Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this clinical article, the authors describe the different manifestations of this inborn error of leucine catabolism and explain pathophysiology, disease diagnosis, and treatment. 3-alpha-methylcrotonylglycinuria appears to be the most common as well as one of the most puzzling of the organoacidopathies. Clinical presentations range from severe neonatal metabolic decompensation and lethal outcome to mostly asymptomatic adults never diagnosed nor treated. This review explains the current knowledge and management of this disease, which can present to pediatricians, neurologists, and internists.
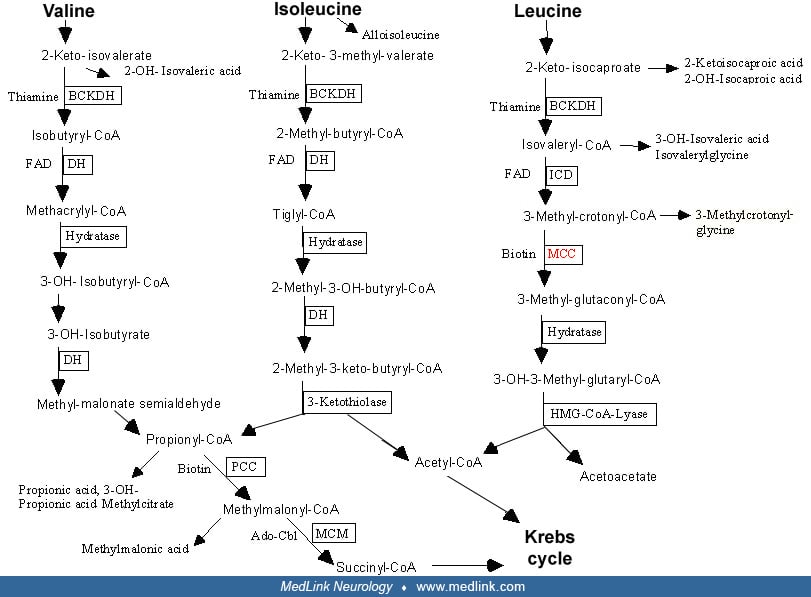
3-alpha-methylcrotonylglycinuria is an inborn error of leucine catabolism due to a deficiency of 3-methylcrotonyl-CoA carboxylase. As the enzyme requires biotin as a cofactor, the isolated enzymatic defect must be differentiated from other forms of methylcrotonylglycinuria caused by deficiencies in the biotin pathway (biotinidase and holocarboxylase synthetase deficiencies). The disease has a wide range of manifestations. Very few affected infants develop severe metabolic crisis, ketoacidosis, and vomiting that may lead to coma and death without appropriate treatment. On the other hand, most individuals remain symptom-free for life. The key metabolites leading to diagnosis are 3-hydroxyisovaleric acid and 3-methylcrotonylglycine in urine and 3-hydroxyisovalerylcarnitine in plasma.
The first case reports of 3-alpha-methylcrotonylglycinuria date from 1970, and it was postulated that 3-methylcrotonyl-CoA carboxylase was deficient (49). The enzyme 3-methylcrotonyl-CoA carboxylase is now recognized to be composed of two different subunits. The respective genes were identified in 2001: Gallardo and coworkers as well as Baumgartner and coworkers localized the gene for the alpha-subunit to 3q25-q27 and the gene for the beta-subunit to 5q12-q13 (04; 19). Several pathologic mutations in both genes have been described. Gallardo and colleagues also reported an unexpectedly high incidence of biochemical markers of this disorder detected in newborn screening programs (19).
