

Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Leukodystrophies are a heterogeneous group of genetic disorders that affect the white matter of the central nervous system, with some entities also affecting the peripheral nervous system. There are over 30 different leukodystrophies, with an overall population incidence of 1 in 7000 live births (60; 49). They are now most commonly grouped based on the initial pattern of central nervous system white matter abnormalities on neuroimaging. Every leukodystrophy is unique in its clinical presentation and pathologic mechanisms, but there is also significant variability within each leukodystrophy, which poses clinical challenges. However, as our knowledge increases, clinical approaches to patients with leukodystrophies and novel therapeutics are constantly being innovated. In recent years, a number of novel therapeutics have been developed and continue to emerge for many leukodystrophies. Hence, early diagnosis with genetic counseling remains one of the cornerstones of patient care. Newborn screening for some of the leukodystrophies has opened new horizons for the disease and increased the need for neurologists to understand and be aware of the need of preventive care for presymptomatic patients.
|
• Leukodystrophies are defined as genetic disorders that primarily affect the white matter predominantly of the central nervous system; these disorders have either glial cell or myelin sheath abnormalities. | |
|
• There are other entities called genetic leukoencephalopathies that also are characterized predominantly with white matter abnormalities but do not meet the criteria for being defined as a leukodystrophy. | |
|
• The pattern of abnormalities on brain MRI is one of the most useful diagnostic tools. | |
|
• Newborn screening and emerging novel therapies such as gene therapy and small molecule therapies affecting change to the CNS are changing the landscape for many leukodystrophies, making it important for neurologist to be aware of many of these disorders. |
The first leukodystrophy was identified in the early 20th century. Both Nissl and Alzheimer coincidentally reported metachromatic staining of the white matter of an adult patient diagnosed with what we now call metachromatic leukodystrophy (05; 42). Globoid cell leukodystrophy, or Krabbe disease, was described in 1916 when Hallervorden suggested that globoid cells may contain kerasin or cerebroside. Biochemical and histochemical studies confirmed the presence of cerebroside in globoid cells (06), and galactocerebroside was the only glycolipid that could produce globoid cells when experimentally injected into the central nervous system of animals. Until the early 1990s, the known leukodystrophies were metachromatic leukodystrophy, Krabbe disease, Canavan disease, Alexander disease, adrenoleukodystrophy, Pelizaeus-Merzbacher disease, and two forms of adult-onset autosomal dominant leukodystrophies (03).
One difficulty has been in differentiating which clinical syndrome qualifies as a leukodystrophy rather than a leukoencephalopathy. Over the years, multiple terminologies, including demyelination, dysmyelination, leukoencephalopathy, and leukodystrophy, have been used for the varied spectrum of clinical and radiological phenotypes with white matter involvement. Most recently, a consensus of experts in the field better defined the terms. Leukodystrophies are defined as “heritable disorders affecting the white matter of the central nervous system with or without peripheral nervous system involvement with common glial cell or myelin sheath abnormalities.” Disorders with significant white matter abnormality that does not meet the inclusion criteria for leukodystrophies are considered genetic leukoencephalopathies (60).
The consensus definition does emphasize that CNS diseases in which neuropathology shows primary involvement of neurons in the cerebral cortex or other gray matter structures, but in which brain MRI also detects significant abnormalities of white matter, should not be characterized as leukodystrophies. For example, disorders like San Filippo disease, Batten disease, etc., which can cause secondary white matter abnormalities, are not defined as leukodystrophies. Similarly, disorders that are primary vasculopathies resulting in white matter abnormalities are also not classified as leukodystrophies (eg, COL4A2-, COL4A1-, and CADASIL-related disorders). Interestingly, the definition does not discriminate based on clinical presentation. Many of the leukodystrophies do not present as such (eg, adrenomyeloneuropathy, PLP1 presenting as spastic paraplegia). Also, the clinical progression does not need to be progressive to meet the definition of leukodystrophy; for example, megalencephalic leukoencephalopathy with subcortical cysts caused by HEPACAM mutations can be characterized by clinical improvements. The disease presentations can also present with a wide spectrum for age of onset with a broader spectrum of disease being recognized, with growing access to genetic testing.
