is needed to manage refractory seizures as well as to improve motor deficits. The timing of surgery has been a challenge with the advent of medical treatment. Hemispherectomy has been the only method of treatment to improve seizure frequency and stabilize motor deficits, though the risk of producing severe deficits exists. Long-term follow-up of patients treated with hemispherectomy, for the most part, has shown positive outcomes.
Seizure outcome after hemispherectomy depends more on surgical technique with complete disconnection than on duration of the disease and degree of histopathological changes (14; 65). However, one study showed that preoperative generalized seizures, contralateral MRI abnormalities, and contralateral EEG discharges were associated with worse postoperative outcomes, whereas early surgery and preoperative immunotherapy were associated with better outcomes (42).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Fernando Cendes MD PhD
Dr. Cendes of the University of Campinas - UNICAMP has no relevant financial relationships to disclose.
See Profile
Jerome Engel Jr MD PhD
Dr. Engel of the David Geffen School of Medicine at the University of California, Los Angeles, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026