





Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Alexander disease is a leukodystrophy that may present at any age. Following the identification of pathogenic variants in the glial fibrillary acidic protein (GFAP) gene as the cause of Alexander disease, an increasing number of patients have been identified. The disease is caused by a combination of the formation of characteristic aggregates, called Rosenthal fibers, and the sequestration of the protein chaperones alpha B-crystallin and HSP27 into Rosenthal fibers. GFAP levels are consistently elevated in the CSF of patients with Alexander disease. The diagnosis is strongly suggested by MRI and confirmed by GFAP gene analysis. Cerebrospinal fluid GFAP levels are an important disease biomarker. Although a commercial assay for GFAP exists, clinical testing is not currently available. Current therapeutic efforts are geared towards reducing the expression of the mutated GFAP allele by using antisense oligonucleotides.
|
• Alexander disease is an autosomal dominant glial cell disease most often caused by de novo heterozygous pathogenic variants in the GFAP gene. | |
|
• In children in particular, seizures, developmental delay, macrocephaly, or an MRI with specific abnormalities typically involving frontal white matter should suggest the diagnosis. | |
|
• Alexander disease is a leukodystrophy in young children but may present as a glial tumor in older patients and bulbar dysfunction in adults. | |
|
• Disease progression is typically slow, with variable rates of disability accumulation and life expectancy. | |
|
• Antisense suppression of GFAP is currently being tested in clinical trials as a treatment for Alexander disease. |
The first reported case of Alexander disease was described as "progressive fibrinoid degeneration of fibrillary astrocytes" in an infant with mental retardation and hydrocephalus (01). The disorder’s defining feature has always been the widespread presence of abnormal astrocytic inclusions within the brain called Rosenthal fibers. The disorder has also been called "megalencephaly associated with hyaline pan-neuropathy," "fibrinoid leukodystrophy," "leukodystrophy with diffuse Rosenthal fiber formation," and "dysmyelinogenic leukodystrophy with megalobarencephaly.
Over time, additional phenotypes have been recognized. For many years, an age-based classification system was used to capture neonatal, infantile, juvenile, and adult-onset forms (48; 07; 02; 23; 44; 50; 25; 54; 34). Although these phenotypes have some differences in clinical and radiographic manifestations, all forms of the disorder are caused by heterozygous, pathogenic variants in the gene for GFAP, a component of astrocytic intermediate filaments (08; 36; 47; 13; 40; 41). As such, the pathologic hallmark is the same for all phenotypes: the presence of widespread and abundant Rosenthal fibers.
The age-based classification system (defined by age at symptom onset) was defined prior to the identification of GFAP as the causative gene. Thus, a study used the age of symptom onset and the GFAP variant site to statistically identify only two clinical types of Alexander disease (45). Type I is characterized by early-onset, seizures, macrocephaly, motor delay, encephalopathy, failure to thrive, paroxysmal deterioration, and typical MRI features. Type II is characterized by later-onset, autonomic dysfunction, ocular movement abnormalities, bulbar symptoms, and atypical MRI features.
Concurrent with the Prust classification (type I vs. type II disease), Yoshida and colleagues proposed a 3-group system based on clinical and imaging features (64). The cerebral form presents with developmental delay, seizures, and macrocephaly with frontally predominant white matter T2 hyperintensities on MRI in addition to other MRI features, whereas the bulbospinal form presents with bulbar dysfunction and long-tract signs that reflect medullary signal abnormalities or atrophy. The authors also described an intermediate form that captures the core features of both the cerebral and bulbospinal forms.
In summary, typical and atypical cases have been reported for each of the classification systems.
|
• The classification systems (neonatal, infantile, juvenile, adult; type I vs. type II; cerebral vs. intermediate vs. bulbospinal) do not entirely overlap as there is considerable clinical and imaging heterogeneity in Alexander disease. For simplicity, typical cases are highlighted in this section. | |
|
• Neonatal disease (younger than 30 days) presents with poor feeding, weak suck, and hypotonia in the first 30 days of life. Seizure, macrocephaly, and hydrocephalus often occur in the first year of life. | |
|
• Infantile/type I/cerebral disease (younger than 2 to 4 years) is typically recognized by the triad of developmental delay, seizures, and macrocephaly. | |
|
• Juvenile cases (2 to 14 years) often have normal developmental milestones and present in the first or second decade with bulbar dysfunction, failure to thrive, short stature, and scoliosis. | |
|
• Adult/type II/bulbospinal disease (older than 14 years) presents with gait and balance difficulties, bulbar symptoms, and dysautonomia. | |
|
• The intermediate type combines features of the cerebral and bulbospinal types and often presents in preteenagers. | |
|
• There is considerable clinical and radiologic variability. |
Infantile/type I/cerebral. This phenotype is perhaps the most commonly recognized form of Alexander disease. The typical features include developmental delay, seizures, and macrocephaly due to megalencephaly (01; 07; 02; 44; 25). Usually, the onset is during the first or second year of life. Of note, the presentation is often insidious, with minor developmental delays in the first 2 years of life or a single provoked or unprovoked seizure, and macrocephaly may be a later finding (27). Thus, a high index of suspicion is needed as a mild clinical presentation may not immediately suggest the diagnosis, especially in the absence of macrocephaly.
