

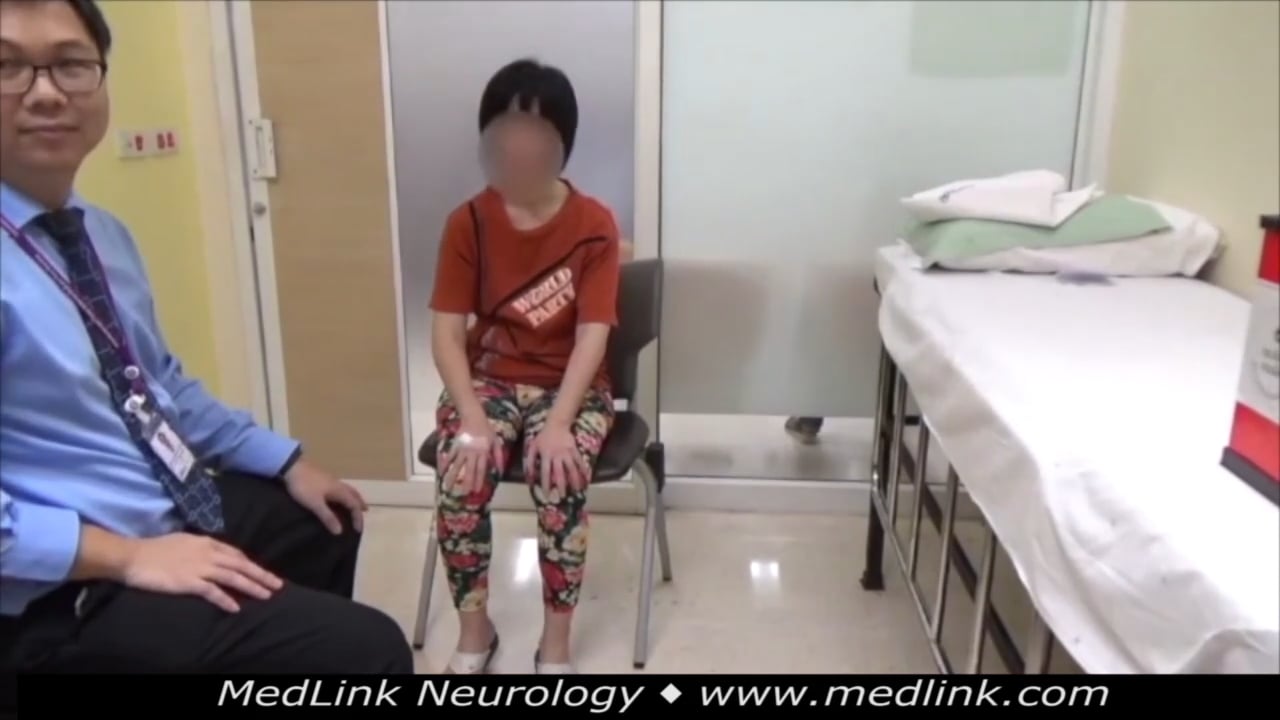




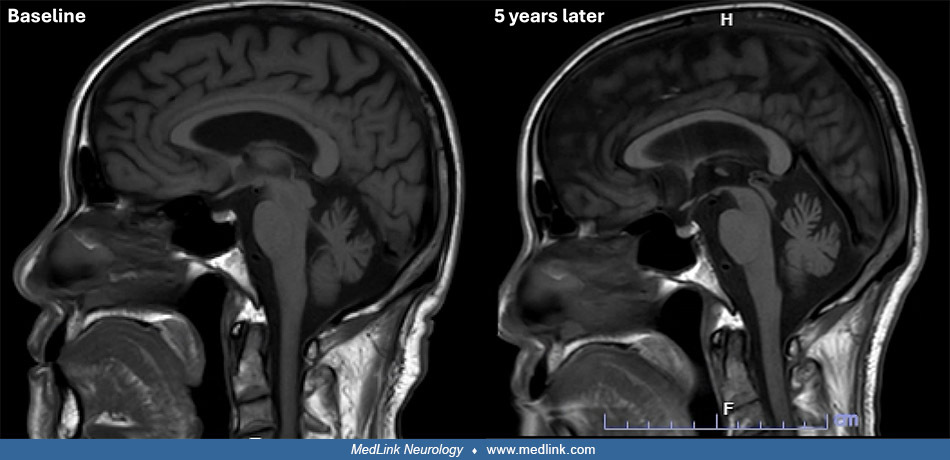
Sleep Disorders
Sudden infant death syndrome
Jul. 05, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The dominant ataxias continue to be better characterized from a genetic perspective. In this article, the author provides an updated review of the autosomal dominant ataxias, including spinocerebellar ataxia syndromes, dentatorubral-pallidoluysian atrophy, and episodic ataxias. To date, the most recently identified are spinocerebellar ataxia type 49 and episodic ataxia type 9. New mutations, as well as the repeat expansions causing these disorders, are described and reviewed. The exact pathogenesis underlying these disorders is not known, but researchers have continued to elucidate the pathways involved in the syndromes based on the genes and differences between mutations or the repeat expansions that are involved. There are currently no disease-modifying therapies; however, a goal for the future is to use the known genetic information to generate new, effective targeted therapeutics.
|
• The autosomal dominant hereditary ataxia syndromes consist of more than 40 known spinocerebellar ataxia syndromes, dentatorubral-pallidoluysian atrophy, and nine episodic ataxia syndromes. There are likely more genetic etiologies that have yet to be discovered. | |
|
• The autosomal dominant spinocerebellar ataxias encompass a wide range of disorders that have a range of phenotypes that can be categorized as ataxia plus extra-cerebellar features, ataxia with pigmentary retinopathy, and almost purely isolated cerebellar ataxia. There is a wide range of age of onset, from childhood to adulthood, for many of the autosomal dominant ataxia syndromes. | |
|
• The spinocerebellar ataxia syndromes are currently classified based on their genetic etiology. The main mechanisms of genetic disruption are via repeat expansions of the genome in either coding or noncoding regions and conventional mutations and translocations within the genome. | |
|
• The family history is a key area to help distinguish between the acquired and the inherited ataxia syndromes. However, even the inherited ataxias can be caused by a de novo mutation. | |
|
• It is difficult to make phenotype/genotype correlations because of the significant overlap of presenting symptoms. | |
|
• Researchers are rapidly learning about the underlying pathophysiology behind the inherited ataxia syndromes. Information based on the genes involved and the pathways they affect is elucidating the basic mechanism of cellular injury in polyglutamine triplet repeat expansion disorders versus single-mutation disorders. | |
|
• There are currently no disease-modifying therapeutic options. Management involves providing symptomatic relief from comorbid conditions and offering genetic counseling. |
Hereditary spinocerebellar degeneration, also referred to as hereditary ataxia, is uncommon. In 1893, Pierre Marie described a clinical condition that he termed “hereditary cerebellar ataxia,” in which cerebellar signs prevailed along with spastic signs. This was later referred to as “Marie’s cerebellar ataxia.” Subsequently, the term was used broadly to describe a variety of hereditary conditions with spinocerebellar manifestations, including the spastic ataxia of Sanger-Brown, Menzel spinocerebellar ataxia with olivopontocerebellar degeneration, the cerebello-olivary degeneration of Holmes, and the olivopontocerebellar degeneration reported by Dejerine and Thomas (55). In 1974, Skre studied the hereditary ataxia diseases in western Norway and chose to consider all these disorders as members of a comprehensive group of diseases called “spinocerebellar ataxias” (249).
