





Epilepsy & Seizures
Hippocampal and parahippocampal seizures
Apr. 22, 2024
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Childhood absence epilepsy is the prototype idiopathic (genetic) generalized epilepsy syndrome of typical absence seizures. It is common in normal school-aged children between the ages of 4 and 10 years (peak age 6 to 7 years). It manifests with severe and frequent (pyknoleptic) typical absence seizures of abrupt onset and termination, lasting around 10 seconds each, many times per day. EEG shows bilateral, synchronous, symmetric spike-and-slow wave discharges, usually 3 Hz, on normal background activity. Females predominate. The prognosis of childhood absence epilepsy syndrome is excellent if properly diagnosed and treated. The focus is on the current understanding of its clinical and EEG phenotype, epidemiology, pathophysiology, genetics, pharmacological treatment, and outcome, along with the differential diagnosis associated with other idiopathic and genetic generalized epilepsies involving absence seizures. The author particularly emphasizes that most of the older and newer antiseizure drugs are contraindicated for the treatment of childhood absence epilepsy.
|
• Childhood absence epilepsy is the prototype idiopathic generalized epilepsy syndrome and is characterized by typical absence seizures of high daily frequency and severe impairment of consciousness in school-aged children. It is associated with generalized spike-and-wave discharges, usually 3 Hz (range 2.5 to 4 Hz). | |
|
• Typical absence seizures with different electroclinical characteristics are also part of the phenotypic expression of other idiopathic-genetic generalized epilepsies of infancy and childhood. | |
|
• The differential diagnosis of childhood absence epilepsy includes other types of epilepsies or syndromes with typical absence seizures manifesting in infancy and childhood. | |
|
• Prognosis is usually excellent for patients diagnosed on strict inclusion and exclusion criteria for the syndrome of childhood absence epilepsy. | |
|
• Ethosuximide and sodium valproate, as monotherapy or combined, are the most effective therapeutic approaches. |
According to Temkin, Poupart was the first to describe absence seizures in 1705 (212). However, Tissot's description of petits accès or petits in 1770 is better known (216). They were called "absences" by Calmeil in 1824 (30), "petit mal" by Esquirol in 1838 (80), and "epilepsia mitior" by Reynolds in 1861 (181). Gowers gave a most accurate description of the absence seizures “without conspicuous convulsions” (101). Friedman reported a long-term favorable prognosis but believed that these absences were not epileptic (89). The frequency of typical absence seizures had also been conspicuous, and Sauer coined the name “pyknolepsy” (from the Greek word pyknos, meaning closely packed, dense, or aggregated) (191). Hyperventilation as a test to introduce absences was first described by Brain in 1924 (174). In the same year, Adie reported on pyknolepsy, a form of epilepsy occurring in children with a good prognosis, and defined it as follows (02):
|
...a disease with an explosive onset between the ages of 4 and 12 years, of frequent short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months, or years, are uninfluenced by antiseizure remedies, do not impede normal and psychical development, and ultimately cease spontaneously never to return. At most, the eyeballs may roll upwards, the lids may flicker, and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations, and lassitude or confusion after the attacks are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present they should be considered as foreign to the more favorable disease. I shall be well satisfied if I have made it appear probable to you that there does exist a form of epilepsy in children which is distinguishable by its clinical features and in which the prognosis is always good.” |
Gibbs, Davis, and Lennox showed that petit mal absences were associated in EEG with a rhythmic 3 Hz discharge of regular spike-and-wave complexes (93). In 1945, Lennox referred to the petit mal triad as absence, myoclonic, and akinetic seizures, and the introduction of trimethadione revolutionized the treatment of absence seizures (131). Video-EEG analysis allowed a precise clinico-EEG correlation of typical absence seizures (173).
The Commission on Classification and Terminology of the International League Against Epilepsy (ILAE) made important progress by accurately defining and differentiating typical absences of idiopathic generalized epilepsies versus atypical absences of symptomatic generalized epilepsies (47). However, all epilepsies with typical absence seizures remained for a long time clustered in the group of "petit mal" and considered as a form of "centrencephalic epilepsy."