A neonatal form is known as well (54; 28). Both sexes are affected (the earlier suggestion of male predominance probably resulted from the small number of cases). The initial signs are often feeding difficulties in the first month of life, typically from a weak suck and hypotonia. Thus, reflux, vomiting, and failure to thrive often precede the more apparent neurologic presentation. Early neurologic features include hyperexcitability and myoclonus. Infants often struggle to obtain early milestones (delayed head control and rolling), and most do not achieve sitting, crawling, or ambulation. If milestones are achieved, infants subsequently lose developmental milestones and develop progressive neurologic regression of motor and language skills as well as spastic quadriparesis. Some infants have a more acute onset of symptoms early in the first year that are often accompanied by hydrocephalus with increased intracranial pressure. In a few cases, megalencephaly may be present from birth. Seizures are common; infantile spasms are rare but have been reported (58). Death usually occurs between 2 and 10 years of age, but children with a more acute onset or exhibiting obstructive hydrocephalus may die in the first or second year of life (54).
Juvenile. The less common juvenile form of Alexander disease generally occurs between the ages of 6 years and the middle teens, but age of onset is variable (48; 07; 44; 25). Such early diagnosed cases challenge the age-based classification system. Typically, the juvenile form manifests as predominant bulbar or pseudobulbar signs, especially dysphagia, vomiting, difficulty talking, ataxia and hyperreflexia, and spastic paraparesis or quadriparesis that is often more severe in the lower extremities. These signs are all slowly progressive and are more important than age of onset for differentiating the juvenile from the infantile form. Cognition often remains intact for much of the illness, and megalencephaly is usually absent. Presentation as a tumor-like lesion, often mistaken for an astrocytoma, is well known (32; 55). Such lesions are commonly (but not exclusively) located in the dorsal medulla (46; 53). In the absence of other clinical and radiological findings of Alexander disease, these patients are frequently diagnosed by biopsy, revealing Rosenthal fibers. Physicians should have a high index of suspicion for Alexander disease in juvenile patients with vomiting or failure to thrive and brainstem lesions in the area postrema and should consider targeted GFAP testing.
Juvenile patients with isolated or predominant brainstem or infratentorial lesions have type II disease. In the absence of seizures, developmental delay, macrocephaly, and frontal white matter abnormalities, they may also be characterized as bulbospinal. Some juveniles may have overlapping cerebral and bulbospinal features (ie, supratentorial and infratentorial involvement) and would be classified as having intermediate disease.
Adult/type II/bulbospinal. The adult-onset form of Alexander disease was thought to be rare (12; 23; 50; 08; 40; 41). Data suggest that it is more common than previously thought (42; 65). Adult-onset Alexander disease must be considered in patients of any age with lower brainstem or cerebellar signs. Pyramidal involvement, cerebellar ataxia, and urinary disturbances are common. Less frequent findings include sleep disorders and dysautonomia (42). When present, palatal myoclonus is strongly suggestive of Alexander disease (51). A presentation with severe vocal cord paralysis during sleep has been described (22). Imaging typically shows symmetric signal abnormalities in the medulla, dorsal medullary mass lesions, or medullary atrophy (even in the absence of signal changes). Cerebellar involvement may also be present. Its pathology closely resembles the pathology of Alexander disease in children, with widespread Rosenthal fibers. However, the adult disorder is varied and has been described in adults from their late teens to 82 years of age, both with and without clinical neurologic symptoms. The presentations are highly variable and sometimes resemble multiple sclerosis (due to spasticity, bladder involvement), a tumor (more common in younger individuals), or spastic paraplegia (due to progressive gait changes) (43). Generally, the adult-onset Alexander disease cases have later onset and longer survival compared to individuals suffering from the others forms of this disorder.
The infantile and juvenile forms are usually sporadic. In adult-onset cases, there is a much greater tendency toward familial incidence with autosomal dominant inheritance (23; 41; 57). Other cases of adult onset are sporadic (08).