Various attempts have been made to classify inherited ataxias. In 1907, Holmes proposed a classification based on pathologic findings, but he did not take into account genetic or clinical features. In 1954, Greenfield classified the inherited ataxias into three categories: (1) predominantly spinal, (2) predominantly cerebellar, and (3) combined spinocerebellar (94). The predominantly spinal category included Friedreich ataxia, abetalipoproteinemia, and hereditary spastic paraplegia. The predominantly cerebellar category included ataxia-telangiectasia, late-onset cervical cerebellar atrophy, and Marinesco-Sjogren-Garland disease. The spinocerebellar group consisted of hereditary spastic ataxia, Ramsay Hunt type 1 syndrome (dyssynergia cerebellaris myoclonica), hereditary periodic ataxia, and the olivopontocerebellar atrophies.
In 1983, Harding proposed a classification based primarily on the biochemical pathogenesis and age of onset of the ataxia (103). She divided those with a known etiology (eg, abetalipoproteinemia, ataxia telangiectasia) from those whose etiology was unknown (eg, Friedreich ataxia, Ramsay Hunt type 1 syndrome [dyssynergia cerebellaris myoclonica], cerebellar ataxia with retinal degeneration). In the latter category, she further subdivided the disorders based on whether the onset occurred early (before 20 years of age) or late (after 20 years of age). In a subsequent article, she refined the classification for the autosomal dominant forms of spinocerebellar degeneration (104). Autosomal dominant cerebellar ataxia type I presents with ataxia and noncerebellar findings (such as pyramidal or extrapyramidal dysfunction and ophthalmoplegia). Autosomal dominant cerebellar ataxia type II represents cerebellar ataxia with pigmentary retinal degeneration. Autosomal dominant cerebellar ataxia type III produces relatively pure cerebellar signs. However, as more has been learned about these disorders, it has become clear that categorizing the disorders on phenotypic characteristics is simplistic. Each spinocerebellar ataxia manifests significant phenotypic heterogeneity, even within the same family. Furthermore, as Harding intimated in her seminal article, her scheme has been modified as specific genetic mutations have been identified (103; 139). The correlation of clinical disease with the underlying genetic defect is enabling more accurate and rational classification of the spinocerebellar degenerations.
Classification of hereditary ataxia is complex, and there is no consensus yet. With advances in genetics, autosomal dominant cerebellar ataxias of Harding are currently termed “spinocerebellar ataxias” with designated numbers from 1 up to almost 50, now in order of genetic discovery. Thus, the spinocerebellar ataxias are referred to as a heterogeneous group of neurodegenerative disorders, primarily affecting the cerebellum, that have autosomal dominant inheritance. Dentatorubral-pallidoluysian atrophy can be included in the spinocerebellar ataxia category but does not have the spinocerebellar ataxia designation. In addition, there are also spinocerebellar ataxias that are inherited in an autosomal recessive manner, and these disorders are grouped under the umbrella of autosomal recessive spinocerebellar ataxias.
|
• Autosomal dominant hereditary ataxias include spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and episodic ataxias. | ||
|
• Harding’s classification remains useful clinically in classifying autosomal dominant cerebellar ataxias into three main categories: | ||
|
- Autosomal dominant cerebellar ataxia type I is associated with extra-cerebellar features. Examples of disorders in this group include SCA1, SCA2, and SCA3. | ||
|
- Autosomal dominant cerebellar ataxia type II is associated with pigmentary retinopathy. This is currently known as SCA7. | ||
|
- Autosomal dominant cerebellar ataxia type III presents with pure cerebellar ataxia. The prototypic disorder in this group is SCA6. | ||
|
• Types of spinocerebellar ataxias have been expanding with advances in genetics. Given clinical and genetic heterogeneity, it is often difficult to predict genotypes (ie, specific types of spinocerebellar ataxias) from clinical phenotypes. | ||
The autosomal dominant hereditary ataxias primarily include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and the episodic ataxias. The dominantly inherited spinocerebellar ataxias have been grouped together because they share, to a greater or lesser degree, degeneration of neurons and selected tracts in the cerebellum and spinal cord and an autosomal dominant inheritance pattern. Their cardinal clinical feature is progressive incoordination of movement, either in the presence or absence of other neurologic signs. However, each spinocerebellar ataxia manifests considerable interfamilial and intrafamilial variability in the clinical presentation and the age of onset. The spinocerebellar ataxia syndromes are currently classified by their genetic etiologies; however, Harding’s clinical classification system from 1993 remains helpful in grouping them based on their clinical presentations.
The autosomal dominant cerebellar ataxias type I group (eg, SCA1, SCA2, SCA3, SCA4, SCA10, SCA12, SCA13, SCA14, SCA17, SCA19/22, SCA20, SCA28, and DRPLA) has complex phenotypes with additional extra-cerebellar features (except pigmentary retinopathy). The extra-cerebellar features include pyramidal signs such as spasticity, hyperreflexia, and extensor plantar response. Extrapyramidal signs such as parkinsonism, dystonia, chorea, tremor, and myoclonus can also occur. Parkinsonism can be a feature of SCA2, SCA3, and SCA17, and many patients have levodopa responsiveness (206). Patients may also have sensory or motor neuropathies. Cognitive decline, depression, brainstem oculomotor problems, deafness, and urinary complaints may be present. Sleep disturbances, such as restless legs and REM sleep disturbances, have also been reported. Although the age of onset is also variable from infancy to adulthood, most spinocerebellar ataxias present in adulthood, in contrast to autosomal recessive ataxias, which generally present earlier in childhood. The initial manifestations in infants are usually hypotonia and abnormal motor development. The symptoms in older children are nystagmus, truncal and gait ataxia, spasticity, extensor plantar response, and intellectual disability. The deep tendon reflexes vary from areflexia to hyperreflexia.
Autosomal dominant cerebellar ataxia type II is less common than types I and III and includes ataxia accompanied by pigmentary retinopathy and vision loss. This category is now known as SCA7.
Autosomal dominant cerebellar ataxia type III was described as a pure cerebellar ataxia (SCA5, SCA6, SCA11, SCA15/16, SCA26, SCA29, SCA30, and SCA31).
