
Neuroimmunology
Balo concentric sclerosis
Jul. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Vogt-Koyanagi-Harada syndrome is an inflammatory condition in which uveitis is the most significant feature of the illness. Neurologists may be called on to diagnose the prodromal phase of the disease in which the predominant symptoms are headache, meningeal signs, and, less often, focal neurologic findings. CSF pleocytosis is also common. In this update, the authors cite data demonstrating that earlier initiation of either glucocorticoids or immunomodulatory measures is associated with a reduction in late visual morbidity and that the neurologic manifestations can include cerebral ischemia.
|
• Vogt-Koyanagi-Harada syndrome is primarily an ocular inflammatory disease, but the prodrome often includes signs and symptoms that bring the patient to a neurologist, including headache, meningeal signs, and CSF pleocytosis. | |
|
• The major targeted tissues are melanin pigment-containing cells, including the retinal pigment epithelium, producing chorioretinitis (uveitis); cutaneous melanocytes, producing vitiligo and poliosis; and the organ of Corti, producing sensorineural hearing loss. | |
|
• The disease is encountered more frequently in persons of Asian, Latin, and Mediterranean descent. |
Vogt-Koyanagi-Harada syndrome is a condition in which targeted pigmentary alterations in various tissues are associated with ocular, cutaneous, aural, and meningeal inflammation. The combination of poliosis (the graying, or whitening, of a shock of scalp hairs, from the Greek for "gray"), and ocular inflammation was noted by the Arab physician Ali Ibn Isa (Ali ben Issa), who died in 1010 (169; 134), and by others including Vogt (219). Both Vogt and Koyanagi described cases that included a combination of bilateral iridocyclitis and non-ocular manifestations, including pigment loss from skin (vitiligo), hair, and lashes (poliosis); hair loss (alopecia); and hearing loss with tinnitus (219; 107). Koyanagi's six cases were described in 1929, but in 1926, Harada had described similar non-ocular manifestations together with posterior uveitis, characterized by exudative retinal detachments, and pleocytosis of the cerebrospinal fluid (66). Moorthy and colleagues credit Babel (23) and Bruno and McPherson (28) with recognizing the unity of the cases described by Vogt, Koyanagi, and Harada (134). Herbort and Mochizuki reviewed the historical development of the syndrome, emphasizing that both Koyanagi and Harada perceived a unity of cases with seemingly disparate multisystem signs and symptoms, whereas Vogt had elegantly described the first case (70).
Most authors agree that the disease can be divided into four stages: (1) prodromal, (2) uveitic, (3) convalescent, and (4) chronic recurrent (126; 134). Although the disease-defining symptoms and signs are ophthalmic, they are rare in the prodromal phase, during which time neurologic symptoms predominate. Therefore, patients may initially present for neurologic care.
Prodromal phase. Central nervous system signs and symptoms are most common during the prodromal phase, which lasts a few days. Symptoms of meningeal irritation are the most common. The patient's hair and skin may be sensitive to touch during the prodromal stage (201).
Central nervous system signs and symptoms may be severe, including alteration of cognitive function, confusional state, and psychosis. Hemiparesis, nonfocal weakness, and fifth, sixth, or seventh cranial nerve palsy are uncommon manifestations (169). Seizures and coma may occur rarely in the early stages (36; 103). Among 87 patients with Vogt-Koyanagi-Harada syndrome in South India, 64% had nonocular symptoms at presentation, and of these, 96% had meningismus (133). Headache lasting several months without other ocular or neurologic findings or symptoms may be a prodromal symptom (106).
CSF pleocytosis is more common than symptomatic meningitis, recorded in the majority of patients who underwent lumbar puncture in several studies (157; 127; 81).
CSF total protein is often normal or mildly elevated, and glucose is usually normal (127; 81). The presence of melanin-laden macrophages in CSF is distinctive (99).
Because CSF pleocytosis can be present in patients with minimal or no neurologic findings, MRI manifestations of meningeal inflammation may provide a sensitive and noninvasive indicator for early diagnosis (65; 123).
Optic nerve swelling has been reported (169), but it more likely to be a sign of optic neuritis than papilledema, as opening pressure is usually normal or only mildly elevated (81; 56).
Transient dizziness and nystagmus observed in about 60% of patients is attributed to labyrinthitis (158), owing to inflammation against inner ear melanocytes (101).
Rare associations with Vogt-Koyanagi-Harada syndrome include ischemic stroke, myelopathy with enhancing lesions in the cervical cord (62), and Guillain-Barré syndrome. These manifestations appear within weeks to months of the initial symptoms (143).
Uveitic phase. Ophthalmic manifestations become apparent during the uveitic phase. The most common symptom is blurred vision, mostly from opacification of the cornea, crystalline lens, or vitreous humor. Reduction of visual acuity may also result from serous detachment of the macula. In about a third of cases, the disease starts symptomatically in one eye, but the second eye becomes involved within days. In a study of 16 patients, all had decreased visual acuity, abnormal color vision, and abnormal visual fields at the initial visit (233).
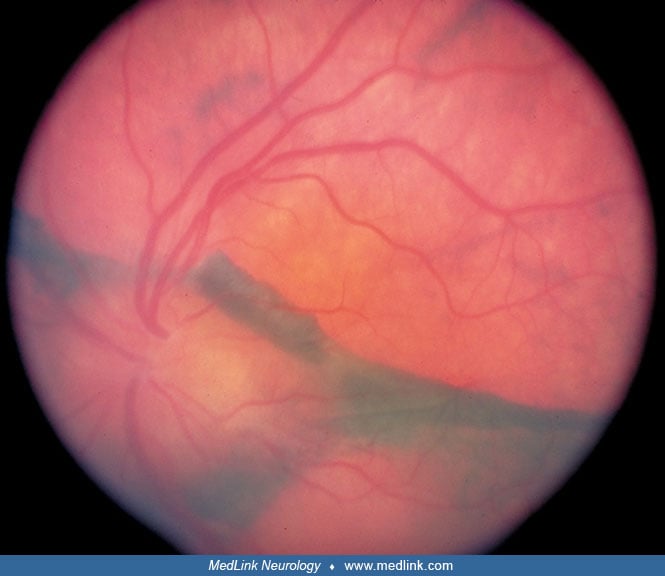
On ophthalmologic examination, a bilateral posterior uveitis is evident, with optic disc, retinal edema, and multifocal serous retinal detachments. The retinal separation occurs between the retinal pigment epithelium and the underlying choroid (97). Non-necrotizing granulomas consisting of epithelioid histiocytes (Dalen-Fuchs nodules) accumulate underneath the retinal pigment epithelium. Choroidal detachment has also been reported (170).