|
Name of disorder |
Mode of inheritance |
Biochemical testing |
Molecular genetics |
|
18q minus syndrome |
Most often de novo deletion; may be inherited | ||
|
Adult-onset leukodystrophy with neuroaxonal spheroids and pigmented glia (including hereditary diffuse leukoencephalopathy with spheroids and pigmentary type of orthochromatic leukodystrophy with pigmented glia) |
AD |
CSF1R | |
|
Aicardi-Goutières syndrome |
Usually AR; may be AD |
In CSF: lymphocytosis, increased INF-alpha, increased pterins In plasma: increased liver enzymes, thrombocytopenia |
TREX 1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1, LSM11, RNU7-1 |
|
Alexander disease |
AD |
GFAP | |
|
Autosomal dominant leukodystrophy with autonomic disease |
AD |
LMNB1 | |
|
Canavan disease |
AR |
In urine and CSF: increased N-acetylaspartic acid |
ASPA |
|
Cerebrotendinous xanthomatosis |
AR |
In plasma: increased cholestanol, normal to low cholesterol, increased lactate In bile, urine, and plasma: increased bile alcohols and glyconjugates In CSF: increased cholestanol and apolipoprotein B |
CYP27A1 |
|
Chloride ion channel 2-related leukoencephalopathy with intramyelinic edema |
AR |
CLCN2 | |
|
eIFB2 -related disorder (vanishing white matter disease or childhood ataxia with central nervous system hypomyelination) |
AR |
In CSF: increased glycine |
EIF2B1-5 |
|
Fucosidosis |
AR |
On urinary oligosaccharide assay: increased fucose-containing glyconjugates In leukocytes and fibroblasts: decreased alpha-fucosidase activity |
FUCA1 |
|
Globoid cell leukodystrophy (Krabbe disease) |
AR |
In leukocytes: decreased galactocerebrosidase activity Plasma: increased psychosine |
GALC |
|
Hypomyelination with atrophy of the basal ganglia and cerebellum |
AD |
TUBB4A | |
|
Hypomyelination with brainstem and spinal cord involvement and leg spasticity |
AR |
DARS1 | |
|
Hypomyelination with congenital cataract |
AR |
FAM126A | |
|
Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation |
AR |
DARS2 | |
|
Leukoencephalopathy with thalamus and brainstem involvement and high lactate |
AR |
EARS2 | |
|
Megalencephalic leukoencephalopathy with subcortical cysts |
AR (MLC1 and MLC2A) |
MLCI, MLC2A, MLC2B | |
|
Metachromatic leukodystrophy |
AR |
In leukocytes and fibroblasts: decreased arylsulfatase activity In urine: increased sulfatides |
ARSA |
|
Oculodentodigital dysplasia |
Usually AD, may be AR |
GJA1 | |
|
Pelizaeus Merzbacher disease |
X-linked |
PLP1 | |
|
Pelizaeus Merzbacher like-disease |
AR |
GJC2 | |
|
Peroxisomal biogenesis disorders (including Zellweger syndrome, neonatal adrenoleukodystrophy and infantile Refsum disease) |
AR |
Plasma VLCFA, phytanic and pristanic acid; plasma and urine pipecolic acid and bile acids help distinguish different forms of peroxisomal disorders |
PEX genes |
|
Pol III-related disorders (4H syndrome: hypomyelination, hypodontia, hypogonadotropic hypogonadism) |
AR |
POLR3A, POLR3B | |
|
Polyglucosan body disease |
AR |
In leukocytes: decreased 1,4 alpha glucan branching enzyme 1 activity Histopathologic examination of muscles, nerve, axillary skin: pathologic polyglucosan accumulation |
GBE1 |
|
RNAse T2 deficient leukoencephalopathy |
AR |
RNASET2 | |
|
Sialic acid storage disorders (Salla disease, infantile sialic acid storage disease, and intermediate form) |
AR |
In urine and fibroblasts: increased free sialic acid |
SLC17A5 |
|
Single enzyme deficiencies of peroxisomal fatty acid beta oxidation (including only D-bifunctional protein deficiency; sterol carrier protein deficiency; peroxisomal acyl-CoA-oxidase deficiency) |
AR |
Plasma VLCFA, phytanic and pristanic acid; plasma and urine pipecolic acid and bile acids help distinguish different forms of peroxisomal disorders |
DBP deficiency: HSD17B4 SCPx deficiency: SCP2 Acyl-Coa-oxidase deficiency: ACOX1 |
|
Sjögren-Larsson syndrome |
AR |
In urine: abnormal metabolites of leukotriene B4 In fibroblasts and leukocytes: decreased fatty aldehyde dehydrogenase activity or of fatty alcohol: NAD reductase activity |
ALDH3A2 |
|