There have been multicenter collaborations in Europe (EUROSCA) and North America (the Clinical Research Consortium for Spinocerebellar Ataxias or CRC-SCA) to obtain natural history and longitudinal data in SCA1, SCA2, SCA3, and SCA6 by using validated outcome measures, especially the Scale for the Assessment and Rating of Ataxia (233; 05; 122; 65). Both cohorts revealed similar findings that among these four most common types of spinocerebellar ataxia, the disease progression, prognosis, and long-term survival were poorest in SCA1, intermediate in SCA2 and SCA3, and best in SCA6.
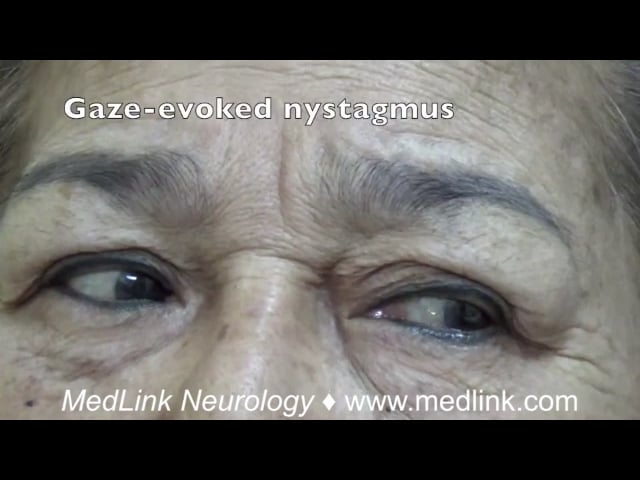
Spinocerebellar ataxia type 1. Patients with spinocerebellar ataxia type 1 can present in adolescence or adulthood with progressive cerebellar and noncerebellar symptoms. The disorder usually presents after 20 years of age, but a juvenile form exists. The cerebellar symptoms include ataxia, gaze-evoked nystagmus, saccadic smooth pursuits, hypermetric saccades, decreased optokinetic nystagmus, and an inability to suppress the vestibulo-ocular reflex.
Noncerebellar symptoms include dysarthria, dysphagia, ophthalmoparesis, pyramidal tract features (eg, spasticity, hyperreflexia, and extensor plantar responses), amyotrophy, muscle atrophy, optic disc pallor, decreased vibratory sensation, choreoathetosis, restless legs syndrome, and bladder dysfunction. Rarely, dystonia may be the initial presentation before ataxia develops (300). Most patients are wheelchair-bound within 15 years of the onset of symptoms. Longitudinal cohort studies revealed that spinocerebellar ataxia type 1 had faster progression and lower 10-year survival, compared to spinocerebellar ataxia type 2, type 3, and type 6 (05; 122; 65). In line with progression of ataxia, cognitive functions, including executive function, speed, attention, visual memory, and theory of mind, are more impaired over time, and the progression was more rapid in spinocerebellar ataxia type 1 compared to types 2, 3, and 6 (180).
Spinocerebellar ataxia type 2. Spinocerebellar atrophy type 2 can present in childhood, adolescence, or adulthood (285). Typically, individuals affected by spinocerebellar ataxia type 2 have progressive ataxia, action or postural tremor, areflexia, extremely slow saccades, ophthalmoplegia, and dysarthria. Slow horizontal saccade is the characteristic oculomotor abnormality in spinocerebellar ataxia type 2 (271). Peripheral neuropathy is more common in late-onset spinocerebellar ataxia type 2. Onset of the disorder in childhood can be characterized initially by bradykinesia and rigidity (235). Spinocerebellar ataxia type 2 can also present as levodopa-responsive familial parkinsonism (209; 206). Parkinsonism may occur with or without prominent ataxia. In some cases, individuals with spinocerebellar ataxia type 2 may present with dystonia without ataxia, parkinsonism, essential tremor, or progressive supranuclear palsy (84; 34). Infants with more than 200 repeat expansions have rarely been reported, and the clinical features include hypotonia, developmental delay, seizures, and retinitis pigmentosa (247). Rufa and colleagues reported a case of adult-onset spinocerebellar ataxia type 2 with retinal degeneration and optic atrophy (224). Therefore, spinocerebellar ataxia type 7 is not the only inherited ataxia associated with retinal changes.
In spinocerebellar ataxia type 2, cramps, facial myokymia, and dementia can occur. There is rarely pyramidal tract involvement or motor neuron features (191). There is mild cognitive impairment in executive functions and verbal memory (22). Sleep disturbances include REM-based sleep disorder (13) and restless legs syndrome.
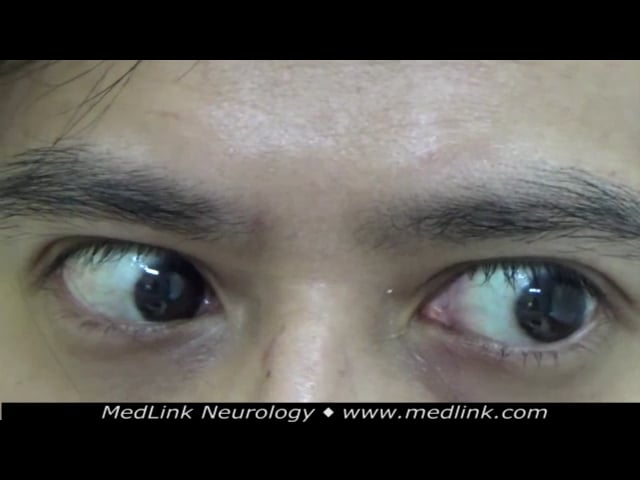
Spinocerebellar ataxia type 3 (Machado-Joseph disease). This disorder was classified into at least three forms according to clinical presentation. However, due to marked phenotypic overlaps, this classification is not commonly used in clinical practice. The most common form, type 2, begins in the third decade of life with progressive ataxia. Over a period of years, the patient develops several ophthalmologic signs (supranuclear ophthalmoparesis, lid retraction with bulging eyes, decreased blinking, double vision, impaired smooth pursuit, and nystagmus), pyramidal signs, and impaired thermal sense.
Various brainstem symptoms can also develop, including dysarthria, dysphonia, facial paresis, and facial fasciculations. In the early-onset form (type 1), the disease begins in childhood and is characterized by dystonia and rigidity. In the late-onset form (type 3), peripheral neuropathy is common, and ataxia is mild (123). Some patients develop external ophthalmoplegia. In the Clinical Research Consortium for Spinocerebellar Ataxia (CRC-SCA) cohort, dystonia was found to be the most common in spinocerebellar ataxia type 3, compared to other common spinocerebellar ataxias, including types 1, 2, and 6 (138). Furthermore, dystonia in spinocerebellar ataxia type 3 was associated with longer CAG repeats and higher Scale for Assessment and Rating of Ataxia scores. Individuals with spinocerebellar ataxia type 3 can have impaired executive functions, including word recall and letter and semantic fluency as well as verbal working memory (268).