In 1989 the Commission on Classification and Terminology of the ILAE recognized the heterogeneity of epilepsies with absence seizures and proposed to distinguish three syndromes of idiopathic generalized epilepsy (childhood absence epilepsy, juvenile absence epilepsy, and juvenile myoclonic epilepsy) (48). Further, it also recognized typical absence seizures in "other idiopathic generalized epilepsies," in "idiopathic generalized epilepsies with specific provocation," and also in a syndrome of cryptogenic generalized epilepsy (epilepsy with myoclonic absences). Panayiotopoulos and colleagues described syndrome-related characterization of absence seizures with video-EEG analysis (171).
In recognition of the diversity of typical absence seizures, the ILAE Task Force proposed four types that are probably of different pathophysiology and syndromic significance: (1) the classical typical absence seizure, (2) myoclonic absence seizures, (3) phantom typical absence seizures, and (4) eyelid myoclonia with absence (79). The task force also recognizes childhood absence epilepsy as a separate epileptic syndrome within idiopathic generalized epilepsies (78; 79). Other syndromes with predominant absence seizures identified by the ILAE Task Force are epilepsy with myoclonic absences, juvenile absence epilepsy, and juvenile myoclonic epilepsy.
All these types of seizures and electroclinical syndromes are mostly maintained in the report of the ILAE Commission on Classification and Terminology (16; 49). However, the latest ILAE position paper for the operational classification of seizure types does not consider phantom absences though “it is recognized that awareness and responsiveness can be at least partially retained during some generalized seizures, for example, with brief absence seizures, including absence seizures with eyelid myoclonias or myoclonic seizures” (85; 86). Furthermore, typical absence seizures designate a syndrome not yet recognized by the ILAE, such as facial (perioral or eyebrow) myoclonia with absences. Eyelid myoclonia with absences (previously known as Jeavons syndrome) has been recognized as a distinct genetic generalized epilepsy syndrome by the ILAE (203). Video-EEG polygraphic recordings have contributed to a better understanding of the various electroclinical characteristics and the close relationship with other types of seizures.
Nomenclature and inclusion and exclusion criteria. Childhood absence epilepsy (pyknolepsy) was defined by the 1989 Commission on Classification and Terminology of the International League Against Epilepsy as follows:
|
Pyknolepsy occurs in children of school age (peak manifestation age 6 to 7 years), with a strong genetic predisposition in otherwise normal children. It appears more frequently in girls than in boys. It is characterized by very frequent (several to many per day) absences. The EEG reveals bilateral, synchronous symmetrical spike-waves, usually 3 Hz, on a normal background activity. During adolescence, generalized tonic-clonic seizures often develop. Otherwise, absences may remit or, more rarely, persist as the only seizure type (48). |
However, this brief definition may be misinterpreted because of some ambiguity in its terminology as illustrated by the fact that many reports consider childhood absence epilepsy any type of epilepsy with onset of absences in childhood (187). Therefore, epidemiology, genetics, age at onset, clinical manifestations, other types of seizures, long-term prognosis, and treatment do not accurately reflect the syndrome of childhood absence epilepsy. A stricter definition of childhood absence epilepsy has been proposed (139; 140; 110; 165), which does not significantly differ from that of the Commission on Classification and Terminology of the International League Against Epilepsy (48):
(1) “Childhood absence epilepsy occurs in children of school age (peak manifestation age 6 to 7 years).” A simplistic but unsatisfactory practice is to label as childhood absence epilepsy any child with onset of daily absences at 10 years or earlier (as early as 2 years of age) (187; 188). The 10 years of age cut off is arbitrary and not a decisive “gold standard” to define what is and what is not childhood absence epilepsy. The arbitrary limit of 10 years is mainly based on the pioneering work of Doose and Janz who found that pyknoleptic absences usually start before 10 years of age, whereas non-pyknoleptic absences usually start after this age (75; 114). Typical absence seizures of recognized syndromes, such as myoclonic absence or juvenile myoclonic epilepsy, may start before the age of 10 years old and even in early childhood. When the onset is in early childhood, the characteristic electroclinical features of the two syndromes are recognized around 3 to 4 years later, when a successful treatment is discontinued and relapses occur. Further, “school age” does not include children younger than 3 to 4 years because absence seizures with onset at this age are heterogeneous; some may constitute discrete genetic entities; they are entirely different to those of childhood absence epilepsy (75; 105; 09; 39; 217); and they are often of bad prognosis (63; 136; 66). Childhood absence epilepsy starting before the age of 4 years and of good prognosis may be a very rare possibility that needs investigation (67; 195; 196).