SOX10-associated PCWH (peripheral demyelinating neuropathy, central demyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease) |
AD |
SOX10 | |
|
X-linked adrenoleukodystrophy |
X-linked |
On plasma VLCFA assay: increased C26:0, ratio of C24:0 to C22:0, ratio of C26:0 to C22:0 |
ABCD1 |
|
| |||
|
• Recognition of red flag clinical symptoms and signs can allow an astute clinician to consider leukodystrophies early in the differential diagnosis of a patient. | |
|
• Delays, stagnation, and regression in neurodevelopment remains an important early presenting symptom of childhood leukodystrophies. | |
|
• Adult-onset leukodystrophies can mimic a variety of conditions, including early-onset dementia and psychiatric illness, multiple sclerosis, and spastic paraplegias. | |
|
• Neuroimaging, although not specific, is a crucial clue for initiating molecular diagnosis of leukodystrophies. |
Patients suspected of having a leukodystrophy require a thorough history (including family history), a thorough general and neurologic examination, brain MRI, and referrals to various subspecialists. Leukodystrophies are rare and clinically heterogeneous with various ages of disease onset. Some have more systemic involvement than others, and some have a more progressive course than others. Due to the clinical variability, it is difficult to create a set of symptoms common among all leukodystrophies. However, there are red flag neurologic features on history and examination that should alert a clinician to initiate a workup for leukodystrophy.
Clinical history. A clinical history or a family history of developmental regression or stagnation, especially in motor development is often the first symptom in many demyelinating leukodystrophies (57). Additionally, a delay in acquisition of milestones can be seen in the hypomyelinating leukodystrophies. Though many of the leukodystrophies are heritable, some have been observed as sporadic cases, especially in adult-onset leukodystrophies (35). Age of onset is also an important factor that may guide diagnosis as some leukodystrophies are more common in adults, such as adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Most patients with ALSP will present with cognitive and frontal lobe-based behavioral symptoms and signs months before motor deficits develop (28). With many leukodystrophies, such as X-linked adrenoleukodystrophy, a patient may present at any age; however, presenting symptoms may be different in each age group. For example, childhood cerebral adrenoleukodystrophy is most common between the ages of 3 and 11 years, whereas adrenomyeloneuropathy is more common among males in their third decade (56).
Examination. Neurologic examination can be revealing and aid in diagnosis. Neurologic signs concerning for a central nervous system pathology, such as appendicular hypertonicity and axial hypotonicity, would indicate the need for neuroimaging. Patients with Aicardi-Goutières syndrome can have axial hypotonia and appendicular hypertonia, along with abnormal eye movements, persistence of the tonic neck reflex, and difficulty with vision (12). Other common neurologic abnormalities seen in patients with leukodystrophies are a combination of spasticity and cerebellar ataxia, as in Krabbe disease and 4H syndrome (44). This can affect all four extremities or only the lower extremities with delays of independent walking. Dystonia may be present when the basal ganglia are involved as in hypomyelination with atrophy of basal ganglia and cerebellum and in RNA polymerase III-related leukodystrophy (51; 45; 04). Tendon reflexes are usually increased, but decreased ankle reflexes compared to knee jerks suggest the presence of a peripheral neuropathy, for example, as seen in metachromatic leukodystrophy (09). The presence of peripheral nerve involvement can be subtle with normal nerve conduction velocity (54). Sensory examination is typically grossly normal, but vibration perception is often reduced, especially when the disease process also affects peripheral nerves (39).