Spinocerebellar ataxia type 4. This disease is also called hereditary ataxia with sensory neuropathy. It was initially believed to cause progressive ataxia, sensory axonal neuropathy, and pyramidal tract abnormalities (79). However, Nagaoka and colleagues describe patients with mutations of the spinocerebellar ataxia type 4 gene loci who do not have an axonal neuropathy or pyramidal tract dysfunction (186). Ouyang and colleagues and Hirano and colleagues later described a group of patients from Japan with a similar phenotype as that described by Nagaoka, localized to the same loci as spinocerebellar ataxia type 4 but a separate gene, PLEKHGA, and named it spinocerebellar ataxia type 31 (203; 108). Spinocerebellar types 4 and 31 are not allelic (69). Symptoms for spinocerebellar ataxia type 4 develop during adulthood.
Spinocerebellar ataxia type 5. This form of inherited ataxia causes slowly progressive cerebellar ataxia, nystagmus, myokymia, and decreased vibratory sensation (216). Symptoms can begin in childhood but most often begin in the third or fourth decade of life. Spinocerebellar ataxia type 5 is rare; at least five families have been reported worldwide (288). Given its relatively pure cerebellar features, spinocerebellar ataxia type 5 may be considered in patients with pure cerebellar ataxia after excluding spinocerebellar ataxia types 6 and 31.
Spinocerebellar ataxia type 6. Spinocerebellar atrophy type 6 is characterized by slowly progressive cerebellar ataxia (162). Other symptoms include dysarthria, oculomotor abnormalities (nystagmus, saccadic intrusions in smooth pursuit, gaze paresis or palsy), dizziness or vertigo, and sensory loss (248). There is a higher incidence of downbeat positioning nystagmus in this type of spinocerebellar ataxia than in other types (303).
Onset usually begins around 50 years of age. There are fewer noncerebellar manifestations compared to spinocerebellar ataxia type 1, type 2, and type 3, but pyramidal tract involvement (hyperreflexia) can be identified late in the course of the disease (253). The disease progresses slowly. Overall, this is usually a milder form of spinocerebellar degeneration (05; 122; 65).
Spinocerebellar ataxia type 7. Unlike most of the spinocerebellar ataxias, type 7 is associated with retinal degeneration or cone-rod dystrophy, optic atrophy, pigmentary retinopathy, and blindness. It also causes the typical spinocerebellar ataxia symptoms of ataxia and bulbar dysfunction. Similar to spinocerebellar ataxia type 2, the earliest findings in type 7 are significantly slowed voluntary and involuntary saccades (197). Rarely, it can present in infancy (12). Marked intrafamilial variability exists in spinocerebellar ataxia type 7. Individuals in the same family may have visual loss without ataxia. In this disorder, visual loss can occur before, at the same, or after the onset of ataxia (171).
Spinocerebellar ataxia type 8. Spinocerebellar ataxia type 8 usually presents in adulthood with slowly progressive dysarthria, swallowing problems, and ataxia of the trunk and extremities (54). Spinocerebellar ataxia type 8 is unique among the dominantly inherited ataxias because dysarthria occurs early in the course of the disease and is disproportionately more severe than the other signs of cerebellar dysfunction. On examination, individuals affected with type 8 may have truncal ataxia, limb incoordination, hyperreflexia, mild sensory loss, and eye movement abnormalities (saccadic smooth pursuit, gaze-evoked nystagmus, and square wave jerks) (54). Age at onset of symptoms ranges from 13 years to 65 years. Some individuals require walking aids if they have had symptoms for more than 20 years. There have been reports of patients with nonataxic phenotypes, including dopa-responsive parkinsonism and atypical parkinsonism (eg, corticobasal syndrome-like or progressive supranuclear-like phenotypes) as well as amyotrophic lateral sclerosis-like phenotype (132; 225).
Spinocerebellar ataxia type 10. Spinocerebellar ataxia type 10 is distinguished by the presence of seizures in addition to cerebellar ataxia. Patients commonly have generalized tonic-clonic seizures or complex partial seizures with or without secondary generalization, but simple focal motor seizures have been reported (04). However, seizures may be absent in patients of Asian ancestry (158). Other associated symptoms include tremor, dysarthria, mild intellectual disability, mood disorders (especially depression and aggressive behavior), polyneuropathy, and ocular dyskinesias (217). Symptoms usually begin in the second decade of life. Nonmotor symptoms include daytime sleepiness, fatigue, depression, and reduced cognitive performance (181).
Rasmussen and colleagues described a family with spinocerebellar ataxia type 10 in which some affected members had non-neurologic abnormalities, including anemia, thrombocytopenia, hepatic dysfunction, and cardiac anomalies (217). Women with spinocerebellar ataxia type 10 can develop initial symptoms or worsen during pregnancy or puerperium (246).
Spinocerebellar ataxia type 11. Spinocerebellar ataxia type 11 is rare and has been reported in five families worldwide. Most reported cases present in early adulthood with cerebellar signs and hyperreflexia. There is no associated movement disorder or sensory deficit (299). A family with onset in early childhood has been reported (149).
Spinocerebellar ataxia type 12. Spinocerebellar ataxia type 12 usually presents in the third or fourth decade with an arm action tremor. The age of onset ranges from 8 years to 55 years. Although tremors can be seen in other spinocerebellar ataxias (type 2, type 3, type 6, and dentatorubral-pallidoluysian atrophy), this is the only spinocerebellar ataxia that has tremor as a common initial sign of disease (198). As it progresses, it can cause head tremor, ataxia, parkinsonism (bradykinesia, rigidity), dysmetria, hyperreflexia, dementia, and abnormal eye movements (109). Tremor in spinocerebellar ataxia type 12 can mimic or be misdiagnosed as essential tremor but generally has lower frequency than essential tremor. Tremor typically involves proximal arms and has high amplitude. Dystonia can coexist. Nonmotor features include agitation, irritability, apathy, anxiety, and depression (39). Patients can have impaired executive function and new learning ability (02).
Spinocerebellar ataxia type 13. Herman-Bert and colleagues reported this ataxia in a French family (107). Almost all of the affected family members developed symptoms in childhood and progressed slowly. The age of onset can vary from infancy to the sixth decade (290). Typical symptoms include ataxia, delay in acquisition of motor and cognitive milestones, mild intellectual disability, dysarthria, and nystagmus. Myoclonus was also found to be a frequent feature in one study (175). Additional abnormalities in some affected individuals include absence seizures, swallowing dysfunction, vertical gaze palsy, torticollis, urinary urgency, and mild dysmorphic features.
Spinocerebellar ataxia type 14. In a Japanese pedigree, spinocerebellar ataxia type 14 is a slowly progressive disease that presents with ataxia, axial myoclonus, and tremor between 12 years and 42 years of age (305). In an American family of Dutch and English heritage, the disease presented as a pure cerebellar ataxia (15). In a French family, affected individuals developed progressive cerebellar ataxia, brisk reflexes, and cognitive impairment but no axial myoclonus (259). For those who develop symptoms in adulthood, the presenting complaint is usually ataxia. For those affected family members who develop symptoms in childhood, the presenting complaint is intermittent tremulous movements of the axial musculature. Other complaints include gaze-evoked nystagmus, saccadic pursuit, ocular overshoot, and decreased deep tendon reflexes. Patients can have complex phenotypes of ataxia in combination with dystonia, parkinsonism, myoclonus, tremor, pyramidal features, or peripheral neuropathy (29). Peripheral neuropathy is rare but has been reported (29). Cognitive impairment is rare and only minimally affected (292). The majority of patients retain the capacity to ambulate.