Inflammation eventually spreads to involve the anterior ocular segment and is manifest by inflammatory cells and flare in the aqueous humor. White-to-yellow greasy inflammatory precipitates (“mutton fat keratoprecipitates”) may be seen on the corneal endothelial surface. Inflammatory nodules may similarly accumulate at the pupil margin (Koeppe nodules) or on the iris surface (Busacca nodules). These are best seen on slit lamp examination. Adhesions between the iris and anterior lens surface (“posterior synechiae”) can rise to an irregular or fixed pupil. Patients with anterior segment inflammation may experience decreased visual acuity and significant photophobia.
Although predominantly inflammatory, the optic disc edema may be due to elevated intracranial pressure or ischemia. Visual field defects may be persistent (146; 145).
Acutely elevated intraocular pressure can occur and is characterized by intense ocular pain, perilimbal conjunctival injection (“ciliary flush”), a mid-dilated pupil, and corneal edema, constituting a medical emergency, warranting prompt referral to an ophthalmologist or emergency department. The elevated intraocular pressure is attributed to plugging of the trabecular meshwork by inflammatory cells, thereby reducing aqueous outflow. Alternatively, the posterior synechiae may impede the flow of aqueous between the ciliary body and cause the iris to bow forward (“iris bombe”) and lead to angle closure (142). Finally, neovascularization of the anterior chamber angle can impede aqueous outflow (72). Long-term use of topical and systemic steroid medications increases the risk of steroid-induced ocular hypertension in the subgroup of patients who are “steroid responders.” The elevated intraocular pressure (glaucoma) may persist beyond the resolution of the uveitic phase. Uveal effusion (abnormal fluid distention of the choroid), younger age, and an increased number of clinical recurrences of ocular inflammation are risk factors for glaucoma (164; 01).
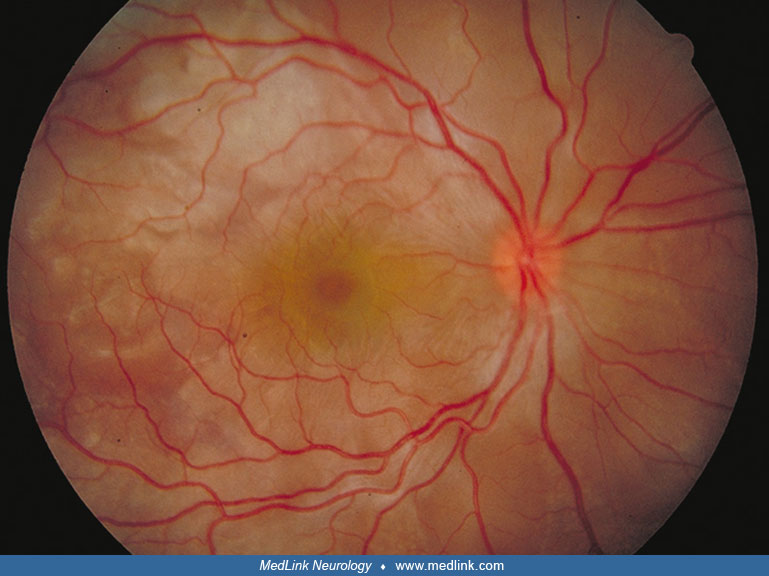
Convalescent phase. This phase lasts several months and is characterized by depigmentation in the skin and choroid. Loss of pigment in the choroid results in a shift of the background fundus color from the usual dusky red to a bright orange, which has been called the "sunset glow fundus." There is often clumping of the retinal pigment in the pigment epithelial layer, which results in dense pigmented accumulations alternating with lighter areas throughout the posterior pole of the fundus. Depigmentation of the skin (vitiligo) and of the hair on the eyebrows, eyelashes, and scalp (poliosis) occur. The vitiligo affects the head, trunk, and sacrum. Cutaneous granulomas have also been reported (214).
Many ophthalmic symptoms improve during the convalescent phase. Visual acuity and color vision may improve as early as 1 to 3 months after presentation, but visual field defects may persist for months or forever. In one study, 30% of patients retained visual field impairment 12 months after treatment onset (233). Patients who develop sunset glow fundus may have long-term reduction in contrast sensitivity (117).
Chronic recurrent phase. After several months of convalescence, a chronic recurrent phase may set in, manifested by low-grade panuveitis punctuated by acute episodes of predominately anterior uveitis. Choroidal or other uveal neovascularization may arise, leading to edema, hemorrhage, or serous retinal detachment. These phenomena may lead to vision loss, macular scarring, and pigmented perivascular atrophy (178).
The presence of DRB1*0405, DRB1*0410, or both, is associated with a more protracted clinical course (83; 84). Patients with HLA-DQA1*0301 and HLA-DQB1*0401 markers are more susceptible to Vogt-Koyanagi-Harada syndrome; HLA-DQA1*0103, 0401, 0501; and HLA-DQB1*0301, 0402, 0601, 0603 may be protective (121).
Early treatment may improve prognosis, decreasing the number of uveitic flares and the risk of subretinal fibrosis (188). In the face of a poor response to corticosteroid therapy, early initiation of immunomodulatory therapy may improve visual outcome (218).
In a study of 23 patients, older age and presence of non-ocular findings were suggestive of higher rate of recurrence (96).
Complications. The neurologic signs and symptoms are most often limited to the prodromal phase of the disease, when cognitive dysfunction and headache characteristically occur.
Ocular complications include cataract (33% to 42%), glaucoma (24% to 27%), posterior synechiae, choroidal neovascular membrane (11%), subretinal scarring (6%), and optic neuropathy (134; 133; 184; 15).