As many of these leukodystrophies cause systemic symptoms, a general examination is also important. Growth is often delayed in systemic disorders, such as mitochondrial diseases; head circumference may be small in patients with Krabbe disease or megalencephalic in those with Canavan disease, Alexander disease, or megalencephalic leukoencephalopathy with subcortical cysts. A complete eye examination is important as findings can help narrow diagnosis. Patients with peroxisomal biogenesis disorders commonly have cataracts, corneal clouding, and findings consistent with glaucoma. Hepatomegaly can also be seen in patients with peroxisomal biogenesis disorders (27). Examination of the mouth can reveal a variety of teeth abnormalities, such as hypodontia, suggesting 4H syndrome (54). Examination of the extremities may reveal xanthomas, indicating cerebrotendinous xanthomatosis (38). Examination of the skin may show increased skin pigmentation, suggesting adrenal insufficiency and X-linked adrenoleukodystrophy (44); ichthyosis would suggest the diagnosis of Sjögren-Larsson syndrome (23).
Neuroimaging. Neuroimaging is key to the diagnosis of a leukodystrophy. By definition, in leukodystrophies, T2-weighted MRI signal hyperintensity in the white matter must be present, but T1-weighted signal may be variable (47; 62). Isointense or hyperintense T1 signal is consistent with a hypomyelinating leukodystrophy. It is also important to note that individual MRIs are generally not sufficient to determine delayed or hypomyelination, especially when done before 1 year of age. Therefore, serial MRIs, at least 6 months apart, are generally recommended (44). Other myelin pathologies, including demyelinating leukodystrophies, cause hypointense T1 signal (47; 61).
It should be stressed that although the pattern on MRI is an extremely valuable diagnostic tool, it does not always necessarily represent the actual pathological process; that is, a hypomyelinating pattern on the MRI may not imply actual hypomyelination. For example, in Pelizaeus-Merzbacher disease, despite the common MRI pattern being concerns for hypomyelination, the actual neuropathology is highly specific to the mutation (22; 31). Also, certain imaging findings are more common in adult white matter disease, such as asymmetrical multifocal white matter irregularities, enhancement, cervical spinal cord engagement, the existence of mass-like lesions, and steroid reactivity (40).
The classically described childhood onset leukodystrophies are progressive, eventually leading to complete dependency for everyday activities and premature death. With more access to genetic testing, a broader spectrum of natural history is being defined in many leukodystrophies. It is important to note that some leukodystrophies, such as megalencephalic leukoencephalopathy with subcortical cysts and leukoencephalopathy, with thalamus and brainstem involvement and high lactate have an improving phenotype; therefore, progressive disease is no longer part of the definition of a leukodystrophy (33; 26). Many leukodystrophies including adrenoleukodystrophy, metachromatic leukodystrophy, Krabbe disease, and cerebrotendinous xanthomatosis have disease modifying therapies available now with a more optimistic outcome, especially if diagnosed in the pre or early symptomatic stages. Even in the later stages, supportive care has extended the lives of many patients with progressive diseases, making it important to ensure appropriate management of the complications associated with many of the leukodystrophies, such as spasticity, seizures, musculoskeletal issues like scoliosis, skin care, urinary incontinence, and lack of ability to verbally communicate (02).
In adult forms of leukodystrophy, behavioral disturbances and psychiatric manifestations often cause loss of employment and conflicts in the family (21). Slowly progressive or even sometimes static psychiatric or cognitive symptoms frequently occur in adults with metachromatic leukodystrophy (64). However, the prognosis in early diagnosed adult onset leukodystrophy with therapeutic options including adult-onset leukoencephalopathy with axonal spheroids and pigmented glia and metachromatic leukodystrophy can be optimistic when diagnosed in the early symptomatic stages.
Overall, the prognosis and complications in each leukodystrophy depends on the early diagnosis and the availability of disease modifying therapies/ interventions.