If diagnosis by age at onset is to be followed, the study population should be correctly defined as “epilepsy with childhood absence seizures” rather than childhood absence epilepsy, which is a subset of it. Therefore, not all typical absence seizures occurring in childhood belong to the syndrome of childhood absence epilepsy. There are idiopathic generalized epilepsies and syndromes that start in infancy, childhood, and adolescence in which typical absence seizures, myoclonic seizures, or generalized tonic-clonic seizures are the only seizure type or the predominant type, and myoclonic seizures or typical absence seizures, alone or combined, may be part of the phenotype, respectively. Table 1 is a practical phenotypical classification of idiopathic generalized epilepsies with typical absence seizures, from infancy to adolescence (53).
|
A. Absence seizures as the only type or the predominant type of seizures. Myoclonic jerks and generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Absence seizures of early onset (under 3 years old) |
|
B. Myoclonic jerks as the only type or the predominant type of seizures. Typical absence seizures and generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Early-onset myoclonic absence epilepsy (under 3 years old) |
|
C. Absence and myoclonic seizures as the predominant types of seizures. Generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Epilepsy with myoclonic absences (previously known as Tassinari syndrome) |
|
D. Generalized tonic-clonic seizures as the only type or the predominant type of seizures. Typical absence seizures and myoclonic jerks may be part of the phenotype. | |
|
|
• Generalized tonic-clonic seizures on waking or only with or without positive response to intermittent photic stimulation and patterns |
|
| |
(2) “Childhood absence epilepsy is characterized by very frequent (several to many per day) absences.” These absences are not only “several to many per day” but also severe according to the ILAE definition:
The hallmark of the typical absence attack is a sudden onset, interruption of ongoing activities, a blank stare, possibly a brief upward rotation of the eyes. If the patient is speaking, speech is slowed or interrupted; if walking, he stands transfixed; if eating, the food will stop on his way to the mouth. Usually, the patient will be unresponsive when spoken to. In some, attacks are aborted when the patient is spoken to. The attack lasts from a few seconds to half a minute and evaporates as rapidly as it commenced (47).
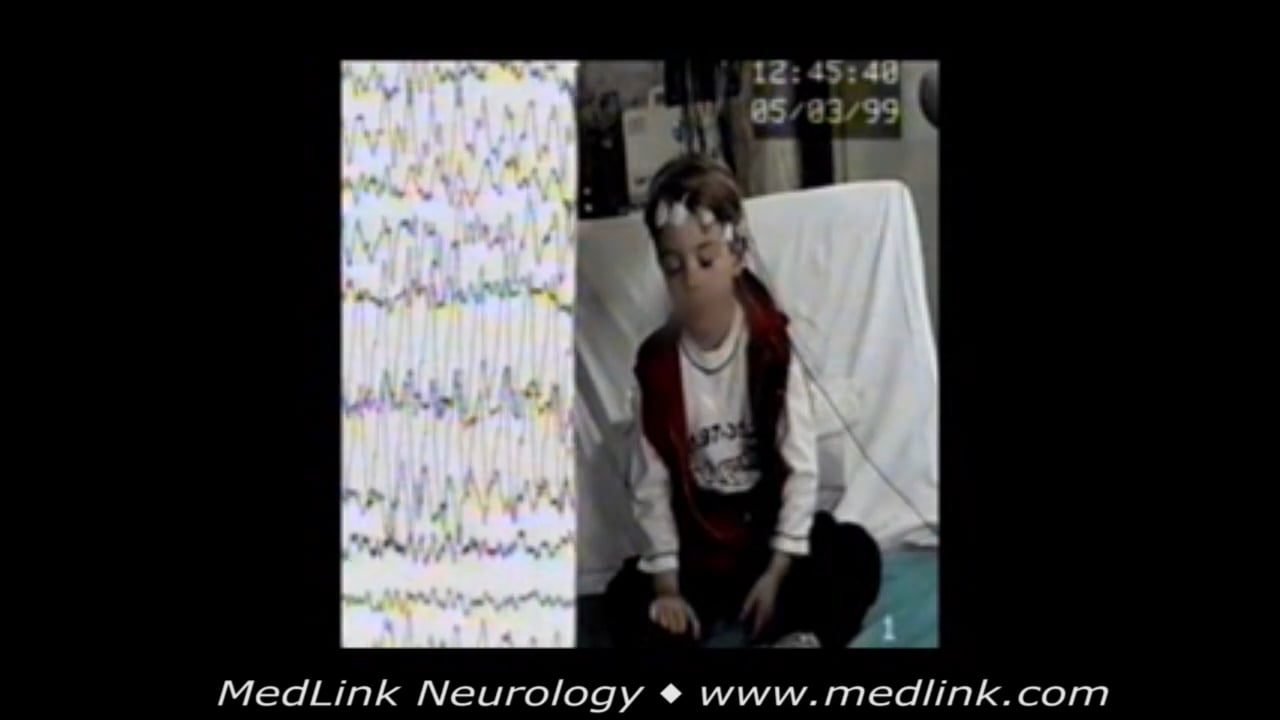
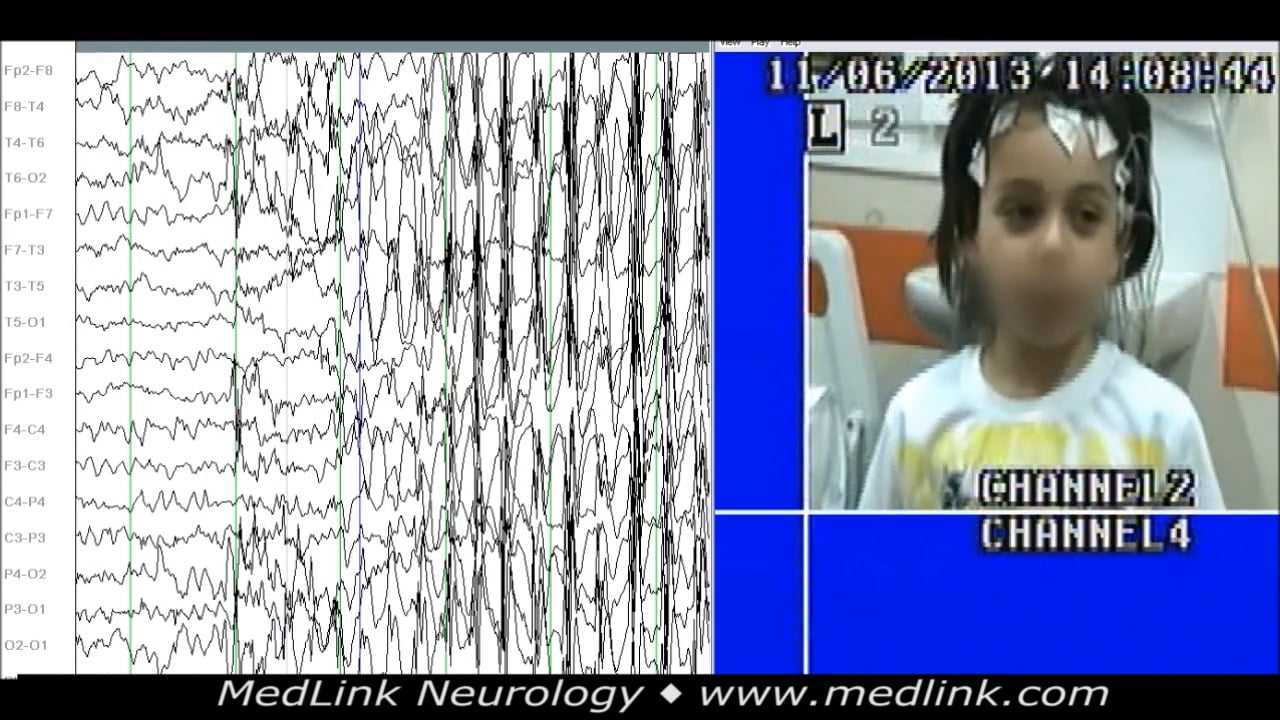
(3) In childhood absence epilepsy “the EEG reveals bilateral, synchronous symmetrical spike-waves, usually 3 Hz, on a normal background activity.” This terminology presumably excludes very brief typical absence seizures and fragmented, asymmetrical, and asynchronous spike-wave discharges of 3 to 5 Hz intradischarge variations during sleep and awake EEG.