Patients with isolated 3-methylcrotonyl-CoA carboxylase deficiency show at least a short period of normal development. Most remain symptom-free throughout their lives, and over time 3-methylcrotonyl-CoA carboxylase deficiency may eventually become classified as a nondisease (02; 21). Comparative analysis of case reports with newborn screening data suggests that without treatment less than 10% of affected individuals ever develop even minor symptoms and less than 1% to 2% have a more severe outcome (47). Reported clinical symptoms vary from mild nonspecific symptoms to a fulminant course with lethality in infancy. In some patients, symptoms have been reported to fit into the spectrum of metabolic diseases similar to other classical organic acidemias or to be rather nonspecific, such as isolated epilepsy or failure to thrive. A neonate who presented in the first day of life and progressed to necrotizing encephalopathy and death at 33 days of age has been described (06). Two patients who presented with metabolic stroke have been reported (38). Metabolic crises occur most frequently between the ages of 6 months to 3 years but have been described in neonates and older children (49). Crises may be triggered by minor infections. Patients present with vomiting, feeding difficulties, lethargy, and apnea with neurologic symptoms, such as muscular hypotonia, hyperreflexia, spasms, and seizures (focal or generalized). A few patients were found to have neutropenia, loss of scalp hair, and fatty changes of the liver reminiscent of Reye syndrome (49). Death is mainly due to cerebral edema (29) or cardiocirculatory arrest (49).
Laboratory tests may reveal a constellation of findings, including hypoglycemia, elevated liver enzymes, hyperammonemia, mild acidosis, moderate ketosis, and very low plasma carnitine (49). Additional patients with the following possibly related clinical presentations were identified: neonatal failure to thrive, isolated intellectual disability, mental retardation, recurrent attacks of status epilepticus, familial hypotonia or hypertonia, lethargy after head trauma, and severe multiple sclerosis (49; 11; 13).
Often, completely asymptomatic individuals were recognized when family members of patients were investigated (20), including women identified because of low acylcarnitine profiles on their children’s newborn screening. These women were asymptomatic or showed only minor symptoms, such as fatigue, weakness, and myopathy. However, some abnormal laboratory findings have been found, including elevated liver enzymes, elevated uric acid, and low free and total carnitine.
The reason for this broad spectrum of clinical manifestations from early death in metabolic crisis to a mostly asymptomatic and benign course remains as yet poorly understood. A study identified much higher consanguinity in symptomatic patients as compared to asymptomatic patients, and furthermore, it identified damaging mutations in other unrelated genes of these individuals. For example, in one case whole exome sequencing identified a homozygous deletion in IGHMBP2, known to cause spinal muscular atrophy, which was more likely to account for the phenotype of the patient (43). This study strengthens the possibility that 3-methylcrotonyl-CoA carboxylase deficiency may be a nondisease altogether.
Isolated 3-methylcrotonyl-CoA carboxylase deficiency has mostly been reported to be unresponsive to biotin. However, two patients were reportedly biotin-responsive (05). The first patient showed progressive psychomotor retardation and seizures, and biotin treatment resulted in a rapid and dramatic improvement of the clinical and biochemical phenotypes. However, psychomotor delay persisted, and the seizures reoccurred (18). The second patient was detected by newborn screening and remained clinically asymptomatic; the biochemical phenotype was also completely corrected by biotin. In both children, heterozygosity for the mutation MCCC1 p.-R385S was found; thus, this variant may have a dominant-negative effect that is responsive to pharmacological doses of biotin. This mutation has been expressed in Escherichia coli, and detailed kinetic studies have been performed (44), supporting the hypothesis of a dominant-negative effect. Other mutations causing 3-alpha-methylcrotonylglycinuria have also been expressed, and null or severely diminished 3-methylcrotonyl-CoA carboxylase (MCC) activity has been demonstrated, but there is no correlation with the clinical phenotype (12; 02).
With the widely varying clinical spectrum of symptoms, it is difficult to make definite statements about the prognosis of 3-methylcrotonylglycinuria. In the first reports, 3-methylcrotonylglycinemia was associated with a poor prognosis as patients succumbed during acute decompensation or suffered severe sequelae (29). It is reasonable to assume that initiation of treatment before severe metabolic decompensation will significantly ameliorate the patient’s prognosis, including potentially normal psychomotor development. A report of a 19-month-old girl who suffered a severe metabolic crisis due to her parents’ nonadherence to medical advice highlights the importance of follow-up programs to improve parental compliance with treatment recommendations (14).