Leukodystrophies, though all with variable underlying etiology, are heritable disorders with glial cell or myelin sheath abnormalities leading to pathology of the white matter of the CNS and, at times, the PNS (60). What is striking is the wide range of etiologies representing a variety of cellular processes and functions. They include lysosomal enzyme deficiencies, inborn errors of intermediate metabolism (lipids or sugars, defects in transport of small molecules across lysosomes and peroxisomes, defects of innate immunity, control of protein translation initiation, and abnormalities of certain structural proteins of cells), and mutations of glial fibrillary acidic protein in Alexander disease.
The pathogenesis of a leukodystrophy varies according to its etiology; therefore, one cannot generalize across the various genetic entities. One approach is to divide the leukodystrophies according to the cell type that is predominantly affected (57). Initially, it was thought that oligodendrocytes are always the type of cell primarily affected; though true in some disorders, such as Pelizaeus-Merzbacher disease, metachromatic leukodystrophy, and Krabbe disease (15), we now know that numerous cell types in the brain can be involved, including oligodendrocytes, astrocytes, and microglia (25). For example, in Alexander disease, the mutated protein, glial fibrillary acidic protein (18), is expressed only in astrocytes. Astrocytes are also prominently involved in megalencephalic leukodystrophy with subcortical cysts and in CACH/VWM (14; 36). Immune cells, including microglia, are dysfunctional in X-linked adrenoleukodystrophy (48), ALSP (29; 28), and likely in Aicardi-Goutières syndrome as well (46). Defects in mitochondrial and cytoplasmic tRNA synthetases have been identified (53; 13; 66), though how these defects cause a leukodystrophy is not yet known. Consequently, in the majority of cases, the complete mechanism is not obvious, but it clearly involves more than one cell type. Axonal dysfunction and death occur in all leukodystrophies and are the main causes of the clinical neurologic dysfunction.
Leukodystrophies are panethnic. There are limited epidemiologic data on the frequency of leukodystrophies, but their overall incidence is estimated to be 1:7500 live births (19). The incidence of individual leukodystrophies varies from 1:17,000 in X-linked adrenoleukodystrophy (17) to 1:40,000-1:100,000 in metachromatic leukodystrophy (34) and 1:500,000 and less in the rarest of these genetic diseases. The lifetime risk for leukodystrophies and genetic leukoencephalopathies in the United Kingdom is 31 per million live births (52).
Reproductive planning in conjunction with genetic counseling is currently the primary method for the prevention of leukodystrophies. For families in whom the molecular defects have been identified in affected members, mutation analysis provides the most accurate method for prenatal diagnosis and carrier identification. When mutation analysis is available, pre-implantation genetic diagnosis is possible.
A number of leukoencephalopathies (diseases that predominantly affect the white matter of the brain) may be confused with and initially diagnosed as leukodystrophies. Therefore, it is important to consider other acquired and genetic disorders that may result in white matter disease on neuroimaging when evaluating a patient for leukodystrophy (Table 2) (60).
Acquired disorders. Acquired disorders can affect the white matter of the brain. Postinfectious, infectious, and autoimmune disorders can result in demyelination without a primary genetic basis. Disorders such as acute disseminated encephalomyelitis, multiple sclerosis, and neuromyelitis optica typically differentiate themselves from the heritable disorders of the white matter by abrupt onset and multiphasic presentations (62). In addition, MRI abnormalities are more likely to be multifocal and patchy (47), with variation or even improvement over time and with treatment. Nevertheless, when evaluating a child with a suspected leukodystrophy, it is always important to exclude potentially treatable or reversible acquired disorders. Central nervous system injury, particularly in the perinatal period, can result in significant white matter signal abnormalities but will typically also be irregular in appearance and may result in a loss of white matter volume, characteristics to help differentiate it from a leukodystrophy (62).