(4) In the syndrome of childhood absence epilepsy or pure childhood absence epilepsy, no seizures other than typical absence seizures are accepted in the phenotype, and, as a rule, remission occurs before or early in adolescence. Otherwise, epilepsy with absences may remit or, more rarely, persist during adolescence and thereafter.
In addition, the ILAE, by accepting epilepsy with myoclonic absences and juvenile myoclonic epilepsy as separate syndromes, probably exclude myoclonic absences and brief mild absences from childhood absence epilepsy (48). It is by this logic that eyelid myoclonia and absences (which are primarily myoclonic) is also an exclusion criterion. Further, by accepting reflex absence seizures as a distinct category, the ILAE indicates that these too may not be part of childhood absence epilepsy. That perioral myoclonia or single violent jerks occurring in the course (not at onset) of typical absence seizures is an exclusion criterion may be debatable; however, their presence indicates worse prognosis (106; 102; 110). The same applies for multiple spikes and fragmented S-PWD that also indicate a bad prognosis, coexistent myoclonic jerks, or generalized tonic-clonic seizures (171; 82; 11). Brief typical absence seizures; myoclonic seizures; generalized tonic-clonic seizures; spanioleptic typical absence seizures; fragmented, asymmetrical, and asynchronous background activity; or photosensitivity are early indicators of a less favorable outcome if included in the phenotype (61; 53). All of these additional electroclinical features may indicate early-onset idiopathic generalized epilepsy; a well-defined syndrome, eg, juvenile absence epilepsy; or an unrecognized syndrome with childhood onset. When treatment is discontinued after 3 to 4 years, children with absence seizures and aberrant features may relapse with generalized tonic-clonic seizures or may even show electroclinical features of a well-defined syndrome, eg, juvenile myoclonic epilepsy (53), which explains the heterogeneous outcome that has been reported for childhood absence epilepsy.
The concise inclusion or exclusion criteria that define the syndrome of childhood absence epilepsy are summarized in Table 2 (139; 140; 141; 110; 165; 61; 53).
|
Childhood absence epilepsy |
Childhood absence epilepsy with aberrant electroclinical features | |
|
Clinical |
• Onset: 4 to 10 years of age (peak: 5 to 7 years of age) |
• Onset: before 3 to 4 years of age or after 10 years of age |
|
EEG |
• Ictal: high-amplitude rhythmic generalized spike-wave discharges and double spike-wave complexes, usually 3 Hz (range 2.5 to 4 Hz). Duration of 4 to 10 seconds, rarely above 20 seconds. |
• Brief (less than 4 seconds) generalized 3 to 4 Hz spike-wave discharges, focal spikes or slow spike-wave complexes, fragmented generalized spike-wave discharges |
|
Prognosis |
Favorable, self-limited before 12 years of age |
Less favorable |
The ILAE epilepsy manual classifies childhood absence epilepsy among epileptic syndromes of childhood (49), and the last ILAE Task Force on Nosology and Definitions classifies childhood absence epilepsy in idiopathic generalized epilepsies, a distinct subgroup of genetic generalized epilepsies (109). The term “genetic generalized epilepsies” includes other syndromes beyond the idiopathic generalized epilepsies.
The classification of childhood absence epilepsy is detailed below.
Childhood absence epilepsy. Childhood absence epilepsy is a genetic generalized epilepsy that should be considered in an otherwise normal child with multiple daily absence seizures associated with 2.5 to 4 Hz generalized spike-and-waves. Absence seizures are provoked by hyperventilation. Between 8 and 12 years of age, the distinction between the clinical syndromes of juvenile absence epilepsy and childhood absence epilepsy depends on the frequency of absence seizures.
A genetic generalized epilepsy is an epilepsy with generalized seizures associated with generalized epileptiform EEG patterns, such as generalized spike wave activity.