Whether of the neonatal, infantile, juvenile, or adult-onset form, Alexander disease is inexorably progressive, but the rate of functional loss is variable (39; 59). It is generally slowest in the adult and juvenile forms, where mental function may be retained for much of the course as the frontal lobes are typically less involved on MRI compared to the neonatal and infantile forms. The major complications in children are seizures, which can contribute to neurologic regression. Some children with the neonatal or infantile form have obstructive hydrocephalus, which may require intervention. All patients with Alexander disease are at risk for respiratory infection, respiratory failure, urinary tract infection, scoliosis, sleep apnea, vomiting, or failure to thrive (from bulbar dysfunction). Patients of all ages may require gastrostomy feeding, tracheostomy, or scoliosis repair, but this is highly variable and depends on the goals of care.
Case 1. A boy was the product of a normal pregnancy and delivery. The parents were nonconsanguineous, there was no family history of neurologic disease, and an older half-sister was healthy. Within the first month of life, he developed nonprojectile vomiting with every feed. Symptoms were attributed to formula intolerance and reflux. He had slow weight gain and two hospitalizations for dehydration in the first several months of life. Around 5 months of age, he was evaluated by his pediatrician for weakness, loss of milestones (loss of social smile and interest in toys), and inability to elevate his head and shoulders while lying prone. His examination was notable for poor head control, decreased global tone, and the inability to sit. Head circumference had remained at the 50th percentile since birth. A brain MRI revealed white matter and basal ganglia T2 hyperintensities and swelling as well as a periventricular rim and brainstem involvement.
After the MRI was obtained, he was confirmed to have a pathogenic variant in GFAP (R239H). Subsequently, he developed profound hypotonia, mixed sleep apnea requiring oxygen, dysphagia, constipation, and feeding intolerance. A gastrotomy tube was placed at 8 months of age. Despite the nutritional support, he continued to have difficulties with feeding intolerance. Although he had several events concerning for seizures, several long-term monitoring electroencephalograms did not have epileptic discharges. The last recorded EEG was normal. By 7 months of age, he developed hydrocephalus, and an endoscopic third ventriculostomy was performed. Two months later, his mental status worsened. An MRI with a flow study revealed mass-like expansion of the optic chiasm and midbrain causing complete obstruction of the cerebral aqueduct and marked narrowing of lower aspect of the floor of the third ventricle above the ventriculostomy. He subsequently underwent a right frontal external ventricular drain placement followed by a ventriculoperitoneal shunt. Despite aggressive management, he died at 18 months of age.
Case 2. An 11-month-old female had a prolonged seizure in the setting of mild congestion (no fever). An MRI was obtained.
She was not treated with antiseizure medications, but she had a second seizure 1 week later and was then started on an antiseizure medication. Genetic testing revealed a pathogenic de novo variant in GFAP. She walked at around 14 months of age. At 2 years of age, she still spoke in single words, and speech therapy was initiated. She had some bulbar symptoms (occasional choking on solids). At around 5 years of age, she developed vomiting and failure to thrive, which were managed with dietary adjustments and medications.
At 7 years old, she remains ambulatory, but her ankles pronate with walking. She is dysarthric but speaks in full sentences, and she attends school where she receives physical, occupational, and speech therapy. Head circumference has been steadily increasing, from the 50th percentile at around 4 years of age to the 95th percentile at 7 years of age.
Case 3. A 10-year-old female presented with intractable vomiting. She had severe motion sickness as a younger child but was otherwise healthy, with normal development, normal head circumference, and no history of seizures. At 9 years of age, her linear growth slowed. At around 10 years of age, emesis began sporadically. Six months later, she was having one to six episodes of vomiting a day. She was ultimately admitted for emesis, weight loss, and dehydration. Due to persistent vomiting, an MRI was obtained, revealing a brainstem lesion.
A de novo pathogenic variant in GFAP was found (R416W). At 17 years of age, her height is at the second percentile. Macrocephaly is absent, and her neurologic examination is normal. Her symptoms and imaging are also consistent with type II and bulbospinal phenotypes.
Case 4. A male in his thirties developed balance and bulbar difficulties. He described “episodes of choking and gasping for air” and “choking on saliva.” These events were attributed to stress. Of note, his uncle was diagnosed with multiple sclerosis in the distant past (45 years ago). At around 48 years of age, he developed shortness of breath and daytime somnolence. Obstructive sleep apnea was suspected, and he was diagnosed with vocal cord paralysis with significantly decreased airflow. An urgent tracheostomy was performed. Three months later, an MRI was obtained. The scan was notable for cerebellar and spinal cord volume loss. Periventricular and basal ganglia changes were also present on MRI.