Increased axial length leading to myopia progression has been described in the Japanese population, occurring as early as 1 month after onset and persisting at 6 months (206; 140).
A 16-year-old Hispanic boy developed new headaches for 2 weeks prior to admission. He also noted blurred vision in his right eye and described images as looking distorted and enlarged when viewed with his right eye. On the day of admission, he was confused and disoriented.
Neurologic examination disclosed mental confusion and mild disorientation in time, but no focal findings. There was moderate nuchal rigidity, and Kernig and Brudzinski signs were present. Visual acuity was 20/60 in the right eye and 20/20 in the left eye. There was no relative afferent pupillary defect. Fundus examination revealed serous elevation of the macula in the right eye and optic disc swelling in both eyes.
MRI showed mild patchy enhancement of the meninges, but no focal signal abnormalities or ventriculomegaly.
Lumbar puncture yielded clear cerebrospinal fluid with a normal opening pressure. There were 60 white blood cells/mm3, of which 95% were lymphocytes and 5% were monocytes. Protein was 56 mg/dl and glucose 40 mg/dl (serum glucose 120 mg/dl). Cytology was negative for malignant or atypical cells. Protein electrophoresis revealed mild elevation of gamma globulin.
He was initially treated with intravenous methylprednisolone 1 gram/day for 3 days, followed by oral prednisone at an initial dose of 60 mg/day, followed by a tapering schedule over the next 3 weeks. His mental status cleared rapidly, and he was discharged to his home after 5 days.
At the first office follow-up visit 2 weeks after discharge, the right eye visual acuity had improved to 20/25 and the symptom of blur and metamorphopsia had resolved. On fundus examination, the optic disc swelling had resolved in both eyes, as had the subretinal fluid in the macula of the right eye. At 3 months after presentation, with corticosteroids discontinued, vision and neurologic status remained normal. The fundus exam showed patches of orange discoloration in the posterior pole and some broad bands of pigment clumping and dark discoloration at the level of the retinal pigment epithelium.
Vogt-Koyanagi-Harada syndrome represents a cell-mediated autoimmune response against melanocytes. Histopathologically, inflammation and loss of melanin-containing cells in the skin and the uvea of the eye have been observed. Melanocytes exist in the meninges and in the inner ear; however, the pathogenesis of hearing loss is unresolved.
The histopathology of Vogt-Koyanagi-Harada syndrome has been sparsely documented. There is granulomatous or non-granulomatous choroidal infiltration, with epithelioid cells that contain cytoplasmic melanin. The choriocapillaris is typically spared (171; 134). Because the available pathologic material comes from enucleated eyes, the only pathology in the central nervous system is lymphocytic infiltration in the leptomeninges along the amputated stump of the optic nerve (171).
Cellular immune mechanisms are probably more important than humoral ones (134). Antibodies to retinal antigens have been detected, but whether they are epiphenomena is uncertain (30).
Melanocytes are likely involved in the meningitic component. Peripheral and cerebrospinal fluid lymphocytes from patients with Vogt-Koyanagi-Harada syndrome exhibit cytotoxicity against melanoma cells and normal melanocytes (128; 154), as have lymphocytes derived from intraocular aqueous humor (155). Macrophages containing melanin granules have been found in the CSF of patients with Vogt-Koyanagi-Harada syndrome but not in patients with other meningeal inflammatory diseases (144).
In one study, T-cells constituted the majority of lymphocytes in aqueous humor; they were more activated than were peripheral blood lymphocytes. Of the lymphokines studied, interleukin-6 appeared to play an important role as an inflammatory mediator (155). Studies of enucleated eyes from a patient with active Vogt-Koyanagi-Harada syndrome showed similar distribution of T-cell subsets in the inflammatory lesion within the uveal tract (94). Studies have also shown that the ratio of T-helper to T-suppressor cells is increased, and that cells expressing surface markers characteristic of early (CD25) and late (CD26) lymphocyte activation are present in the inflammatory lesion. Also, a class 2 major histocompatibility complex was expressed in the choroidal melanocytes, and in the endothelium of the choriocapillaris, indicating a probable primary role for the melanocyte in the pathogenesis of the lesion (187).
The antigens MART-1/Melan-A (200), peptidase (104), P-36 (153), tyrosinase (230; 60), IgG anti-lens epithelium-derived growth factor (229), and KU-MEL-1 (161) have also been implicated.
In a study of T-helper cells from nine patients with Vogt-Koyanagi-Harada disease versus nine controls, peripheral blood mononuclear cells from patients produced significantly higher levels of mRNA only for interferon-gamma and not for interleukin-2 or interleukin-4 (79). Levels of interferon-gamma and interleukin-2 were significantly increased in the stimulated cell culture supernatant of patients as compared with controls. In a study of 128 patients, IgE levels were correlated with retinal disease severity (89).
One study described three patients who developed Vogt-Koyanagi-Harada syndrome shortly after skin injuries (180). All three patients developed vitiligo in the injured areas as the skin healed, and the uveitis began at the same time. The authors suggest that the onset of Vogt-Koyanagi-Harada syndrome following skin injury supports the idea that the disease involves systemic sensitization to shared melanocytic antigens (180). Another study described a 68-year-old Chinese woman who developed uveitis followed by generalized cutaneous inflammation (erythroderma) 6 months later and by vitiligo 6 months after that. This is the first report of cutaneous inflammation preceding vitiligo (226).
A Vogt-Koyanagi-Harada–like syndrome has been described in patients with metastatic melanoma following initiation of checkpoint inhibition or protein kinase inhibition therapy (58; 242; 37; 27).
A similar syndrome has also been described following vaccination (43; 195; 138), suggesting an exuberant immune reaction as a possible trigger. During the 2020 COVID-19 pandemic, there were several reports of a Vogt-Koyanagi-Harada–like syndrome associated with COVID-19 vaccination (105; 09; 40; 93; 100) and infection (241). One case report described a relapse of VKH following COVID-19 vaccination (141).