On the other hand, almost all patients identified by newborn screening appear never to develop symptoms, even without treatment. The lack of genetic or biochemical markers to differentiate these individuals from those who are at risk for decompensation has led to controversy about the benefit of newborn screening for 3-methylcrotonylglycinuria (22; 47; 02; 43; 16; 53).
Gruenert and coworkers reported comprehensive clinical, biochemical, enzymatic, and molecular data on 88 individuals, which is the largest cohort published to date (21). The study confirmed the genetic heterogeneity of 3-methylcrotonylglycinuria and underlined the difficulty in predicting the clinical phenotype because there is no genotype-phenotype correlation and also residual enzyme activity cannot predict the clinical course. Arnold and colleagues reported similar findings in a cohort of 35 children diagnosed by newborn screening; they found an inverse correlation between initial metabolite levels and residual enzyme activity but no relationship with clinical (especially neurologic) outcome (02). The study by Shepard and colleagues even suggests that other independent causative diseases are likely and should be looked for in children with 3-methylcrotonyl-CoA carboxylase deficiency who develop symptoms (43).
A 9-year-old boy first developed infantile spasms (Blitz-Nick-Salaam convulsions) at three weeks of the age. The diagnosis of 3-methylcrotonylglycinuria was unfortunately delayed until two years of age when urine analysis revealed massive 3-methylcrotonylglycinuria and 3-hydroxyisovaleric aciduria, suggesting MCC deficiency. Biochemical findings showed partial MCC deficiency in cultured fibroblasts. Molecular genetic studies revealed a heterozygote missense mutation, MCCC1 p.-R385S, converting arginine to serine in a highly conserved region of the MCCC1 gene.
Interestingly, biotin therapy in this patient led to a dramatic decrease in the frequency of seizures, disappearance of hypsarrhythmia, as well as near normalization of organic aciduria. Four months later, a protein-restricted diet was introduced, and the boy remained clinically and metabolically stable. However, severe psychomotor delay persisted, and the seizures partially reoccurred.
At nine years of age, the boy is severely intellectually disabled and exhibits a significant delay in gross motor performance. Generalized hypotonia with lack of head control and tetraparesis is observed. Deep tendon reflexes cannot be elicited. The boy does not sit independently and cannot get up on his own. He is able to sit and walk a few steps supported under both arms. The patient does smile responsively and recognizes his parents. He has no speech development, and anger or pain are expressed by crying. Developmental milestones at nine years of age are less than one year of life according to the Denver developmental assessment. EEG shows mild to moderate background abnormalities and recurrent focal abnormalities suspicious for epileptic abnormalities. Treatment with valproate and vigabatrine does control seizures most of the time; however, especially during the courses of an infection, frequent brief multifocal predominantly clonic seizures occur. MRI studies revealed a predominantly fronto-parietal atrophy of the brain.
This is the first patient with MCC deficiency due to a heterozygous mutation and who demonstrates a substantial and sustained clinical and biochemical response to therapeutical doses of biotin (18).
3-alpha-methylcrotonylglycinuria is an inborn error of leucine catabolism due to deficiency of 3-methylcrotonyl-CoA carboxylase (EC 6.4.1.4) (OMIM 210200).
The disease usually shows an autosomal-recessive pattern of inheritance. In two patients, however, clinical and biochemical relevant 3-methylcrotonyl-CoA carboxylase deficiency was caused by a heterozygote MCC p.R385S (05). This mutation appears to exert a dominant negative influence on the wild type allele and, thus, on 3-methylcrotonyl-CoA carboxylase activity corresponding to a potentially autosomal dominant pattern of inheritance.