He was ultimately confirmed as having a pathogenic variant of GFAP at 48 years of age. A year later, he began to walk with a cane. At the age of 51, he was hospitalized for a febrile urinary tract infection. At the age of 54, he started to use a powered mobility device. He also developed an oxygen requirement. His examination was notable for a tracheostomy, dysarthria, difficulty with secretions, abnormal pursuits with left-beating nystagmus, weakness in hip flexors and extensors, abnormal balance with cerebellar signs, brisk reflexes, and extensor plantar responses. He died at 57 years of age.
|
• Alexander disease is caused by a gain-of-function mutation in GFAP. | |
|
• The accumulation of aberrant GFAP leads to the formation of Rosenthal fibers. |
Alexander disease is a caused by a heterozygous, dominant pathogenic variant in the gene for the astrocytic intermediate filament glial fibrillary acidic protein GFAP (08; 36; 47; 13; 33; 40; 41; 52; 34). To date, there are more than approximately 180 distinct pathogenic variants. The great majority of pathogenic variants occur in exon 1, exon 4, or exon 8. The severity of disease does not strictly parallel the particular variant present, but in some instances, a general correlation exists (47; 17; 37).
Almost all cases of childhood Alexander disease (neonatal and infantile) are sporadic and are believed to occur de novo in one of the alleles for GFAP (08; 47; 13; 30). Most pathogenic variants are missense, although other types have been reported (https://www.ncbi.nlm.nih.gov/clinvar/?gr=0&term=gfap%5Bgene%5D&redir=gene). An in-frame deletion of exon 5 has been described, suggesting abnormal secondary structure or solubility characteristics analogous to the presumed pathology of the more commonly reported GFAP missense variants (15). For some individuals, no GFAP variants have been identified by sequencing (08; 47). Although this has led some to hypothesize that there may be additional causes of Alexander disease, aberrant GFAP splicing affecting other isoforms of GFAP have also been implicated (21). Adult-onset cases are more frequently inherited as autosomal dominant heterozygous pathogenic variants in GFAP (08; 40; 41). However, the rare occurrence of affected siblings has been reported for the infantile form and for a set of siblings with either juvenile or adult-onset disease (63; 12). Alexander disease has also been reported in two sets of monozygotic twins (33; 52). For most families, the disorder originates from a de novo germline mutation; however, affected siblings due to presumed gonadal mosaicism and individuals with somatic mosaicism have also been observed (16).
How the pathogenic variants in GFAP lead to the disease and the pathogenesis of this disorder are poorly understood. They seem to be associated with a marked increased GFAP turnover in the cell (38). All clinical forms are characterized pathologically by the presence of widely distributed Rosenthal fibers and by greatly increased numbers of astrocytic intermediate filaments. In the infantile form especially, the brain exhibits a pronounced lack of myelin. The brain in infantile cases is heavy for the age and shows the greatest involvement bilaterally in the frontal lobes, where the white matter is discolored, shrunken, and may show cystic degeneration and cavitation. The lesions frequently extend to involve the subcortical U or arcuate fibers. The basal ganglia are also affected early, but relative sparing of the occipital lobes and cerebellum occurs. Children with severe hydrocephalus have extensive Rosenthal fiber involvement of the periaqueductal area, which may cause obstruction. The neurons are generally not affected, but in advanced cases there may be some axonal loss. In juvenile cases, the greatest involvement is in the brainstem, with plentiful Rosenthal fibers and less severe, but variable, hypomyelination or demyelination. Adult-onset cases may show axonal loss with brainstem and medullary shrinkage, cerebellar involvement, and scattered Rosenthal fibers.
Rosenthal fibers are cytoplasmic inclusions that occur only in astrocytes. Under the light microscope, Rosenthal fibers appear as round or elongated hyaline bodies that are stained with eosin, Luxol Fast blue, Heidenhain hematoxylin, and phosphotungstic acid-hematoxylin. In the electron microscope, Rosenthal fibers are electron dense, osmiophilic, granular, nonmembrane-bound bodies up to 50 microns or more in size.
The Rosenthal fibers are surrounded by and intimately related to dense aggregates of glial intermediate filaments that often penetrate into the Rosenthal fibers. In Alexander disease, Rosenthal fibers are present both in astrocytic processes and end-feet, and also in perikarya, where they are small and frequently multiple. They are characteristically found in subpial, subependymal, and perivascular locations in Alexander disease, as well as diffusely in the white matter. In addition to the astrocytes with Rosenthal fibers, other astrocytes may be enlarged and have a bizarre appearance, and there may be a fibrillary gliosis. The particular GFAP variant present does not appear to have any effect on the appearance or qualities of the Rosenthal fibers.