Spinocerebellar ataxia type 15/16. Storey and colleagues reported this inherited ataxia in an Anglo-Celtic family from Australia. It is one of the pure cerebellar ataxias (262). It has variable presentation in mid-childhood to adulthood. The most common symptoms are gait ataxia, dysarthria, nystagmus, and limb ataxia (272). Other features include action tremor, truncal ataxia, pyramidal signs, dysphagia, dysmetric saccades, and impaired smooth pursuits. The majority of individuals are only mildly impaired, and all have been able to remain ambulatory. The clinical symptoms progress slowly over many years in most patients. Gaze-evoked nystagmus and brisk reflexes occur in those who are most severely affected.
In 2001, Miyoshi described a similar phenotype in a Japanese family, with the addition of head tremor (174), and this was named spinocerebellar ataxia type 16. In 2006, the gene was localized to the same area as spinocerebellar ataxia type 15 (173).
Spinocerebellar ataxia type 17. Affected individuals with spinocerebellar ataxia type 17 develop progressive cerebellar ataxia, dementia, and psychosis (83). Other associated symptoms may include choreoathetosis, rigidity, parkinsonism, and seizures. The phenotype may be similar to Huntington disease and, thus, considered one of the Huntington disease phenocopies and called Huntington disease-like 4 (HDL-4). Spinocerebellar ataxia type 17 is the second most common Huntington disease phenocopy after C9orf72 disease, at least in Europe (106). Rarely, focal dystonia can be the presenting sign (99). Onset is usually in the third to fifth decades of life but can range from 3 to 60 years of age (274). However, a sporadic trinucleotide repeat expansion in the spinocerebellar ataxia type 17 allele has been reported to cause infantile progressive cerebellar ataxia and mental deterioration (136).
Spinocerebellar ataxia type 18. A multigenerational American family of Irish descent with gait difficulties and both motor and sensory neuropathies was described (16). The disorder is also called autosomal dominant sensorimotor neuropathy with ataxia (SMNA). The primary presentation was gait difficulties in the second and third decades. Associated symptoms in the family included pyramidal signs, muscle weakness and atrophy, and decreased vibration and proprioception.
Spinocerebellar ataxia type 19/22. A Dutch family has been described with this relatively mild progressive ataxia syndrome (230). In addition to ataxia, affected individuals may have a peripheral neuropathy, pyramidal signs, postural tremor and myoclonus as well as cognitive impairment affecting executive and visuospatial functions. Epilepsy manifesting as generalized tonic-clonic seizures or myoclonia and parkinsonism has been described (112).
In 2003, a single, 4-generation Chinese pedigree was described for a slowly progressive, pure cerebellar ataxia (51). All affected individuals had gait ataxia. Other features included dysarthria and hyporeflexia. Age at onset of clinical symptoms ranged from 10 to 46 years. This disorder was initially thought to be unique and was called spinocerebellar ataxia type 22. However, it was later discovered to be the same affected gene that causes spinocerebellar ataxia type 19 (67; 141).
Spinocerebellar ataxia type 20. A single family with spinocerebellar ataxia type 20 has been described (135). Unique among the spinocerebellar ataxias, spinocerebellar ataxia type 20 typically presents with dysarthria. Additional features include palatal myoclonus, dysphonia, hypermetric saccades, and mild pyramidal signs.
Spinocerebellar ataxia type 21. Spinocerebellar ataxia type 21 was originally reported in a 4-generation French family (61), and later more affected individuals in the same family were studied (56). The age at onset is typically in early childhood but can be up to 61 years of age. Characteristic clinical features include mild-to-moderate cognitive impairment or intellectual disability and slow progression. Other features are clumsiness, gait and limb ataxia, dysarthria, hyporeflexia, bradykinesia, pyramidal signs, and postural tremor (275). Eye movements are relatively preserved, but there can be intermittent microsaccadic pursuit, square wave jerks, and slow saccades. There are no pyramidal signs. Anticipation has been observed.
Spinocerebellar ataxia type 23. Spinocerebellar ataxia type 23 is a rare form of spinocerebellar ataxia (152; 73). It was described in a Dutch family, clinically characterized by a late-onset (older than 40 years of age), slowly progressive, head and upper limb tremor, and sensory neuropathy (07).
Spinocerebellar ataxia type 24. This disorder is autosomal recessive and has been recategorized as autosomal recessive cerebellar ataxia type 4.
Spinocerebellar ataxia type 25. A single French family with spinocerebellar ataxia type 25 has been reported (258). This syndrome demonstrates significant intrafamilial phenotypic variability. Some individuals presented with cerebellar ataxia and a sensory neuropathy in infancy; others manifested prominent sensory neuropathy with mild ataxia at an older age. In three of seven individuals, vomiting and gastrointestinal features were the presenting problems. Based on this family, spinocerebellar ataxia type 25 shows reduced penetrance.
Spinocerebellar ataxia type 26. Yu and associates described a Norwegian family with affected members with pure cerebellar ataxia with dysarthria, with an age of onset ranging from 26 to 60 years (308).
Spinocerebellar ataxia type 27A. Fourteen patients of a Dutch pedigree were reported to have childhood-onset postural tremor and a slowly progressive ataxia evolving from young adulthood (20). Dyskinesia was often present as were cognitive and behavioral difficulties. Early-onset tremor is common in reported cases, whereas gait ataxia presents later (95). Other features include dysarthria, nystagmus, psychiatric symptoms, and cognitive impairment. Some individuals may have an episodic nature of ataxia (38).
Spinocerebellar ataxia type 27B. This disease has been studied in patients from various independent cohorts, including French Canadian, Quebec, German, Australian, and Indian populations (211; 214; 215). This is a relatively common form of late-onset spinocerebellar ataxia. The age at onset typically falls in the sixth decade, although it can vary between the third and ninth decades (211; 295). The clinical presentation involves a slowly progressive pancerebellar syndrome, downbeat nystagmus, gaze-evoked horizontal nystagmus, sensory impairment, dysautonomia, and vestibular dysfunction. Initially, patients may have an episodic nature of ataxia before it progresses into a more continuous and progressive course of cerebellar features (211). Notably, cognitive impairment is not commonly observed in spinocerebellar ataxia type 27B (295).