3-methylcrotonyl-CoA carboxylase belongs to a group of enzymes called biotin-dependent carboxylases, which are widely distributed in nature. Beside 3-methylcrotonyl-CoA carboxylase, two other members of this family are involved in human diseases: propionyl-CoA carboxylase (EC 6.4.1.3 activity is impaired in patients affected by propionic aciduria). Defective pyruvate carboxylase (EC 6.4.1.1) results in private carboxylase deficiency. Studies in pseudomonas aeruginosa have unraveled the crystal structures of 3-methylcrotonyl-CoA carboxylase and revealed unanticipated features in the architecture of the holoenzymes, particularly important structural differences between 3-methylcrotonyl-CoA carboxylase and propionyl-CoA carboxylase despite strong sequence homologies between these two enzymes (25; 52).
Biotin-dependent carboxylases have two distinct enzymatic activities and catalyze their reactions in two steps. In the first, a biotin carboxylase component catalyzes the MgATP-dependent carboxylation of the N10 atom of the biotin cofactor using bicarbonate as the CO2 donor while biotin is covalently linked through an amide bond to a lysine side chain in the biotin carboxyl carrier protein component. In the second step of the reaction, a carboxyltransferase component catalyzes the CO2 transfer from carboxybiotin to the acceptor of the carboxyl group (52).
The human 3-methylcrotonyl-CoA carboxylase protein has been described (08). It comprises a larger alpha subunit and a smaller beta subunit, which form alpha6beta6 dodecamers (04; 19; 24). The alpha subunit is encoded by the MCCC1 (formerly MCCA, 19 exons, 3q25-q27) gene and contains biotin carboxylase and biotin carboxylase carrier domains. The beta unit is coded by the MCCC2 gene (formerly MCCB, 17 exons, 5q12-q13) and contains the carboxyltransferase domain (04; 19; 24).
The enzyme is located in the inner membrane of mitochondria. It is also expressed in human fibroblasts and lymphocytes.
Major advances in the understanding of the molecular mechanism of this disease have been made. Baumgartner and coworkers were able to demonstrate the dominant negative effect of the mutated allele MCCC1 p.-R385S, ie, its ability to reduce the activity of the MCCA wild type in a compound heterozygous state (05; 03). Additionally, the cleavage sites and the targeting signals of methylcrotonyl-CoA carboxylase have been identified by site-directed mutagenesis, an important prerequisite to understand the functional relevance of mutations (46). Cryptic exon activation by disruption of an exon splice enhancer by a particular point mutation (c.1054G> A mutation in exon 11 of MCCB gene) has been described as another mechanism (48).
One mutation with a clinically completely asymptomatic phenotype has been identified in the Amish population in Lancaster County of Pennsylvania (USA) and could be traced back to their Swiss ancestors (04; 19). Two series of mutations in the MCCA and MCCC2 locus have been reported (10; 35).
In patients, the activity of 3-methylcrotonyl-CoA carboxylase measured in fibroblasts is severely reduced to 0% to 12% of normal without response to biotin (49). Due to the enzymatic block, 3-methylcrotonyl-CoA accumulates and is further hydrated to 3-hydroxyisovaleryl-CoA by crotonase and deacylated to 3-hydroxyisovaleric acid.
The other important pathway is the conjugation with glycine by glycine-N-acylase to form 3-methylcrotonylglycine. Usually, the excretion via 3-hydroxyisovaleric acid is more prominent, but there are also patients with opposite proportions reflecting a different activity of glycine N-acylase (37). Secondary severe carnitine depletion, which is known to develop in many patients, is caused by urinary excretion of acylcarnitine metabolites. The most characteristic acylcarnitine is 3-hydroxyisovalerylcarnitine formed from the conversion of 3-methylcrotonyl-CoA to methylcrotonyl carnitine and then to 3-hydroxyisovalerylcarnitine (41).
A first MCCA-deficient skin fibroblast transcriptome was published pointing to mitochondrial dysfunction, decreased antioxidant response, and disruption of energy homeostasis as possible mechanisms of pathophysiology (55).