Investigators have identified a subset of pathologically activated monocytes with gene expression indicative of inflammation, antiviral activity, and pathologic activation (74). Peripheral blood mononuclear cells in Vogt-Koyanagi-Harada syndrome have also been shown to express unique RNA profiles (63). Genome-wide association analysis has also identified new loci associated with Vogt-Koyanagi-Harada syndrome risk (176). Studies have identified a set of hub genes that may contribute (223). Proteomic profiling demonstrated increased levels of carbonic anhydrase 2 and Ras-related protein Rap-1b (247) and C1QB (246) in plasma exosomes.
Studies have shown differences in gut microbiota in patients with Vogt-Koyanagi-Harada syndrome compared to healthy controls and other noninfectious anterior uveitides (114; 113; 222; 117).
In a case report, Maehira and colleagues observed different severities of inflammation between the eyes of a single patient with significantly different axial lengths (125).
Using a retinal pigment epithelium model derived from Vogt-Koyanagi-Harada peripheral mononuclear cells, one study observed barrier and phagocytic dysfunction as well as depigmentation compared to normal controls, suggesting dysfunction at the level of the retinal pigment epithelium (115). In the same study, treatment with a molecule targeting early growth response gene 2 restored barrier and phagocytic function in the pathological retinal pigment epithelium cells, suggesting a pathological role and a possible target for future pharmaceutical treatment. Patients with Vogt-Koyanagi-Harada syndrome were also shown to harbor a mutation in membrane palmitoylated protein 2 (MPP2), associated with cytokine and cellular barrier dysfunction (122). Possible contributory polymorphisms in PRKCD, CARD9 (249), ZC3HAV1 (227), OR11H1 (116), and LRRC30 dysregulation (245) have also been identified. Bioinformatics analysis has identified interferon gamma and interleukin 6 as key mediators of Vogt-Koyanagi-Harada syndrome and as possible targets for future therapeutics (33).
Vogt-Koyanagi-Harada syndrome is most common among more heavily pigmented populations including Asians, Hispanics, Native Americans, and Indians. In a series of 65 cases from southern California, 78% were Hispanic, 10% Asian, 6% of African descent, and 3% of European ancestry (134). The racial breakdown of patients seen at the National Institutes of Health was 13% Hispanic, 50% Caucasian (European), and 35% of African descent. Among the Caucasians, the majority were of Native American ancestry (156). Native Americans with Vogt-Koyanagi-Harada syndrome in Oklahoma were more likely to be younger and have fewer neurologic or systemic symptoms, but they were more likely to develop cataracts (185). In a Spanish study of 112 patients, 61.6% were Caucasian and 30.4% were Hispanic (22).
In Japan, the annual incidence was estimated to be 6.5 per million and the prevalence was 15.5 per million. Females were slightly more commonly affected than males, and the peak age at presentation had a median age of 42.3 years (137).
In Asian countries, Vogt-Koyanagi-Harada disease represented the largest proportion of panuveitis cases (220; 25). On the other hand, in Saudi Arabia, only 2.5% of 200 patients with uveitis had Vogt-Koyanagi-Harada syndrome (85).
A small retrospective study in Puerto Rico suggested that the incidence of Vogt-Koyanagi-Harada syndrome may follow a seasonal pattern, with 50% of cases presenting in the Fall (17).
No means of prevention are known.
Sympathetic ophthalmia is an autoimmune condition that closely mimics Vogt-Koyanagi-Harada syndrome. A bilateral non-necrotizing granulomatous uveitis, it is an autoimmune response against uveal autoantigens exposed by penetrating trauma or surgery. Non-ocular manifestations are far fewer in sympathetic ophthalmia than in Vogt-Koyanagi-Harada syndrome (232). A negative history of ocular trauma or surgery makes a diagnosis of sympathetic ophthalmia unlikely.
Lymphoma can present simultaneously in the eye and in the brain as a space-occupying lesion or diffuse meningeal infiltration (102; 134; 20; 225). Subretinal deposits, as well as vitreous cells, may be seen. The vitritis may initially respond to corticosteroid therapy but will recur once treatment is stopped. Multiple serous retinal detachments have also been reported (112). Acute lymphoblastic leukemia (68) and lymphoma (136) may present as Vogt-Koyanagi-Harada syndrome.
Sarcoidosis is a granulomatous cause of panuveitis. Ocular sarcoidosis tends to produce retinal perivenous sheathing, called "candle-wax drippings," whereas the serous retinal detachment characteristic of Vogt-Koyanagi-Harada syndrome is not encountered. The neurologic findings in sarcoidosis are focal, often involving the cranial nerves, whereas headache and cognitive impairment are most common in Vogt-Koyanagi-Harada syndrome. Both conditions may have chronic CSF pleocytosis (134).
A rare, immune complex–mediated choroidal vasculitis in systemic lupus erythematosus mimics Vogt-Koyanagi-Harada syndrome by causing serous retinal detachments (86; 134). Patients often exhibit systemic manifestations of systemic lupus erythematosus.
Infectious posterior uveitis, including syphilis and tuberculosis, can present as subretinal nodules, posterior or anterior uveitis, and serous retinal detachment. Infection must be ruled out prior to starting corticosteroid or other immunomodulating therapy (120; 02; 18). Acute retinal necrosis (ARN) presents as uveitis and retinal vasculitis and is caused by herpes simplex virus or varicella zoster virus. It is diagnosed with viral PCR of aqueous or vitreous fluid (252).
Other diseases in the differential diagnosis of Vogt-Koyanagi-Harada syndrome include acute multifocal placoid pigment epitheliopathy, multiple evanescent white dot syndrome, posterior scleritis (192), Behcet disease (95), and bilateral diffuse melanocytic proliferation (39). The two features that most reliably distinguish Vogt-Koyanagi-Harada syndrome from these other forms of uveitis are exudative retinal detachment in the acute uveitic phase and sunset glow fundus in the late phases (179; 45).
Central serous retinopathy typically presents with subretinal fluid and may be recurrent. Central serous retinopathy may be distinguished from Vogt-Koyanagi-Harada syndrome by the lack of uveitis and characteristic leakage pattern on fluorescein angiography. Importantly, central serous retinopathy is typically exacerbated by systemic corticosteroid treatment (150).