Spinocerebellar ataxia type 28. This is a juvenile-onset, slowly progressive, autosomal dominant cerebellar ataxia. The mean age at onset is 19.5 years, and the symptoms of gait incoordination and gaze-evoked nystagmus may present at onset; some patients may develop slow saccades, ophthalmoparesis, and, often, ptosis (27). Deep tendon reflexes in the lower limbs were increased in 80% of the cases. Dysphagia and neurocognitive deficits affecting memory and attention domains have also been reported (265). Pathogenic variants in the AFG3L2 gene, which underlie spinocerebellar ataxia type 28, are associated with a broad phenotypic spectrum. In addition to spinocerebellar ataxia type 28, other AFG3L2-related neurologic disorders include optic atrophy type 12, inherited in an autosomal dominant manner, as well as spastic ataxia type 5 and AFG3L2-related autosomal recessive spinocerebellar ataxia, both of which are inherited in an autosomal dominant manner (40).
Spinocerebellar ataxia type 29. This is an early-onset nonprogressive ataxia and vermian hypoplasia (254). Patients typically have nonprogressive early-onset ataxia and gait difficulties in infancy and early childhood, hypotonia, motor and language delay, and mild-to-moderate cognitive impairment (309). A de novo missense variant, p.Tyr567Cys, located in the IP3-binding pocket (ARM1 domain) of the ITPR1 gene, was reported in a patient with spinocerebellar ataxia type 29 presenting with neurodevelopmental disorder and cerebral palsy (200). Previously reported cases of spinocerebellar ataxia type 29 associated with cerebral palsy were also related to variants in or near the IP3 binding pocket, suggesting a genotype-phenotype correlation.
Spinocerebellar ataxia type 30. Spinocerebellar ataxia type 30, thus far, appears to be a primarily cerebellar type ataxia. Storey and colleagues described a family from Australia with a slowly progressive, late-onset, relatively pure cerebellar ataxia disorder (261).
Spinocerebellar ataxia type 31. Spinocerebellar ataxia type 31 has been described in multiple families in Japan. The clinical features are a late onset with mostly cerebellar ataxia features and variable hearing loss of unknown significance. Initial genetic studies led to a similar location as spinocerebellar ataxia type 4, but Ouyang and colleagues and Hirano and colleagues later described a group of patients from Japan with a similar phenotype as Nagaoka that led to the same loci but a separate gene, PLEKHGA; it was named spinocerebellar ataxia type 31 (203; 108). A natural history study demonstrated a mean age at onset of ataxia of 58.5 years and a protracted clinical course with a mean age at which patients became wheelchair-bound and mean age at death of 79.4 and 88.5 years, respectively (189).
Spinocerebellar ataxia type 32. This disorder has been described in only one family (127). The clinical features, in addition to progressive ataxia, include infertility (especially in males due to azoospermia) as well as cognitive impairment in individuals with onset before the age of 40 years.
Spinocerebellar ataxia type 34. This disorder was originally reported in a French-Canadian kindred and called erythrokeratodermia with ataxia (89). Dermatologic findings, which developed in childhood, included erythema, hyperkeratosis, papulosquamous lesions, and ichthyosis. These lesions disappeared during or by the third decade. Progressive ataxia developed around the fourth decade. Later, this disorder was also reported in two Japanese families. The clinical features in these patients, in addition to progressive ataxia, included eye movement abnormalities (eg, horizontal gaze nystagmus and supranuclear gaze palsy, more prominent in vertical gaze) as well as dysarthria and pyramidal features (204). The age of onset varied from the second to sixth decades.
Spinocerebellar ataxia type 35. This was originally reported in a 4-generation Chinese family and later in multiple families of Han-Chinese descent as well as Asian, European, and Hispanic populations (287; 276; 148). Clinical features include progressive gait and limb ataxia, scanning speech, dysarthria, and impaired hand dexterity. Some patients also have saccadic pursuits and intention hand tremor (97).
Spinocerebellar ataxia type 36. This disorder was originally reported in multiple families in Japan and Spain (115; 264). Subsequently, it has also been reported in other regions, including France, Germany, China, and the United States (142; 278). However, this is a rare form of autosomal dominant cerebellar ataxia. Clinical features include adult- or late-onset limb and gait ataxia as well as characteristic sensorineural hearing loss, tongue fasciculation, and atrophy.
Spinocerebellar ataxia type 37. This disorder has an onset in adulthood. Initial presentation includes falls, dysarthria, and clumsiness. Cerebellar features are slowly progressive. Characteristic abnormal vertical eye movements are hypermetric vertical saccade and saccadic pursuit, especially during downgaze (242).
Spinocerebellar ataxia type 38. This disorder has been reported in three unrelated Italian families and one French family (64). Clinical features include adult-onset, slowly progressive gait and limb ataxia, dysarthria, slow saccades, and nystagmus. Some patients have peripheral neuropathy.
Spinocerebellar ataxia type 40. This has been reported in one family from Hong Kong (277). The age of onset is early 40s. Clinical presentation, in addition to slowly progressive cerebellar ataxia, includes ocular dysmetria, spastic paraparesis, and pyramidal features.
Spinocerebellar ataxia type 41. A 40-year-old Caucasian man of European descent was reported (80). He presented with progressive gait ataxia and imbalance for 2 years. There was no family history. There were no other associated features reported in this patient.
Spinocerebellar ataxia type 42. This has been reported in families from France, Japan, and China (47; 133; 145). The age at onset varies from 9 to 78 years. Clinical features include slowly progressive cerebellar ataxia and dysarthria. Eye movement abnormalities include saccadic pursuits and nystagmus. Impaired vibration sense was found in some patients.
Spinocerebellar ataxia type 43. This has been reported in a 5-generation Belgian family (58). Clinical features are late-onset cerebellar ataxia associated with sensorimotor axonal neuropathy. Some affected individuals have pectus carinatum.
Spinocerebellar ataxia type 44. This has been reported in three families from the United Kingdom (291). Two of these families had adult-onset cerebellar ataxia; the other family presented with juvenile-onset cerebellar ataxia with intellectual disability.
Spinocerebellar ataxia type 45. This was reported in two families, one family with six affected family members in two generations and the other with only one affected individual (194). Clinical features include late-onset, relatively pure cerebellar ataxia manifesting as limb and gait ataxia, dysarthria, and downbeat nystagmus.
Spinocerebellar ataxia type 46. This was originally reported in a Dutch kindred in 1995 (282). Subsequent follow-up report with gene identification from whole-exome sequencing was then published 22 years later (194). Affected individuals have adult-onset cerebellar ataxia and sensory axonal neuropathy. Eye movement abnormalities, such as gaze-evoked nystagmus, square wave jerks, saccadic pursuits, slow saccades, and hypermetric saccades can also be seen.
Spinocerebellar ataxia type 47. Gennarino and colleagues reported two early- and adult-onset forms of ataxia related to PUM1 mutations leading to reduction of Pumilio1 protein (88). In the adult-onset form, patients present during the third to fifth decade with slowly progressive gait and limb ataxia and dysarthria. Some patients also have diplopia. In the early-onset form, patients have developmental disability and seizures in addition to progressive ataxia.