The incidence of 3-methylcrotonylglycinuria is difficult to estimate without a broad registration of patients or large-scale newborn screening databases. It appears to range between 1:30,000 and 1:70,000, making 3-alpha-methylcrotonylglycinemia the most frequent organic aciduria known to date (42; 01). In countries where 3-methylcrotonylglycinuria is not part of the routine newborn screening, the diagnosis can be easily missed. Gallardo was the first to report an unexpectedly high incidence through newborn screening. An incidence of 1:64,000 live births was reported in North Carolina (17); 3-alpha-methylcrotonylglycinuria is also frequently found in newborn screening programs in Germany (42), Taiwan (36), California (30), Korea (28), Israel (39), China (32), and Slovenia (45).
3-alpha-methylcrotonylglycinuria is easily identified in newborn screening programs using electrospray tandem mass spectrometry. Early treatment can be initiated before the first metabolic decompensation occurs, and this early treatment may help ensure a good long-term prognosis (49; 02). However, it remains unclear whether and which continuous treatment with low-protein diet, carnitine, and glycine supplementation has the potential to prevent the clinical course in severely affected patients, and conversely, whether treatment is at all necessary in the great majority of asymptomatic patients. In a retrospective analysis from the metabolic unit in Boston, all patients affected by 3-alpha-methylcrotonylglycinuria came to medical attention by newborn screening, and no patient was diagnosed by clinical symptoms (31). Considering the high number of apparently healthy individuals incidentally detected by extended newborn screening programs, it is uncertain whether this condition warrants inclusion into newborn screening programs (53). It has, therefore, been excluded from the panels of inborn metabolic diseases that are screened for in Germany and Israel (22; 39). It is not yet possible to differentiate patients with a low risk for developing disease from those with a high risk (02; 43; 16).
A status report about newborn screening worldwide has been published and shows the divergent landscape: 3MCC is part of the USA RUSP, but not of the Canadian panel; newborn screening has meanwhile been discontinued in New Zealand due to lack of clinical benefit for the child (50). It is screened for in Japan, Korea, Philippines, Singapore, Sri Lanka, Taiwan, and Thailand.
The acute metabolic decompensation can be similar to that in other organoacidopathies, fatty acid oxidation disorders, or urea cycle disorders.
Persistently elevated 3-hydroxy-isovalerylcarnitine in newborns detected by newborn screening programs can also be caused by maternal 3-alpha-methylcrotonylglycinuria (20; 15). Additionally, carriers of one deleterious mutation, mostly in the MCCA gene, may show elevated metabolites suggestive for 3-methylcrotonyl-CoA carboxylase deficiency in newborn screening or specific metabolic investigations (34).
The catabolic pathway of leucine comprises six enzymes, each of which can be affected by monogenic defects.
MSUD and isovaleric acidemia (deficiency of isovaleryl-CoA dehydrogenase) are described in separate clinical summaries. Organoacidopathies can best be differentiated by their specific patterns in gas chromatography-mass spectrometry analysis of nonvolatile urinary organic acids and their associated abnormalities in an acylcarnitine profile.
As biotin is the cofactor of all carboxylases (propionyl-CoA carboxylase, 3-methylcrotonyl-CoA carboxylase, acetyl-CoA carboxylase, and pyruvate carboxylase), deficiencies in the biotin pathway result in multiple carboxylase deficiency (biotinidase and holocarboxylase synthetase deficiency). These can be distinguished from 3-methylcrotonylcarboxylase deficiency by the presence of additional pathological metabolites, such as 3-hydroxypropionic acid, methylcitric acid, and lactic acid. The definitive diagnosis is by enzyme assay or mutation analysis.
3-methylcrotonylglycinuria should be considered not only in patients with typical concomitants of organic acidurias but also in patients presenting with Reye syndrome, hypoglycemia, and unexplained encephalopathies. As in other organic acidurias, the clinical presentation can be nonspecific, so differentiation by laboratory workup is essential. The best way to distinguish the organic acidurias is through analysis of urinary nonvolatile organic acids by gas chromatography-mass spectrometry and acylcarnitine profiles by tandem mass spectrometry (23; 56). Newborn screening of acylcarnitine profiles using tandem mass spectrometry leads to early diagnosis and initiation of therapy (42; 36; 28). Nevertheless, the inclusion of 3-methylcrotonylglycinuria into national newborn screening programs remains controversial, even though the condition is included in the recommended uniform screening panel core conditions in the United States and some European countries (27); more information can be accessed at the following site:www.hrsa.gov/advisory-committees/heritable-disorders/rusp). Other countries, such as Germany, have excluded it from the national screening panel because of the high percentage (greater than 90%) of individuals who will remain asymptomatic (47).