A Vogt-Koyanagi-Harada-like syndrome has been described, with protein kinase inhibitors (197; 207; 216) and antineoplastic immune checkpoint inhibitors, even after their discontinuation (148; 211; 41; 78; 221; 250; 228). Of the adverse reactions reported in the WHO international pharmacovigilance database, immune checkpoint inhibitors and protein kinase inhibitors were associated with a Vogt-Koyanagi-Harada-like syndrome (21). In 2025, a consensus statement defined “immune-related Vogt-Koyanagi-Harada-like syndrome” (irVKHLS) as “classic for VKH” with concurrent use of an immune checkpoint inhibitor (31).
A case of Vogt-Koyanagi-Harada–like syndrome preceding melanoma-associated retinopathy has been reported in a patient with metastatic melanoma (148).
Vogt-Koyanagi-Harada syndrome has been described after COVID-19 infection and vaccination (149; 190; 241; 238).
Vogt-Koyanagi-Harada syndrome has been described in patients with autoimmune diseases, including ulcerative colitis (54), IgA nephropathy (244), and erosive seronegative polyarthritis (159), suggesting an association with autoimmunity. A case of Vogt-Koyanagi-Harada disease has been reported as a complication of treating hepatitis C with interferon alpha-2b and ribavirin combination therapy (203).
Vogt-Koyanagi-Harada syndrome is associated with HLA-DR4 positivity (83; 84; 194; 224; 175; 186) and HLA-DQ4 (83% vs. 32% in controls). Among variants, HLA-DQA1*0301 positivity is associated with the Vogt-Koyanagi-Harada syndrome phenotype, whereas HLA-DQB1*0604 is considered protective. The DRB1*0405 or DRB1*0410 variants portend to a longer disease course (83; 84). One epigenetic association study of 160 patients identified methylation in HLA-DRB1 and HLA-DQA1, as well as several non-HLA methylation sites (199).
Seven of 62 clones from patients with Vogt-Koyanagi-Harada disease that were reactive with tyrosinase or tyrosinase-related protein showed proliferative responses to peptides that match the motif of the strong binding site for HLADRB1*0405 (60). Weisz and colleagues determined human leukocyte antigen specificities in 25 Hispanic patients living in Southern California and compared them to 217 healthy Hispanic control subjects (224). HLA-DR4 was present in 56% of the patients and in 29% of the control subjects. HLA-DR1 was present in 36% of the patients and in 9% of the control subjects. Combinations of HLA-DR4 and HLA-DR1 characterized 84% of patients but only 35% of control subjects. These findings point to a common immunogenic predisposition to Vogt-Koyanagi-Harada syndrome among different racial groups that are differentially susceptible and that there may be a common epitope shared by DR1 and DR4 that is involved in the pathogenesis of the disease.
In studies examining BRAF-mutant melanoma patients treated with BRAF kinase inhibitors and MEK kinase inhibitors (16), or checkpoint inhibitors for other malignancies (209), the incidence of Vogt-Koyanagi-Harada-like syndrome was higher in those with HLA-DRB1*04 and HLA-DRB1*04:05, respectively.
Diagnostic criteria. Consensus criteria developed in 1978 by the American Uveitis Society were updated following the First International Workshop on Vogt-Koyanagi-Harada Disease in 1999. This newer set of criteria recognizes that Vogt-Koyanagi-Harada has distinct features in different stages of its natural history and establishes criteria for early diagnosis when many of the characteristic cutaneous findings have not yet occurred (183) (Table 1).
Complete Vogt-Koyanagi-Harada disease (criteria 1 to 5 must be present) | ||
1. No history of penetrating ocular trauma or surgery preceding the initial onset of uveitis | ||
2. No clinical or laboratory evidence suggestive of other ocular disease entities | ||
3. Bilateral ocular involvement (a or b must be met, depending on the stage of disease when the patient is examined). | ||
4. Neurologic and auditory findings (may have resolved by time of examination) | ||
- meningismus | ||
5. Integumentary finding (not preceding onset of central nervous system or ocular disease) | ||
- alopecia | ||
Early manifestations of disease | ||
• There must be evidence of a diffuse choroiditis (with or without anterior uveitis, vitreous inflammatory reaction, or optic disc hyperemia), which may manifest as one of the following: | ||
- focal areas of subretinal fluid | ||
• With equivocal fundus findings, both of the following must be present as well: | ||
- focal areas of delay in choroidal perfusion, multifocal areas of pinpoint leakage, large placoid areas of delay in choroidal perfusion, pooling within subretinal fluid, and optic nerve staining by fluorescein angiography | ||
- diffuse choroidal thickening, without evidence of posterior scleritis by ultrasonography | ||
Late manifestations of disease | ||
• History suggestive of prior presence of findings | ||
• Ocular depigmentation (either of the following manifestations is sufficient): | ||
- sunset glow fundus | ||
• Other ocular signs: | ||
- nummular chorioretinal depigmented scars | ||
Incomplete Vogt-Koyanagi-Harada disease (criteria 1 to 3 and either 4 or 5 must be present) | ||
Probable Vogt-Koyanagi-Harada disease (isolated ocular disease; criteria 1 to 3 must be present) | ||
| ||
New diagnostic criteria were proposed in 2018 using retrospective data from 1257 Chinese patients in China (234). The criteria consisted of a 3-class model grouping patients into early-phase disease, late-phase disease, or non-Vogt-Koyanagi-Harada uveitis. They compared 37 variables, 21 of which were deemed important in the diagnosis of Vogt-Koyanagi-Harada disease as they were associated with high-specificity or high-positive rates (Table 2). The Diagnostic Criteria for Vogt-Koyanagi-Harada Disease showed higher sensitivity (94.6% vs. 71.9%) and negative predictive value (94.3% vs. 76.6%) than the revised diagnostic criteria. The positive predictive value showed no difference when compared to the revised diagnostic criteria. As these criteria were developed retrospectively, they need to be evaluated in prospective studies for further investigation (234).