Spinocerebellar ataxia type 48. This was reported in a Spanish family (86). Affected individuals had late-onset, slowly progressive cerebellar ataxia, dysarthria, and psychiatric features (eg, anxiety and organic personality disorder) as well as cognitive features (eg, anosognosia and executive dysfunction). Psychiatric symptoms, especially anxiety, were the initial presentation in over half of the reported patients, even before the onset of cerebellar ataxia. The combination of cerebellar, cognitive, and psychiatric features is called cerebellar cognitive affective syndrome (CCAS). In addition to CCAS, patients can also have chorea, parkinsonism, dystonia, urinary incontinence due to sphincter disturbance, and epilepsy (57; 147). However, there are no pyramidal features and peripheral nervous system involvement (147).
Spinocerebellar ataxia type 49. This was reported in a Spanish family from Menorca (44). Affected individuals had ataxia, horizontal and vertical gaze-evoked nystagmus, dysarthria, and pyramidal features, especially hyperreflexia and sensory polyneuropathy. Age at onset varied from 12 to 60 years.
Spinocerebellar ataxia type 50. This was reported in a large European family (48). Affected individuals had late-onset cerebellar ataxia; eye movement abnormalities, including oculomotor apraxia and downbeat nystagmus; cognitive impairment; and variable neurologic features, such as myoclonus, tremor, chorea, dystonia, and hearing loss. The natural history was slowly progressive. The mean age at onset was 50.7 years (range 34–71). Subsequently, there was a case report of the infantile-onset form of SCA50 in a 6-year-old girl who developed unsteadiness and irritability at the age of 21 months (234).
Spinocerebellar ataxia type 51. This was originally reported in two Chinese families, with 22 and two affected individuals, respectively (269). An additional case was later identified through the UK Biobank, although this individual also had a CAG repeat expansion in the CACNA1A gene, consistent with spinocerebellar ataxia type 6 (74), making the role of the THAP11 repeat expansion in this case unclear. Overall, there have been a limited number of reported cases. Affected individuals typically present with ataxia, dysarthria, and nystagmus (in two of six patients) (269). One patient had bilateral upper limb rest tremor. The age at onset varied from 4 to 51 years, with a median age of onset of 34 years in the larger family with 22 affected family members. Although CAA interruptions within the repeat tract may influence disease expression, their role in pathogenesis remains unclear (293; 223). Further studies across diverse populations are needed to define the phenotypic spectrum and genotype-phenotype correlations. This condition is now officially recognized and assigned by OMIM (#620947) as being caused by CAG repeat expansion in the THAP11 gene.
Dentatorubral-pallidoluysian atrophy. Dentatorubral-pallidoluysian atrophy is named for the pathologic findings associated with the disease. It can cause a wide range of clinical symptoms, including ataxia, choreoathetosis, dystonia, ballism, myoclonus, epilepsy, and dementia (267). It presents anytime from childhood to late adult life, but the clinical presentation differs between the childhood-onset (younger than 20 years) and adult-onset forms (28). The childhood form causes ataxia, progressive myoclonus, and epilepsy. The adult form commonly causes ataxia, psychiatric disease (hallucinations, delusions), dementia, and choreoathetosis. Thus, the adult form of dentatorubral-pallidoluysian atrophy is similar to spinocerebellar ataxia type 17. In the late-onset form, ataxia can occur years before dementia or chorea develops (302). Most patients also have oculomotor abnormalities. A Haw River syndrome reported in an African-American family in North Carolina represents the same disorder (24).
Episodic ataxias. Episodic ataxia type 1 is a rare disorder that causes brief episodes of ataxia, tremor, and dysarthria that last for seconds to minutes, but prolonged attack has been reported (50). The episodes can also be associated with diplopia or vertigo (101). Patients usually experience interictal hand and facial myokymia. Nevertheless, absence of ataxia or myokymia does not exclude this disorder (312). Acetazolamide may be an effective treatment for episodes of ataxia and myokymia. There is a large amount of clinical heterogeneity among patients with episodic ataxia type 1. Graves and colleagues observed the variable phenotypes in monozygotic twins, thereby implicating a role for nongenetic influences on the variable phenotypes (93).
Episodic ataxia type 2 presents in childhood or early adulthood with episodes of ataxia and nystagmus that last for minutes to days. Other associated features include nausea, vertigo, and diplopia. Between episodes, affected individuals show signs of cerebellar dysfunction, such as nystagmus and mild ataxia (124). Cases presenting with recurrent dizziness or vertigo have been reported (150). Individuals with episodic ataxia type 2 do not have myokymia. In episodic ataxia type 1 and type 2, sudden movement, startling, intercurrent infections, stress, prolonged exercise, and alcohol can trigger attacks (101). Patients may also get ictal or interictal migraine headaches. Familial episodic ataxia with interictal nystagmus may be difficult to differentiate from dominantly inherited spinocerebellar atrophy syndromes because some of the patients with episodic ataxia develop progressive cerebellar ataxia (08).
Episodic ataxia type 3 was described in a single large Canadian family with episodic vertigo, tinnitus, and ataxia, with episodes typically lasting minutes (257).
Episodic ataxia type 4, also referred to as periodic vestibulocerebellar ataxia (PATX), is a syndrome that causes ataxia, vertigo, tinnitus, and interictal abnormalities in smooth pursuit and the vestibulo-ocular reflex (52). The attacks last for hours, and patients do not respond to acetazolamide.
Episodic ataxia type 5 has a broad range of clinical phenotypes and is caused by different mutations in the same gene. The gene encodes a calcium channel that is involved in epilepsy syndromes in addition to episodic ataxia (72).
Episodic ataxia type 6 was identified in a single child with episodic and progressive ataxia as well as episodes of hemiplegia and seizures (125). De Vries and colleagues described a Dutch family with milder symptoms with the same affected gene (62). The affected family members’ symptoms resembled a posterior circulation stroke with nausea, vomiting, vertigo, and slurred speech that lasted for only hours, as compared to days in the case described by Jen and colleagues.
Episodic ataxia type 7 was identified in a family with associated symptoms of vertigo, migraine, and weakness in some affected members (131).
Episodic ataxia type 8 was identified in a 3-generation Irish family with 13 affected individuals (42). The onset is in early childhood. During an attack, patients have unsteadiness, generalized weakness, and slurred speech. The attack may be triggered by fatigue or stress, lasts minutes to hours, and occurs every several months up to twice a day.
Episodic ataxia type 9 was originally described in 2010 (146), and there have been over 20 patients reported since then (238). Affected individuals have neonatal or infantile-onset tonic or generalized tonic-clonic seizures. Seizures typically subside or are under control with antiseizure medications after the age of 18 months. Episodic ataxia develops at later ages, after the improvement of seizures. The attacks usually last minutes up to several hours and may be triggered by minor head trauma or sleep deprivation. The frequency of episodic ataxia episodes is weekly or monthly in most patients. However, some patients may have only a few episodes per year, although each episode can last several weeks. Some patients have developmental delay, intellectual disability, or autistic features.