Primary neuronal disorders. Primary neuronal disorders are often associated with significant white matter abnormalities (55). The infantile variants of disorders like GM1 gangliosidosis, GM2 gangliosidosis, and neuronal ceroid lipofuscinosis may have significant white matter abnormalities because the primarily neuronal disease disrupts the normal myelination process. Later onset variants of these disorders eventually affect the cerebral white matter, and mild signal abnormalities arise related to Wallerian degeneration with loss of axons and myelin. In other disorders, such as MCT8-related disorder (65), myelination can be extremely delayed but progress over time to become almost complete. These are not considered primary white matter disorders. Primary neuronal disorders may, thus, be confused with leukodystrophies (47). When features such as significant encephalopathy and seizures are present in a child with leukoencephalopathy, primary neuronal disorders should be considered along with leukodystrophies.
Disorders affecting white and gray matter. Some disorders affect the white and gray matter indiscriminately and are not leukodystrophies. When evaluating a patient with abnormal white matter on neuroimaging, it is important to carefully evaluate for associated cortical lesions. Examples include mitochondrial disorders, such as MELAS (mitochondrial encephalopathy with lactic acidosis and seizures) and POLG1-associated mitochondrial encephalopathy, or familial hemophagocytic lymphohistiocytosis, which results in both white and gray matter injury (68).
Vasculopathies. Vasculopathies can result in white matter signal abnormalities but are not leukodystrophies. Genetic abnormalities of the vessel wall, such as CADASIL (cerebral autosomal dominant arteriopathy with subcortical infants), and defects in COL4A1, such as in autosomal dominant porencephaly, can give multifocal and, in the end, confluent signal abnormalities in the periventricular and deep white matter, typically with multifocal abnormalities in the central gray matter structures and brainstem (07). Cathepsin A-related arteriopathy with strokes and leukoencephalopathy also has been described in adult patients to show extensive white matter disease (11; 20). However, because they are not primary glial cell disorders, they are arbitrarily not considered leukodystrophies.
|
3-hydroxy-3-methylglutaryl-CoA lyase deficiency | |
|
• Untreated propionic aciduria | |
|
BH4-deficient hyperphenylalaninemia C | |
|
• Alpha mannosidosis | |
|
Fabry disease | |
|
• Carbamoyl phosphate synthetase I deficiency | |
|
Wilson disease | |
|
| |
For further information and assistance in unsolved leukodystrophies, please also see the Myelin Disorders Bioregistry Project at www.myelindisorders.org.
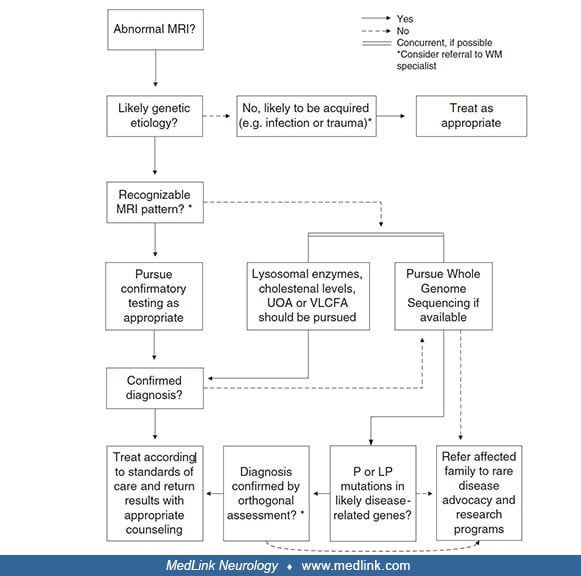
The approach to diagnosis is heavily based on clinical features, advanced neuroimaging, and laboratory testing, including ancillary biochemical tests and genetic testing. Pattern recognition in advanced imaging plays an important role in diagnosis, and the diagnostic algorithm based on imaging is an important tool for clinicians (Schiffman and van der Knaap 2009; 44).
Clinical and laboratory tests that can aid in further narrowing the diagnosis are: ophthalmologic examination; biochemical tests – very long chain fatty acids (plasma), lactate (blood), pyruvate (blood), amino acids (blood), sulfatides (urine), organic acids (urine), lysosomal enzymes; CSF analysis; neurophysiologic testing, such as visual evoked potentials and nerve conduction studies; and genetic testing.