A. No history of penetrating ocular trauma or intraocular surgery preceding the initial onset of uveitis | ||
B. Bilateral ocular involvement (time interval between the two eyes should be 2 weeks) | ||
C. No evidence of infectious uveitis or accompanying systemic rheumatic diseases or evidence suggestive of other ocular disease entities | ||
D. Early-phase Vogt-Koyanagi-Harada disease: | ||
1. Signs of diffuse choroiditis and exudative retinal detachment | ||
2. Serous retinal detachment on optical coherence tomography or B-scan ultrasonography | ||
3. Choroidal thickening on enhanced depth imaging-optical coherence tomography (B-scan can be used where enhanced depth imaging-optical coherence tomography not available, though it is not as precise) | ||
4. Early punctate staining and late subretinal dye pooling on fluorescence fundus angiography | ||
5. Hyperfluorescence of the optic disc on fluorescence fundus angiography | ||
Definite diagnosis: | ||
Variant 1: In patients presenting with A+B+C+ D(1) | ||
Variant 2: In patients without clinically visible exudative retinal detachment, ie, A+B+C+ D(2) + D(3) or A + B + C + D(4) | ||
Variant 3: In patients already treated with systemic corticosteroids or combined with other immunosuppressive agents, a history of typical appearances of variant 1 or 2, and A+B+C+ D(5) | ||
E. Late-phase Vogt-Koyanagi-Harada disease: | ||
1. Signs of definite sunset glow fundus or retinal pigment epithelium clumping/migration | ||
2. Signs of bilateral recurrent granulomatous anterior uveitis | ||
3. Signs of Dalen-Fuchs nodules or multifocal chorioretinal atrophy | ||
4. Window defects/moth-eaten fluorescence on fluorescence fundus angiography | ||
5. Previous history of characteristic findings corresponding to diagnosis of early-phase Vogt-Koyanagi-Harada disease | ||
Definite diagnosis: | ||
Variant 1: In patients presenting with A+B+C+ E(1) + E(2) | ||
Variant 2: In patients without sunset glow fundus or visible pigment alternations due to early and appropriate treatment, ie, A+B+C+ E(2) + E(3) or A+B+C+ E(2) + E(4) | ||
Variant 3: In patients with significant media opacity, ie, A+B+C+ E(2) + E(5) | ||
Criteria are evolving to include modern ophthalmic imaging findings. Indocyanine green angiography is a diagnostic imaging procedure that examines blood flow through the choroid. The fluorescent molecule indocyanine green is infused intravenously, and a series of fluorescent photographs are taken with a specialized fundus camera. Fluorescein angiography is a technique similar to indocyanine green angiography but evaluates the retinal circulation in addition to the choroid. Optical coherence tomography is a noninvasive imaging technique used to generate a cross-section of the retina and choroid.
In 2021, the Standardization of Uveitis Nomenclature (SUN) Working Group published its classification criteria of early and late-stage Vogt-Koyanagi-Harada disease, which utilized machine learning to determine a set of classification criteria that minimized misclassification of Vogt-Koyanagi-Harada disease with other forms of panuveitis (69). Over 1000 cases of panuveitis were evaluated, including 156 cases of early-stage Vogt-Koyanagi-Harada disease and 103 cases of late-stage Vogt-Koyanagi-Harada disease. The 95% confidence interval for overall accuracy was between 89.0% and 96.8%.
Criteria | ||
1. Evidence of Harada disease | ||
a. Serous (exudative) retinal detachment AND (b) and/or (c) | ||
b. Multiloculated appearance on fluorescein angiogram OR | ||
c. Septae on optical coherence tomogram | ||
OR | ||
2. Panuveitisa with two or more of the following neurologic symptoms or signsb | ||
a. Headache OR | ||
b. Tinnitus OR | ||
c. Dysacusis OR | ||
d. Meningismus OR | ||
e. Cerebrospinal fluid pleocytosis | ||
AND | ||
3. No history of penetrating ocular trauma or vitreoretinal surgery prior to disease onset | ||
Exclusions | ||
1. Positive serology for syphilis using a treponemal test | ||
2. Evidence for sarcoidosis (either bilateral hilar adenopathy on chest imaging or tissue biopsy demonstrating noncaseating granulomata) | ||
a Uveitis should have evidence of choroidal involvement on clinical examination, fluorescein angiography, indocyanine green angiography, or optical coherence tomography, including enhanced depth imaging of the choroid. | ||
b Onset of neurologic symptoms and signs and onset of the uveitis should occur within 4 weeks of each other. | ||
| ||
Criteria | ||
1. History of early-stage Vogt-Koyanagi-Harada disease | ||
AND 2 and/or 3 | ||
2. Sunset glow fundus | ||
OR | ||
3. Uveitis* AND one or more of the following cutaneous findings | ||
a. Vitiligo OR | ||
b. Poliosis OR | ||
c. Alopecia | ||
Exclusions | ||
1. Positive serology for syphilis using a treponemal test | ||
2. Evidence for sarcoidosis (either bilateral hilar adenopathy on chest imaging or tissue biopsy demonstrating noncaseating granulomata) | ||
* Uveitis may be (1) chronic anterior uveitis; (2) anterior and intermediate uveitis; or (3) panuveitis with multifocal choroiditis (“Dalen Fuchs–like nodules”) | ||
| ||
Ophthalmic ancillary testing. During the acute uveitic stage, indocyanine green angiography demonstrates multifocal areas of delayed choroidal filling. In the late stages, peripapillary and multifocal choroidal pinpoint leakage can be seen. Indocyanine green may pool late in the areas of serous retinal detachment. Features on pre-treatment ICG angiography may also provide prognostic value (212).
Fluorescein angiography demonstrates similar punctate hyperfluorescence in the choroid and retinal pigment epithelium, with progressive staining in areas of active choroiditis in the later phase of the angiogram. Dye can similarly accumulate in the areas of serous retinal detachments. This pattern of hyperfluorescence is said to give a “moth-eaten” appearance. Optic nerve head leakage is found in 70% of cases (134).
Due to differences in protein binding and wavelengths used, there are differences between indocyanine green angiography and fluorescein angiography leakage patterns. Indocyanine green angiography is advantageous for evaluating the choroid and retinal pigment epithelium, with less interference from the retinal pigment epithelium (67). Both fluorescein angiography and ICG angiography can detect hyperpermeability and dilated choroidal veins, which are features of choroidal venous overload in early stage disease (152).