Rosenthal fibers are not unique to Alexander disease because they also sometimes occur, but with a more localized distribution, in other conditions (astrocytomas, chronic glial scars, tuberous sclerosis and hamartomas, multiple sclerosis, and Alzheimer disease). Although Rosenthal fibers may be perivascular in these other conditions, subependymal and subpial collections are not generally found. The widespread and characteristic distribution of Rosenthal fibers throughout the neuraxis is unique to Alexander disease.
Much about the composition of Rosenthal fibers has been identified, but their relation to the myelin deficits in Alexander disease is unclear, and no difference has been noted between the composition of Rosenthal fibers in Alexander disease and in other conditions. Rosenthal fibers can be difficult to label immunocytochemically at the light microscope level, probably due to poor penetrability. However, at the electron microscope level, Rosenthal fibers in Alexander disease are immunoreactive for GFAP (26; 56). They also contain ubiquitin and two small heat shock proteins, alphaB-crystallin and HSP27 (56). AlphaB-crystallin has been purified from isolated Rosenthal fibers and appears to be a major constituent. Because elevated levels of these heat shock proteins can occur in astrocytes without Rosenthal fibers and in other glia, their presence is not unique to Rosenthal fibers or to Alexander disease. Heat shock proteins are believed to function as chaperones and increase in reaction to stress (20). Animal studies in mouse models that over-express GFAP crossed with Cryab (alphaB-crystallin) knockout mice show that AlphaB-crystallin plays a critical role in tempering Alexander disease pathology. In Alexander disease, some unidentified stress (possibly caused by the mutated GFAP) leads to up-regulation of these heat shock proteins and to the formation of the Rosenthal fibers. In addition, Rosenthal fibers in Alexander disease are immunoreactive for advanced glycation and lipid peroxidation end products, which suggests a role for oxidative stress (09).
Alexander disease is considered an example of a positive dominant disease. Evidence suggests that the basic pathology results from a combination of overexpression of GFAP in certain brain regions and the toxic effect of the mutated protein (35). Various models suggest that to eliminate toxicity, one has to reduce the expression of mutated allele to a level as close as possible to zero (35). The basis of the myelin defect in Alexander disease is not known. It is particularly severe in the infantile form and in areas of white matter with abundant Rosenthal fibers. It can be considered, in large part, as a deficiency of normal myelination or hypomyelination, rather than demyelination. Little evidence exists of macrophage or microglial reaction or of sudanophilia, indicating that myelin destruction is not likely to be the primary process.
One hypothesis on the cause of hypomyelination posits that the involvement of the astrocytes by the underlying abnormality interferes with their supportive role in myelination, and that the timing of this involvement in infantile cases coincides with the time of frontal lobe myelination. This is consistent with the more normal presence of myelin in the lower neuraxis and posterior cerebrum and the frontal predominance of hypomyelination in young cases. The same hypothesis can also be invoked to explain the less severe degree of a lack of myelin in the juvenile and adult-onset forms because myelin may have already been formed before the astrocytic involvement became severe.
It has also been proposed that a defect is located in the blood-brain barrier, which could be caused by the astrocytic abnormality. Increased endothelial pinocytic activity has been reported as an early finding in the periventricular frontal lobe and basal ganglia of infantile cases, areas that are affected early in the disease process. Likewise, early in the course of the infantile form, CT contrast enhancement is often present in the same areas that also show increased density in unenhanced scans as well as intense Rosenthal fiber deposition. This suggests that the presence of Rosenthal fibers and the increased vascularity and pinocytic activity may be related. In contrast to other intermediate filament disorders, the GFAP pathogenic variants appear to cause a gain of function (30). Astrocytes derived from Alexander disease patient-induced pluripotent cells inhibit proliferation of human oligodendrocyte progenitor cells in coculture and reduce their myelination potential (29). The secreted glycoprotein CHI3L plays a role in that noncell autonomous inhibitory effect (29).
Alexander disease is a rare disorder; the most recognized subtype is the infantile form. One study reported a birth prevalence for infantile Alexander disease of 1 in 1.3 million births (04). The later presentations are less commonly identified, perhaps due to milder symptoms or lack of genetic testing in individuals with subtle MRI features. A Japanese survey estimated a prevalence of 1 in 2.7 million (64). None of the forms show an association with any particular ethnic group.