In 3-methylcrotonyl-CoA carboxylase deficiency, the urinary organic acid profile reveals high excretion of 3-hydroxyisovaleric acid (500 to 7000 mmol/mol creatinine, compared with 1 to 20 mmol/mol creatinine in healthy individuals and 50 to 200 mmol/mol creatinine in patients with severe ketosis from other causes) as well as 3-methylcrotonylglycine (50 to 4000 mmol/mol creatinine, normally not detectable) (49). Elevations of 3-hydroxybutyric, acetoacetic, and dicarboxylic acids may be present in ketotic patients, but there is no elevation of 3-hydroxypropionic, methylcitric, or lactic acids that is present in multiple carboxylase deficiency. Examination of the carnitine status may reveal a low total carnitine level, an elevated ratio of esterified to free carnitine, and a high elevation of 3-hydroxyisovalerylcarnitine in plasma or dried blood spots. 3-hydroxyisovalerylcarnitine can be readily detected in newborn screening. 3-methylcrotonylcarnitine is sometimes but not always present. Two cases have been reported of patients with 3-alpha-methylcrotonylglycinuria deficiency in fibroblasts and homozygous mutations in the MCCB gene who did not have elevated 3-methylcrotonylglycine levels and had only moderate elevations of 3-hydroxyisovaleric acid (54).
The definitive diagnosis is made by identification of pathogenic mutations and/or assay of enzyme activity in fibroblasts or leukocytes with significant reduction of enzyme activity of 0% to 12% of control activity and, except in rare instances (05), no response to biotin (07). It is important to exclude multiple carboxylase deficiency up to demonstrating normal enzyme activities of propionyl-CoA carboxylase, pyruvate carboxylase, and biotinidase. An example of a work-up in an asymptomatic girl has been published (09).
Prenatal diagnosis is possible by stable isotope dilution analysis of amniotic fluid, which shows elevated 3-hydroxyisovaleric acid by enzyme activity assay in cultivated amniocytes or chorionic villi material (26). With the description of the gene locus, molecular prenatal diagnosis has also become possible in families with known mutations. In view of the likely nature as a benign variant, it is, however, difficult to envision indications for prenatal diagnosis for 3-methylcrotonyl-CoA carboxylase deficiency.
A diagnostic algorithm for elevation of C5OH acylcarnitine in newborn screening has been published by the American College of Medical Genetics (available at www.acmg.net), as well as a Delphi-based clinical practice protocol for the diagnosis and management of 3-alpha-methylcrotonylglycinuria (01).
Because data regarding the correlation between the degree of metabolite elevation and definite diagnosis and metabolic risk are sparse, the evaluation of the significance of persistently elevated metabolites and the decision about possible metabolic treatment has to be done in the clinical context of the individual patient and the pattern of abnormal metabolites in close discussion between clinicians and the diagnostic laboratory. Mutation analysis is now the most practical approach in most cases to confirm the diagnosis, and enzyme studies may only be necessary in selected cases.
As discussed, there has been considerable and even increasing uncertainty about the clinical relevance of 3-methylcrotonyl-CoA carboxylase deficiency (43; 53). The main objective for patients is to prevent metabolic decompensation and crisis. During intercurrent illnesses up to acute decompensation, 3-methylcrotonylglycinemia is treated like other organic acidemias. Measures include increasing the provision of energy by 20% to 100% above the recommended daily requirements using carbohydrate (dextrose 20%) and fat (intralipid 20%) via oral, nasogastric, or intravenous routes. Insulin can be administered to avoid hyperglycemia and to support intracellular glucose uptake. The intake of natural protein is stopped for 24 to 48 hours and then is reintroduced gradually as tolerated (23). As mentioned below, augmented doses of glycine and L-carnitine are recommended (49). The New England Consortium of Metabolic Programs has created a list of emergency recommendations of the management of the acute illness in 3-alpha-methylcrotonylglycinuria patients, which can be found at: newenglandconsortium.org.