Melanin accumulation may be seen with near-infrared photography with corresponding hyper-reflectivity on optical coherence tomography. These lesions may resolve with treatment (130). Spectral domain optical coherence tomography (sdOCT) is another noninvasive diagnostic modality used to assess the retina, retinal pigment epithelium, and choroid. This technique can be used to monitor retinal and choroidal thickness, as well as the extent of subretinal fluid. It is routinely performed in the office, and acquisition is rapid, thus, making it one of the most commonly used imaging modalities to monitor disease activity (88; 10; 87; 50; 202; 139) and predict recurrence (73).
Swept-source and enhanced depth imaging optical coherence tomography have also been used to evaluate increased choroidal thickness in acute Vogt-Koyanagi-Harada syndrome. Swept-source optical coherence tomography has better resolution for the choroid than enhanced depth imaging optical coherence tomography and can be used to follow resolution of inflammation after treatment with steroids (174; 210; 231). Optical coherence tomography can identify development of focal choroidal excavations (251). Fibrin membranous structures on OCT were associated with younger age, greater disease severity, and cumulative steroid dose but did not affect final visual outcome (135). Optical density of subretinal fluid has not been shown to be helpful in differentiating Vogt-Koyanagi-Harada syndrome from other causes of serous retinal detachment (49; 237).
Optical coherence tomography angiography is a noninvasive imaging technique that provides an angiogram of the retinal and choroidal vasculature without the need for intravascular fluorophores. Optical coherence tomography angiography has shown multiple dark foci at the level of the choriocapillaris corresponding with hypocyanescence on indocyanine green angiography, indicating hypoperfusion and ischemia. Microvascular density is significantly decreased in eyes with sunset glow fundus (51). Flow defects, as well as vascular density, have been shown to worsen in acute disease (240; 42) and improve with glucocorticoid treatment (174; 75; 76). Further, optical coherence tomography angiography has demonstrated changes in choroidal thickness and blood flow in active and remitting disease (53). Longer duration disease is associated with a greater decrease in retinal and choroidal vascular density (64). It has also demonstrated flow voids preceding clinically relapsing disease (82). Thus, optical coherence tomography angiography can be used to detect subclinical disease activity as well as monitor response to treatment. Persistent flow voids in the convalescent phase were associated with worse final visual acuity (24). The presence of optic nerve swelling is associated with lower retinal arteriolar blood flow and oxygen saturation (06), as well as lower peripapillary vessel densities (90). Reduced retinal blood flow during the acute phase is associated with greater risk of developing sunset glow fundus (04).
Anterior segment optical coherence tomography may show scleral thickening and ciliochroidal effusion, both of which improve with treatment (160).
Ultrasound biomicroscopy may show ciliochoroidal detachment (20%) and unclear ciliary processes (15%), which are characteristic in the uveitic phase. These changes improve with treatment but may be observed in recurrent uveitis (163).
Adaptive optics scanning laser ophthalmoscopy, an emerging technology, may be a useful indicator measuring photoreceptor density during active disease and recovery (173). Laser speckle flowgraphy (03; 06) and retinal oximetry has also been used to evaluate choroidal perfusion in Vogt-Koyanagi-Harada syndrome (05). Retromode scanning laser ophthalmoscopy may offer an alternative to optical coherence tomography and ICG angiography by scanning a larger area without the need for dye (239).
Neurologic ancillary testing. Lumbar puncture yields diagnostically useful information in the form of lymphocytic pleocytosis in a high percentage of patients, if fluid is sampled within the first 8 weeks. There may be a lag in the development of pleocytosis, as the percentage of patients with this finding rises from 80% in the first week of symptoms to 97% by the third week (201; 134). Other authors questioned the necessity of CSF examination and found that fluorescein angiography along with clinical examination were diagnostic even in patients without cerebrospinal fluid pleocytosis (215).
Though MRI is often obtained for workup of neurologic symptoms, findings are often nonspecific related to leptomeningeal inflammation. Le described a case of Vogt-Koyanagi-Harada syndrome presenting with bilateral optic nerve swelling and leptomeningeal enhancement (111). Ikeda and Tsukuguchi report a case of Vogt-Koyanagi-Harada syndrome with multiple hyperintense signal abnormalities in the cerebral white matter. One patient had high signal abnormalities in the periventricular cerebral white matter (77).
MRI may show choroidal thickening, indicating a concurrent ocular process (92; 134; 172). Theta-range slowing of the EEG is often present, but this is so nonspecific as to be relatively unhelpful in establishing the diagnosis of Vogt-Koyanagi-Harada syndrome.
Other ancillary testing. In small series, the measurement of individual cell populations may be useful in differentiating Vogt-Koyanagi-Harada syndrome from other uveitic syndromes (95) and measuring response to treatment.
High-dose systemic corticosteroid treatment is recommended during the uveitic stage, with slow dosage tapering over the subsequent 3 to 6 months. Treatment is generally monitored by ophthalmologic parameters, and patients should be under the care of an ophthalmologist for this. High-dose corticosteroid treatment can induce central serous choroidopathy in patients with Vogt-Koyanagi-Harada as with other forms of chronic uveitis and this must be considered as possibly causative if a patient has worsening of visual acuity with treatment (191).
The neurologic problems are generally limited to the prodromal and early uveitic stages. Specific management of these problems is seldom an issue. Recurrences during corticosteroid tapering may become refractory to treatment and require the use of cytotoxic agents such as cyclophosphamide, chlorambucil, or azathioprine, or cytostatic agents such as cyclosporine (134).
Case reports and case series have shown stable control achieved with mycophenolate mofetil (07), rituximab (44), infliximab (253), tofacitinib (46), azathioprine (166), and adalimumab (198; 109; 208; 193; 55; 207). Small series have demonstrated improved visual acuity outcomes in patients treated early with immunomodulatory therapy (167; 124). Large reviews suggest that steroidal and nonsteroidal dual therapy given at onset is better than corticosteroid monotherapy alone (71; 165; 235). Biologics have shown promise in achieving control in refractory and recurrent disease (26; 236; 193).