Clinical context. This syndrome is characterized by onset of frequent absence seizures between the ages of 2 to 12 years (peak 5 to 6 years of age). Both sexes are equally affected. Antecedent and birth history is normal. A previous history of febrile seizures may occur (seen in 15% to 20% of cases). Neurologic examination and head size are normal. Development and cognition are typically normal. Attention deficit hyperactivity disorder and learning difficulty may occur. Seizures are typically self-limiting. Self-limiting means having a high likelihood of seizures spontaneously remitting at a predictable age.
|
|
Caution. Onset of absence seizures less than 4 years, then consider glucose transporter disorders. |
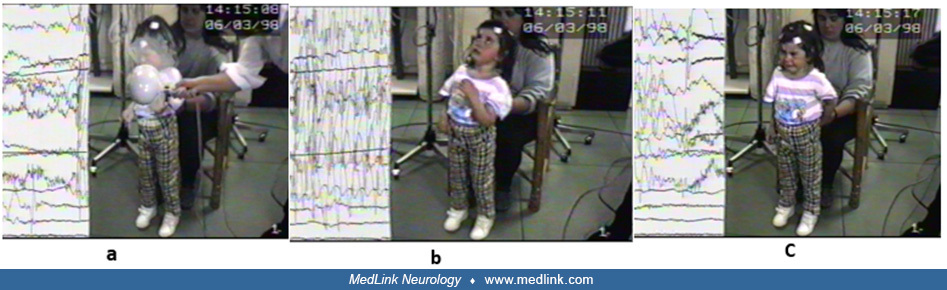
Mandatory epileptic seizures. Absence seizures in childhood absence epilepsy are typically frequent (multiple daily) and brief (average 10 seconds). Awareness and responsiveness are usually severely impaired, but the child may be more responsive towards the end of the seizure.
|
|
Caution. Absence seizures longer than 45 seconds or with postictal phase should not occur. Then consider focal impaired awareness seizures (eg, due to structural brain abnormality). |
|
|
Caution. The presence of awareness during absence seizures. Then childhood absence epilepsy is unlikely, other syndromes with absences should be considered. |
|
|
Caution. If onset or offset of absences is not abrupt, or absences occur less than daily. Then consider other syndromes with absences. |
Generalized tonic-clonic seizures rarely precede or occur during periods of frequent absences but may occur later with evolution to other idiopathic generalized syndromes. Such cases may be part of childhood absence epilepsy in general, but not the syndrome childhood absence epilepsy with no aberrant electroclinical features (Table 2).
|
|
Caution. Generalized convulsive seizures before adolescence? Then consider other epilepsy syndromes. |
|
|
Exclusionary. Any other seizure type is not expected in this syndrome. |
EEG background. The background is normal. Occipital intermittent rhythmic delta activity (OIRDA) may be seen in a third of children with childhood absence epilepsy, at a frequency of 2.5 to 4 Hz and may have a notched appearance.
|
|
Caution. Focal slowing seen consistently in one area consider structural brain abnormality. |
|
|
Caution. Generalized slowing is not seen. |
Interictal EEG. Generalized spike-and-wave, or fragments of generalized spike-and-wave, are seen in the interictal EEG. These are brief (usually less than 2 seconds) and occur most commonly seen in sleep.
|
|
Caution. Although focal spikes (as fragments of generalized spike-and-wave) can occur, if they consistently arise in one area, then consider structural brain abnormality. |
Activation. EEG abnormality and absence seizures are provoked by hyperventilation. If hyperventilation is poorly performed, generalized spike-and-wave may not be triggered. Intermittent photic stimulation triggers generalized spike-and-wave in a small proportion of individuals but does not induce seizures.
EEG abnormality is enhanced by sleep deprivation and by sleep. Generalized spike-and-wave often becomes fragmented with sleep deprivation or in sleep. Fragmented generalized spike-and-wave can appear focal or multi-focal but usually is not consistently seen in one area. The morphology of the focal spike-and-wave typically appears similar to the generalized spike-and-wave.
|
|
Caution. Where hyperventilation is performed well for 3 minutes, and no generalized spike-and-wave is seen in an untreated patient, childhood absence epilepsy is unlikely. |
Ictal EEG. Regular 3 Hz generalized spike-and-wave occurs associated with absence seizures. Polyspike-and-wave can occur in the ictal EEG.
|
|
Caution. Slow spike-and-wave (less than 2.5 Hz) is exclusionary. |
Imaging. Neuroimaging is normal. If the electroclinical diagnosis of childhood absence epilepsy is established, and there are no atypical features, neuroimaging is not required.