The cause of de novo genetic pathogenic variants is unknown; thus, no form of prevention is available. Although prenatal testing can be performed in families where the spontaneous pathogenic variant in the GFAP gene is identified, the utility of this testing is likely to be extremely limited because familial cases of Alexander disease are so rare.
Megalencephaly or macrocephaly. Canavan disease, especially the neonatal presentation, is clinically similar to Alexander disease as both disorders manifest as developmental delay, hypotonia, and macrocephaly (the latter is not universally present in Alexander disease, but it is typically present in Canavan disease). Diffuse white matter involvement is present on MRI in Canavan disease. Although many patients with Alexander disease have frontally predominant white matter changes, diffuse involvement (extending to the occipital subcortical U-fibers) may also occur in Alexander disease. As opposed to Alexander disease, in which vision is preserved, optic atrophy occurs in Canavan disease, and children lose the ability to fix and follow. The disorders can be distinguished based on biochemistry (elevated N-acetylaspartic acid) in urine. MR spectroscopy shows elevated N-acetylaspartic acid in affected white matter in Canavan disease.
Like Alexander disease, Tay-Sachs disease (HEXA disorders) presents in neonates with an exaggerated startle, weakness and progressive loss of motor skills, seizures, and macrocephaly. Tay-Sachs disease can be clinically distinguished from Alexander disease by the classic retinal “cherry-red spot” and biochemically by enzymatic activity of beta-hexosaminidase A and B.
Infants with gross motor delay or regression, seizures, failure to thrive, and progressive macrocephaly may have glutaric acidemia type I. Elevations in glutaric acid and other metabolites raise concern for this disorder.
Multiple sulfatase deficiency has overlapping clinical features, namely developmental delay and neurologic regression, seizures, macrocephaly, and failure to thrive. However, multiple sulfatase deficiency has systemic findings (hepatosplenomegaly, bone disease, cardiac involvement, and ichthyosis) and laboratory abnormalities (decreased sulfatase, increased urinary sulfatides, and increased urinary glycosaminoglycans) as well as prominence of perivascular spaces on MRI (all absent in Alexander disease).
Megalencephalic leukoencephalopathy with subcortical cysts also presents with macrocephaly, gross motor delay, and seizures, but imaging typically reveals subcortical cysts in the anterior temporal lobes (as well as frontoparietal regions), which are not typical of Alexander disease.
Although not a common feature of childhood ataxia with central nervous system hypomyelination (vanishing white matter), 10% of children with advanced disease have macrocephaly (19).
Benign familial macrocephaly and the rare cerebral gigantism (Sotos syndrome) should also be considered, although these disorders lack the other clinical and imaging features of Alexander disease.
In summary, disorders accompanied by megalencephaly, such as Canavan disease, Tay-Sachs disease, glutaric acidemia type I, multiple sulfatase deficiency, megalencephalic leukoencephalopathy with subcortical cysts, vanishing white matter disease, and Sotos syndrome, need to be ruled out. Although there is some overlap in clinical features, these disorders can often be distinguished by the presence of systemic symptoms, specific examination abnormalities, biochemical testing, and imaging features. In addition, biochemical testing (available for some of the disorders) can be obtained to inform the need for targeted versus broad genetic testing. These other leukodystrophies do not manifest either Rosenthal fibers or pathogenic variants in GFAP.
Developmental delay or regression. Alexander disease in children needs to be differentiated from nonprogressive disorders that may give rise to developmental delay and intellectual disability, as well as other leukodystrophies and mitochondrial, metabolic, and genetic etiologies.
Area postrema syndrome. Alexander disease should be considered in juveniles with anorexia, vomiting, hiccups, and weight loss with an area postrema syndrome (46; 53). This may be difficult to distinguish clinically or radiographically from neuromyelitis optica, an acquired neuroinflammatory disorder, although serum testing is available for neuromyelitis optica.
Gait abnormalities or spastic paraparesis. Adults with Alexander disease typically present with gait or balance abnormalities and bladder dysfunction, raising concern for multiple sclerosis, particularly primary progressive multiple sclerosis (31). Imaging and lumbar punctures can be used to distinguish the disorders. Symmetric periventricular and basal ganglia changes, along with medullary atrophy and, for some, cerebellar involvement, are typical of adult-onset Alexander disease.
Diffuse leukodystrophy. Although frontally predominant white matter T2 signal hyperintensities are typical of Alexander disease on MRI, some children have diffuse white matter involvement similar to Canavan disease and other disorders.
Periventricular rim. Periventricular disease often mimics multiple sclerosis, although Alexander disease is more likely to be symmetric and lacks lesions or Dawson fingers.