In a clinical trial of 110 patients, initial treatment with cyclosporine showed similar efficacy compared to adalimumab when combined with a low-dose oral corticosteroid (248), although in some markets cyclosporine may be considered more cost-effective (38). Subgroup analysis of Vogt-Koyanagi-Harada syndrome patients from the First-line Antimetabolites as Steroid-sparing Treatment Uveitis (FAST) trial showed greater reduction in central retinal thickness and serous retinal detachment with methotrexate compared to mycophenolate. However, there was no significant difference in final visual acuity outcome (08).
In case reports and one small prospective study, intravitreal dexamethasone implant (29; 32; 48) or intravitreal fluocinolone acetonide implant combined with intravitreal methotrexate (168) has shown efficacy. In a small series, suprachoroidal steroid injections were shown to be an effective adjunct to oral steroid therapy in treating serious retinal detachment (205). These local therapies may be a safe alternative for patients who develop morbidity or adverse events with systemic treatment.
Vitrectomy to drain subretinal fluid has been described (47). However, in a small case series, three of five eyes with Vogt-Koyanagi-Harada syndrome that underwent vitrectomy had recurrence of serous retinal detachment that did not respond to medical therapy and required repeat surgery (182).
A study of 97 consecutive patients presenting in Riyadh, Saudi Arabia, included 13 children and 84 adults (204). Sixty-one percent (eight patients) of those younger than 14 years of age (children) required cataract surgery, as compared with 17% (14 patients) of the adults. Sixty-one percent of the children had final visual acuity equal to or worse than 20/200, whereas only 26% of adults had that severe visual consequence. Chronic recurrent disease is associated with worse outcomes in children (11).
Visual acuity outcomes in children are generally good (12; 35). Children with chronic-recurrent Vogt-Koyanagi-Harada syndrome are at higher risk of developing late complications including cataract, glaucoma, and choroidal neovascular membranes. Of 98 patients with Vogt-Koyanagi-Harada syndrome seen at the Aravind Eye Hospital in Madurai, India, between 1993 and 1995, three were children (under 16 years of age), and all required cataract surgery (181). Visual outcome was good, despite the eventual development of retinal pigment epithelial atrophy in two children and glaucoma in one child.
The presence of posterior synechiae at diagnosis is associated with an increased risk of recurrent anterior uveitis (118). Patients with Vogt-Koyanagi-Harada syndrome who develop a bullous retinal detachment have an eight times higher risk of developing subretinal fibrosis (61). Late presentation, recurrent inflammation, and iris and angle scarring were associated with greater development of secondary glaucoma (14; 243).
Patients with hyperreflective outer nuclear layer or bacillary layer detachment had more severe disease in the acute phase and a higher rate of recurrence within the first 6 months (108), and greater risk of subretinal fibrosis and sunset glow fundus (129), although there does not seem to be a significant relationship between any imaging-based biomarker and long-term visual prognosis (139; 162; 202).
Small series have demonstrated improved visual acuity outcomes in patients treated early with immunomodulatory therapy (167; 119; 52). However, some have observed increased adverse events in patients who start immunomodulatory therapy immediately and have proposed early initiation of immunomodulation based on stratified risk (91). Initiation of treatment preceding anterior segment inflammation (a late presentation) was associated with fewer long-term complications (06). More aggressive treatment with intravenous methylprednisolone compared to conventional dose oral corticosteroids showed fewer recurrences and better long-term visual outcomes (131). This may be a pulse of intravenous corticosteroids followed by an oral taper (57). Finally, it has also been shown that patients whose initial corticosteroid treatment was continued for 6 months or longer have significantly fewer late recurrences of ocular inflammation than patients initially treated for less than 6 months (110). Delayed treatment and greater severity at presentation were associated with greater risk of treatment-refractory disease (59).
Patients on high-dose corticosteroid or immunomodulatory therapy are at increased risk of opportunistic infections. Some of these infections may also manifest as posterior uveitis, complicating the management and clinical picture. Cases of tuberculosis uveitis and cytomegalovirus retinitis during therapy for Vogt-Koyanagi-Harada syndrome have been described (98; 19). Development of COVID-19 has also been described during immunosuppressive therapy (151), although some authors suggest that the antiviral properties of cyclosporine as a treatment agent may reduce this risk (147).
Reversal of iris depigmentation with treatment has been described (213).
Control of uveitis, better vision, and treatment-free survival were associated with higher quality of life scores (196).
The course of uveitis during and after pregnancy was studied in a cohort of 50 women and 76 pregnancies. Thirty-three pregnancies were in women with Vogt-Koyanagi-Harada disease, and these women characteristically had flare-ups in the early part of the pregnancy. Postpartum flare-up was frequent, especially among patients with Behçet disease (177; 189).
Several drugs used in the treatment Vogt-Koyanagi-Harada syndrome are contraindicated during the first trimester of pregnancy, if not throughout pregnancy. Difficult judgments as to corticosteroid and cytotoxic drug usage must be made if a pregnant woman develops Vogt-Koyanagi-Harada syndrome because it is a disease with a high risk of causing severe visual disability if untreated. A limited experience with successful treatment of three pregnant women with Vogt-Koyanagi-Harada using topical and systemic corticosteroids without obvious infant complication has been reported (132). A similar case described pulse steroids administered at 34 weeks, complicated by threatened preterm labor and gestational diabetes, but ultimately resulted in normal delivery and favorable maternal and fetal outcome (217). A case of azathioprine added to high-dose corticosteroid therapy in the first trimester of pregnancy has been described, with good maternal-fetal outcome (80). In a small case series of 10 Saudi Arabian women treated with corticosteroids and cyclosporine, one was complicated by abortion, one with hypertension, and two with premature rupture of membranes (13). In an Indian study, three of nine pregnant women maintained with azathioprine experienced a flare-up during pregnancy and were managed by supplemental systemic corticosteroids (34).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Homer Chiang MD
Dr. Chiang of Kaiser Permanente Santa Clara Homestead Medical Center has no relevant financial relationships to disclose.
See Profile
Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Child Neurology
May. 12, 2026
Neuroimmunology
Apr. 26, 2026
General Child Neurology
Apr. 24, 2026