Genetics. It has a complex pattern of inheritance. In children who have absences with onset before 4 years of age, 10% have GLUT1 deficiency, which is caused by a mutation in SLC2A1 (205; 08). Other genes linked to this syndrome include GABRG2 and CACNA1A.
Family history of seizures/epilepsy. Up to 20% of patients may have a first-degree relative with seizures. When a family history is present, the affected family members typically have childhood absence epilepsy or a related genetic or idiopathic generalized epilepsy, eg, juvenile absence epilepsy, juvenile myoclonic epilepsy, or, less commonly, genetic epilepsy with febrile seizures plus.
A genetic generalized epilepsy is an epilepsy with generalized seizures associated with generalized epileptiform EEG patterns, such as generalized spike wave activity.
|
Differential diagnoses. | |
|
• Juvenile absence epilepsy: Consider if there are infrequent (eg, once daily) absence seizures in a child who is 8 years of age or older. | |
|
• Epilepsy with eyelid myoclonias: Consider if there is repetitive, rhythmic, fast greater than 4 Hz jerks of the eyelids, with upward deviation of the eyeballs, and with head extension; seizures are often very frequent and induced by eye closure (voluntary or on command) and photic stimulation. | |
|
• Epilepsy with myoclonic absences: Consider if there are 3 Hz myoclonic jerks of upper limbs with tonic abduction. | |
|
• Nonepileptic disorders such as daydreaming and inattention | |
|
| |
According to the position paper of the ILAE Commission for Classification and Terminology, childhood absence epilepsy is an idiopathic or genetic self-limited epilepsy (192). The 2017 ILAE classification suggested that the term “genetic generalized epilepsies” be used for the broad group of epilepsies with generalized seizure types and generalized spike-waves based on a presumed genetic etiology arising from twin and family research study data. It was suggested that the term “idiopathic generalized epilepsy” be reserved for the four syndromes: childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and generalized tonic-clonic seizures alone.
The four idiopathic generalized epilepsy syndromes continue to be regarded as a special group under the umbrella term “idiopathic generalized epilepsy syndromes” in the 2022 ILAE classification and are regarded as a distinct subgroup of genetic generalized epilepsies (109). The term “genetic generalized epilepsies” includes other syndromes beyond the idiopathic generalized epilepsy syndromes, such as epilepsy with myoclonic absences and epilepsy with eyelid myoclonia (previously known as Jeavons syndrome). The recognition of the idiopathic generalized epilepsy syndromes as a distinct subgroup of the genetic generalized epilepsies remains helpful as they carry prognostic and therapeutic implications.
Childhood absence epilepsy occurs in an otherwise normal child, with daily absence seizures associated with 2.5 to 4 Hz generalized spike-waves at seizure onset. Age at onset is typically 4 to 10 years (range 2 to 13 years). Absence seizures are provoked by hyperventilation (109). Neurologic examination is normal. Development and cognition are typically normal. Attention deficit hyperactivity disorder and learning difficulties may occur. Seizures are brief but may occur in clusters. Incontinence and loss of postural control can be seen. Seizures typically occur multiple times a day but are often under-recognized.
Generalized tonic-clonic seizures rarely precede or occur during the period of frequent absence seizures in childhood. More commonly, they begin in adolescence, often after the resolution of absence seizures, and may herald evolution to another idiopathic generalized epilepsy syndrome. Neurologic examination is normal. Development and cognition are typically normal, but subtle specific learning difficulties and attention deficit hyperactivity disorder may be present. Background EEG is normal. Fragmented generalized spike-waves can appear focal or multifocal but are not consistently seen in one area. The morphology of the focal spike-wave is similar to the generalized spike-wave. Polyspike-waves may be seen in drowsiness and sleep only, but not during wakefulness. Ictal EEG is characterized by regular 3 Hz (range 2.5 to 4 Hz) generalized spike-waves in the first second of seizure onset with absence seizures. Disorganized discharges, defined by brief (less than 1 second) or transient interruptions in the ictal rhythm or waveforms of different frequency or morphology, are significantly less common than in juvenile absence epilepsy. Epilepsy remits in 60% of children, often within 2 years of onset or by early adolescence.
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125