Gray matter involvement. Given the symmetry of basal ganglia and brainstem T2 hyperintensities and swelling in Alexander disease, a mitochondrial disorder is often considered. Basal ganglia involvement may be present in Alexander disease of all ages, including later-onset forms in which the frontal white matter may be spared.
Brainstem lesions. Symmetric and asymmetric lesions, typically in the dorsal medulla and accompanied by an area postrema syndrome, raise concern for Alexander disease. One approach may include rapid sequencing of GFAP in an individual with medullary lesions prior to biopsy. If a brain biopsy is performed, Alexander disease must be differentiated from other entities associated with Rosenthal fibers, such as hamartomas and tumors.
Seizures and epilepsy. Seizures are typically convulsive, either provoked or unprovoked, with focal or generalized EEG abnormalities.
Gastrointestinal reflux disease. Reflux is common and is likely due to hypotonia and bulbar dysfunction.
Dysautonomia. Syncope and dizziness are common in adult-onset/type II/bulbospinal disease due to bulbar dysfunction.
Sleep apnea. Bulbar dysfunction can cause sleep apnea.
Palatal myoclonus. Palatal myoclonus, vocal cord paralysis, and spastic dysphonia are more common in adult/type II/bulbospinal disease but are not universally present.
Urinary dysfunction. Urinary tract infections and incontinence can be caused by an upper motor neuron bladder.
Spasticity. Tone is often variable (hypotonia and hypertonia). Management should include physical therapy and stretching; medications, such as muscle relaxants; or injectable focal therapy (Botox or phenol injections).
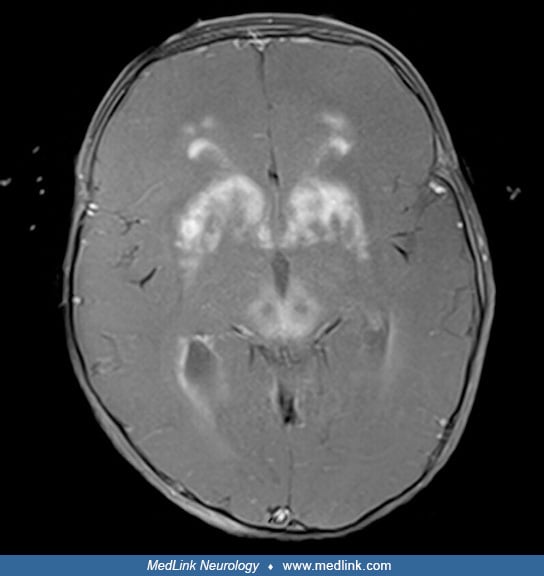
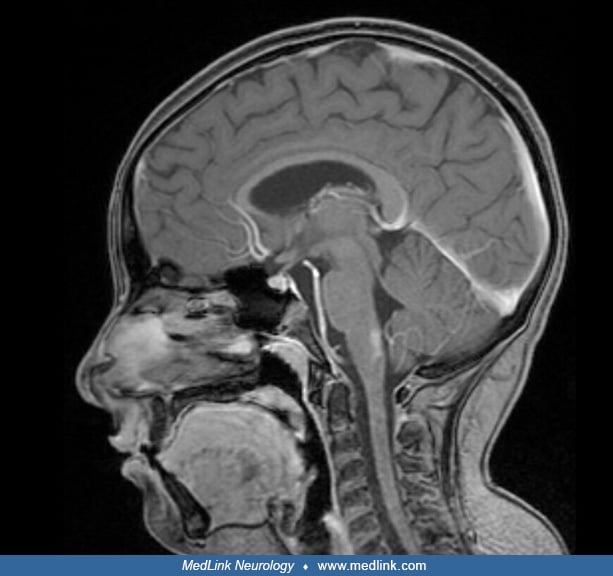
Almost all cases of Alexander disease can now be diagnosed by genetic analysis for pathogenic variants in GFAP. In addition, the MRI is often diagnostic (49). The typical MRI abnormalities include extensive cerebral white matter changes with frontal predominance, a periventricular rim with high signal on T1-weighted images and low signal on T2-weighted images, abnormalities of basal ganglia and thalami, brainstem abnormalities, and contrast enhancement of particular gray and white matter structures (60). The presence of these findings varies among affected individuals. For example, bilateral frontal predominance of white matter involvement with symmetric basal ganglia and dorsal medullary signal abnormalities, along with relative sparing of the occipital lobe and cerebellum, support a diagnosis of infantile/type I/cerebral Alexander disease. Lower brainstem and cervical atrophy are very common in adult/type II/bulbospinal Alexander disease and may be the sole abnormality on brain MRI in patients with the adult-onset form (49; 10). In addition to medullary atrophy, the cervicothoracic spinal cord may also be atrophic with shrunken pyramids (14). Juvenile/type II and other forms may involve signal change on FLAIR in the dorsal medulla and spinal cord as well as abnormalities in the middle cerebellar peduncle and pia (14). Less common MRI variants include predominantly posterior fossa lesions, especially multiple tumor-like brainstem lesions (32; 53), asymmetrical frontal white matter and basal ganglia abnormalities as well as ventricular garlands (62; 61), or even focal central lesions (05). When the lesions are in the brainstem, magnetic resonance spectroscopy may help differentiate between a glioma and Alexander disease (11).