Patients with significant metabolic symptoms should be supplied with an emergency card, letter, or bracelet containing instructions for emergency measures and phone numbers. Logistics of emergency therapeutic measures should be regularly reviewed with the family and the primary care physicians.
As leucine is an essential amino acid, the catabolic pathway is challenged by increased protein intake or increased endogenous protein degradation (49). No randomized controlled studies of leucine-restricted diets have been published, thus, the efficacy of such diets to prevent or ameliorate symptoms remains doubtful. Therefore, decisions regarding diets in symptomatic children or mothers have to be individualized based on severity of symptoms, response to a diet trial, or other factors (01).
Carnitine depletion has been described in 3-methylcrotonyl-CoA carboxylase deficiency; therefore, L-carnitine levels should be monitored in plasma, aiming at normal levels of free carnitine (51). Supplemental carnitine should be initiated if free carnitine is very low. In previously symptomatic patients L-carnitine in a dosage of 40 to 100 mg/kg per day is used to prevent carnitine depletion during intercurrent illness (01).
In addition, detoxification is possible with glycine via 3-methylcrotonylglycine, which has been shown to be effective in well-documented cases. It seems to be especially effective during a crisis, but chronic prophylaxis is also a possibility (40). Therapeutic guidelines recommend a dosage of 150 mg/kg per day of glycine while the patient is stable. The dosage can be augmented up to 600 mg/kg per day during metabolic crisis. However, no randomized clinical trials have been published and a panel of metabolic specialists from Canada and the United States did not recommend the use of glycine (01).
The widely used anticonvulsant drug valproate has been shown to inhibit 3-methylcrotonyl-CoA carboxylase and should not be considered in patients with inborn errors of leucine degradation (33).
With the widely varying clinical spectrum of symptoms, it is difficult to make definite statements about the prognosis of 3-methylcrotonylglycinuria. In the first reports, 3-methylcrotonylglycinemia was associated with a poor prognosis as patients succumbed during acute decompensation or suffered severe sequelae (49). It was reasonable to assume that initiation of treatment before severe metabolic decompensation would significantly ameliorate the patient’s prognosis up to a completely normal psychomotor development.
On the other hand, almost all “patients” identified by newborn screening never develop symptoms, even without treatment. The lack of genetic or biochemical markers to differentiate these individuals from those who are at risk for decompensation has led to controversy about the benefit of newborn screening for 3-methylcrotonylglycinuria (22; 47; 02; 43; 16; 53).
Women with methylcrotonylglycinuria were identified for the first time by detection of 3-hydroxyisovalerylcarnitine in the newborn screening passed on to their babies (20; 15). These women did not show any significant clinical symptoms during pregnancy, labor, or childbed; and their children were healthy.
Patients may be at risk for metabolic decompensation by catabolism induced by fasting, anesthesia, or a surgical procedure. In such situations it is important to meet increased energy requirements by intravenous administration of glucose (23). Supplementation with glycine or carnitine should be continued or increased. No information is available about the particular side effects of drugs normally used to induce anesthesia in patients with methylcrotonylglycinuria.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Friederike Hoerster MD
Dr. Hoerster of University Children's Hospital in Heidelberg, Germany, has no relevant financial relationships to disclose.
See Profile
Georg F Hoffmann MD
Dr. Hoffmann of the University Center for Child and Adolescent Medicine in Heidelberg has no relevant financial relationships to disclose.
See Profile
Deepa S Rajan MD
Dr. Rajan of UPMC Children's Hospital of Pittsburgh has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026