Vanderver and colleagues showed that genomic sequencing (of patients and both parents) as a first-line test in patients with pediatric white matter disease not only decreases the time to diagnosis but also increases the overall diagnostic yield from 21% to 56% when compared to standard-of-care diagnostic testing. Additionally, one study found exome sequencing to have a diagnostic yield of 82.9%, emphasizing its importance in reaching a faster diagnosis (69). With this in mind, a revised diagnostic algorithm was proposed. It is important to note that biochemical testing is still the cornerstone of testing to identify treatable entities in a timely manner(59).
P = pathogenic, LP = likely pathogenic, UOA = urine organic acids, VLCFA = very long chain fatty acids. (From: Vanderver A, Bernard G, Helman G, et al. Randomized clinical trial of first-line genome sequencing in pediatric whit...
It is important to note that earlier interventions, when available, lead to improved outcomes. Hence, newborn screening can potentially play a crucial role in early diagnosis and management of certain leukodystrophies. Through the Secretary of Health and Human Services, the federal government recommended that X-linked adrenoleukodystrophy be added to the Recommended Uniform Screening Panel (RUSP) (24). Of note, while the RUSP is federally recommended, the implementation of newborn screening is state-based. Many states are currently screening for X-linked adrenoleukodystrophy. The U.S. Advisory Committee on Heritable Disorders in Newborns and Children decided with a 10 to 3 vote to propose including infantile Krabbe disease in the list of recommended newborn screenings. The development of a screening technique for metachromatic leukodystrophy using dried blood spots from newborns is currently in process (30). The challenges of balancing the need for early diagnoses as new therapeutics evolve with the challenge of addressing pseudodeficiency alleles, variants of uncertain significance in genetic screening, and long-term financial and ethical considerations will need to be assessed, discussed, and evaluated as conditions are added to the newborn screening process.
In select leukodystrophies, therapies exist to slow disease progression and perhaps change the natural history of the disease. Currently, leukodystrophies with disease modifying therapies include X-linked adrenoleukodystrophy, metachromatic leukodystrophy, cerebrotendinous xanthomatosis, and Krabbe disease; however, early detection is imperative to make a meaningful difference in outcomes (44).
In X-linked adrenoleukodystrophy, hematopoietic stem cell transplantation (HSCT) is the treatment of choice in patients with cerebral adrenoleukodystrophy. There may also be a role for HSCT in juvenile pre and early symptomatic patients with metachromatic leukodystrophy and in presymptomatic Krabbe disease (10; 63), where there is increased survival noted.
It is not clear how transplant prevents progression of X-linked adrenoleukodystrophy, but the effect is likely related to immunological modification. However, in lysosomal diseases, the therapeutic efficacy of transplantation relies on the migration of donor bone marrow-derived cells of the monocyte-macrophage lineage into diseased target organs, where they replace the resident enzyme-deficient population and become a local and steady source of the missing enzyme (43). On the other hand, in metachromatic leukodystrophy, there is no enzymatic cross-correction; rather, inflammatory and immune modulation likely explains the therapeutic effect (67). In select metachromatic leukodystrophy patients, such as in the very early stage of the juvenile and adult forms of leukodystrophy, transplantation has been reported to delay onset and slow progression of the disease (10). Clinical experience has shown no efficacy of transplantation if performed in symptomatic patients with early-onset disease or in advanced stages for patients with late-onset disease. Unfortunately, considering these selection criteria, only a minority of metachromatic leukodystrophy patients can benefit from transplantation, which can be further limited by the availability of HLA-compatible donors (43). In addition, transplantation is associated with fairly high morbidity and mortality due to conditioning regimens, rejection, failure of engraftment, and graft-versus-host disease. However, with the advent of reduced-intensity conditioning and availability of umbilical cord blood, mortality and morbidity have been reduced, and the umbilical cord blood allows for a more readily available stem cell source (58).