Routine studies on blood, urine, and cerebrospinal fluid are normal in Alexander disease, but GFAP is elevated in the CSF of most patients and sometimes in the blood as well (24; 03). A clinical test is available for plasma GFAP; however, it has not been validated for longitudinal use.
EEG may present slow waves in the frontal region but is not diagnostic (44). EEG may be normal or may reveal focal deficits or generalized spikes and waves.
In patients with typical clinical and imaging features of Alexander disease, with or without a family history, targeted GFAP testing can be obtained. For patients with various or atypical clinical or imaging features, additional biochemical testing may be informative. The absence of urinary organic acid abnormalities or elevated N-acetylaspartic acid and a normal leukocyte hexosaminidase facilitate the dismissal of other megaloencephalic disorders considered, such as glutaric aciduria type I, Canavan disease, and Tay-Sachs disease. Furthermore, in patients for whom the diagnosis is not apparent, panel testing or whole exome sequencing can be obtained.
|
• No disease-modifying therapy is available; however, clinical trials are underway to determine whether suppression of GFAP by antisense oligonucleotides can reduce neurologic and other complications. | |
|
• Antiseizure medications are frequently prescribed for younger patients. | |
|
• All patients benefit from physical, occupational, speech, and feeding therapies. | |
|
• Feeding and nutritional support are recommended. | |
|
• Many older individuals benefit from respiratory support (sleep apnea). | |
|
• Scoliosis in teenagers and adults is often progressive. |
No specific treatment is available. The care of patients with Alexander disease is entirely supportive and dictated by the current condition and any complications that have arisen. Treatment for seizure prevention is recommended, even if seizures are considered provoked by illness, as some children experience developmental regression after prolonged seizures. Many children have gastrointestinal complications, including failure to thrive, reflux, constipation, abdominal pain, and vomiting. These symptoms can be managed with nutritional support, hydration, and various medication strategies. Assisted feeding procedures often become necessary as the disease progresses. Current evidence suggests that reducing the expression of the mutated GFAP allele by using antisense oligonucleotides to reverse the neuropathology may be the most direct therapeutic approach (18). However, at present, the antisense oligonucleotides reduce the expression of both the wild type and the mutated allele (35).
Life expectancy has been estimated in type I and type II disease. Median survival in type I disease was 17.3 years (standard deviation 3.6 years) compared to 25.0 years (standard deviation 1.9 years) for type II disease (45). However, there is considerable variability given the disease heterogeneity (areas of disease involvement). Neonatal patients often fail to achieve sitting and ambulation and have the shortest life expectancies. Type I/cerebral patients have considerable variability in disease progression: some never achieve ambulation, others walk but lose milestones within the first 5 years of life, and others experience delays but achieve disease stability into or beyond adolescence (39). Morbidity and mortality can be reduced with aggressive and proactive symptom management.
Seizures in children are often responsive to first-line antiepileptic medications, although refractory epilepsy can occur.
Gastrointestinal complications (reflux, constipation, and vomiting) can impact quality of life.
Failure to thrive and short stature are common.
Limited information is available on pregnancy in Alexander disease. Pregnant women should be monitored for known complications of Alexander disease that are also common in pregnancy, such as gastrointestinal symptoms (worsening reflux, constipation, and vomiting), respiratory involvement (treatment for sleep apnea), and bladder dysfunction (upper motor neuron bladder).
With pre-evaluation and caution concerning the possibilities of seizures, gastroesophageal reflux, vomiting (with possible aspiration), and respiratory-pharyngeal muscle weakness, anesthesia can be safe (06).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Amy T Waldman MD MSCE
Dr. Waldman of Children's Hospital of Philadelphia Research Institute served as principal investigator in a clinical trial and received research support from Ionis Pharmaceuticals.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026