Gene therapy and leukodystrophies. Gene therapy includes introduction of a functional gene and can include in-vivo or ex-vivo approaches. In-vivo approaches include introduction of a modified gene with the help of a vector; ex-vivo approaches involve autologous cells taken from the patient, modified in the lab to introduce a functional gene, and then reintroducing the cell back into the patient. Currently, viral vectors are most commonly used intruding lenti-viruses for ex-vivo approaches and Adeno-associated viruses used for in-vivo approaches, being most common. Ex-vivo gene therapy, which negates the need for a matched donor and the risk of rejection altogether, is now approved for patients with X-linked adrenoleukodystrophy and metachromatic leukodystrophy (50; 16).
Elivaldogene autotemcel, eli-cel, a lentiviral vector containing manufactured ABCD1 complementary deoxyribonucleic acid (DNA) was granted accelerated approval by the U.S. Food and Drug Administration (FDA) in 2022 for the treatment of boys ages 4 to 17 years with early active cerebral adrenoleukodystrophy. Currently, this is administered in select centers for patients who do not have a matched donor for hematopoietic stem cell transplantation (HSCT). The risk for myelodysplastic syndrome was noted in these patients, and the long-term risks and the comparative efficacy of this to HSCT are still being studied.
Atidarsagene autotemcel was approved by the U.S. FDA and is now indicated for the treatment of children with presymptomatic late infantile (PSLI), presymptomatic early juvenile (PSEJ), or early symptomatic early juvenile (ESEJ) metachromatic leukodystrophy (MLD) (32).
Other disease modifying therapies for leukodystrophies. In cerebrotendinous xanthomatosis, early treatment with chenodeoxycholic acid has shown to slow the progression of both the neurologic and nonneurologic disease (41). This therapy corrects the abnormalities in bile acid metabolism, restoring lipid levels in the blood, urine, and cerebrospinal fluid to normal. Adjunctive therapy with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors may offer supplementary support (01).
Leukodystrophies | Disease modifying treatments |
X-adrenoleukodystrophy | 1. HSCT/UCBT for pre and early symptomatic patients with a Loes score less than 9 and a matched donor |
Metachromatic leukodystrophy | 1. Ex-vivo gene therapy, atidarsagene autotemcel for presymptomatic late infantile and pre or early symptomatic juvenile patients |
Cerebrotendinous xanthomatosis | 1. Chenodeoxycholic acid, inhibitors of HMG-CoA reductase |
In patients with advanced neurologic signs, symptomatic and supportive therapy are important in improving the quality of life of patients (02):
• Respiratory physiotherapy and adequate control of pulmonary infections, which are frequent as a consequence of dysphagia | |
• Adequate support of calories, vitamins, and minerals, which may require, in the advanced stages, a nasogastric tube or a permanent gastrostomy | |
• Antiepileptic drugs to control seizures, which may not be clinically evident and, therefore, may require electroencephalographic monitoring | |
• Adjustment of school environment and counseling to maintain contact with family and friends and activities | |
• Psychiatric intervention and medications to control psychosis and behavioral symptoms |
In general, once a patient has been diagnosed with a leukodystrophy, the patient should be referred to a center with expertise in managing patients with leukodystrophies.
In conclusion, the horizon of therapeutic options for many of the leukodystrophies is rapidly becoming optimistic, increasing the critical nature of early diagnosis and referral to a center with expertise in managing these patients.
Children with leukodystrophy often require anesthesia during imaging procedures or for surgical procedures (37; 08). These children are at special risk of airway complications with sedation and anesthesia because of poor pharyngeal muscle control, copious oral secretions, and the prevalence of gastroesophageal reflux and seizure disorders. Therefore, it is recommended that when needed, these patients be referred to centers with expertise in treating children with leukodystrophies.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Deepa S Rajan MD
Dr. Rajan of UPMC Children's Hospital of Pittsburgh has no relevant financial relationships to disclose.
See ProfileEsraa A Ali MBBS
Dr. Ali of Children’s Hospital of Pittsburgh has no relevant financial relationships to disclose.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Peripheral Neuropathies
May. 12, 2026
Neurogenetic Disorders
May. 08, 2026