Most of the spinocerebellar ataxia syndromes are degenerative. Generally, the syndromes caused by mutations versus repeat expansions of the genome appear to be more stable and less progressive in their clinical course. Mobility is progressively limited until the patient becomes completely bedridden. Urinary incontinence and swallowing problems may occur. Dementia is associated with spinocerebellar degeneration in xeroderma pigmentosum and in some of the autosomal dominant spinocerebellar atrophy syndromes.
|
• Common spinocerebellar ataxias are mainly due to CAG trinucleotide repeat expansions in coding regions. These include SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17. | |
|
• There are also other genetic bases of spinocerebellar ataxias, including repeat expansions (including trinucleotide and nontrinucleotide repeat expansions) in noncoding regions, as well as point mutations. | |
|
• Most spinocerebellar ataxias with repeat expansions usually have anticipation, a phenomenon where successive generations have earlier ages at onset and more severe diseases due to more expansion of nucleotide repeats. |
When known, the etiology of autosomal dominant inherited ataxias is always a pathogenic mutation or expansion of a gene. For this reason, please see the "Pathogenesis and pathophysiology" section for etiology.
For each of the nonmetabolic causes of inherited ataxia. Table 1 lists each disease with its gene locus, gene product, and causative mutation.
|
Name |
Chromosome |
Gene Product |
Mutation |
Clinical Presentation |
|
SCA1 |
6p23 |
Ataxin-1 |
CAG repeat expansion in ATXN1 gene |
Ataxia, pyramidal signs, neuropathy, dysphagia, restless legs |
|
SCA2 |
12q24.1 |
Ataxin-2 |
CAG repeat expansion in ATXN2 gene |
Ataxia, slow saccades, neuropathy, restless legs |
|
SCA3 |
14q32.1 |
Ataxin-3 |
CAG repeat expansion in ATXN3 gene |
Ataxia, pyramidal signs, ophthalmoplegia, neuropathy, dystonia, restless legs |
|
SCA4 |
16q22.1 |
Zinc finger homeobox protein 3 |
ZFHX3 |
Ataxia, sensory neuropathy, autonomic dysfunction, slow saccades |
|
SCA5 |
11p13 |
Beta III spectrin |
SPTBN2 gene |
Almost purely cerebellar ataxia |
|
SCA6 |
19p13.1 |
Calcium channel alpha-1a subunit |
CAG repeat expansion in CACNA1A gene |
Almost purely cerebellar ataxia |
|
SCA7 |
3p14 |
Ataxin-7 |
CAG repeat expansion in ATXN7 gene |
Ataxia, visual loss, ophthalmoplegia |
|
SCA8 |
13q21 |
Ataxin-8 |
CTA/CAG bidirectional repeat expansion of one coding (ATXN8) and one noncoding gene (ATXN8OS) |
Ataxia, sensory neuropathy, spasticity |
|
SCA10 |
22q13 |
Ataxin-10 |
ATTCT noncoding repeat expansion in ATXN10 gene |
Ataxia, epilepsy |
|
SCA11 |
15q15.2 |
Tau tubulin kinase 2 |
TTBK2 gene |
Almost purely cerebellar ataxia |
|
SCA12 |
5q32 |
Serine/threonine protein phosphatase 2A, regulatory subunit B |
Noncoding CAG expansion in PPP2R2B gene |
Ataxia, tremor |
|
SCA13 |
19q13.3-4 |
Kv3.3 voltage-gated potassium channel |
KCNC3 gene |
Ataxia, intellectual disability |
|
SCA14 |
19q13.4 |
Protein kinase C gamma |
PRKCG gene |
Ataxia, myoclonus, dystonia, sensory loss |
|
SCA15/16 |
3p26-p25 |
Inositol 1,4,5-triphosphate (IP3) receptor |
ITPR1 gene |
Almost purely cerebellar ataxia |
|
SCA17 |
6q27 |
TATA-binding protein |
CAG/CAA repeat expansion in TBP gene |
Ataxia, dystonia, chorea, dementia, psychiatric abnormalities |
|
SCA18 |
7q22-q32 |
Interferon-related developmental regulator 1 |
IFRD1 gene |
Ataxia, sensory neuropathy, neurogenic muscle atrophy |
|
SCA19/22 |
1p21-q12 (284) |
Kv4.3 voltage-gated potassium channel |
KCND3 gene |
Ataxia, myoclonus, cognitive impairment |
|
SCA20 |
11 |
Unknown |
Unknown |
Ataxia, dysphonia |
|
SCA21 |
7p21.3-p15.1 |
Transmembrane protein of unknown function |
TMEM240 gene |
Ataxia, parkinsonism |
|
SCA23 |
20p13-12.3 |
Prodynorphin |
Missense mutations in PDYN gene |
Ataxia, tremor, sensory neuropathy |
|
SCA25 |
2p21-p13 |
Polyribonucleotide nucleotidyltransferase PNPase 1 |
PNPT1 gene |
Ataxia, sensory neuropathy |
|
SCA26 |
19p13.3 |
Eukaryotic elongation factor 2 |
eEF2 gene |
Almost purely cerebellar ataxia |
|
SCA27A |
13q34 |
Fibroblast growth factor 14 |
Mutation or translocation in coding region of FGF14 gene |
Ataxia, tremor, dyskinesias, cognitive and behavioral disturbances |
|
SCA27B |
13q33.1 |
Fibroblast growth factor 14 |
Intronic GAA repeat expansion in FGF14 gene |
Late-onset ataxia, pancerebellar syndrome, downbeat nystagmus, sensory impairment, autonomic dysfunction |
|
SCA28 |
18p11.22-q11.2 |
ATP-dependent mitochondrial matrix-AAA (m-AAA) protease |
Mutation in coding region of AFG3L2 gene |
Ataxia, ophthalmoparesis |
|
SCA29 |
3p26 |
Inositol 1,4,5-triphosphate (IP3) receptor |
ITPR1 gene |
Early-onset nonprogressive ataxia, cognitive difficulties |
|
SCA30 |
4q34.3-q35.1 |
Unknown |
Unknown |
Almost purely cerebellar ataxia |
|
SCA31 |
16q21 |
Brain expressed associated with NEDD4 type 1 |
TGGAA repeat expansion in intron of BEAN1 gene |
Late-onset ataxia, variable hearing loss |
|
SCA32 |
7q32-33 |
Unknown |
Unknown |
Progressive ataxia, azoospermia, cognitive impairment when onset younger than 40 years |
|
SCA34 |
6q14.1 |
Very long-chain fatty acid elongase 4 |
ELOVL4 gene |
Skin lesions, eg, erythema, hyperkeratosis in childhood, ataxia |
|
SCA35 |
20p13 |
Transglutaminase 6 |
TGM6 gene |
Progressive ataxia, pyramidal features, intention hand tremor |
|
SCA36 |
20p13 |
Nop56p, a component of small nucleolar ribonucleoprotein |
GGCCTG repeat expansion in the first intron of NOP56 gene |
Late-onset ataxia, hearing loss, tongue fasciculation, and atrophy |
|
SCA37 |
1p32.2 |
DAB1, the reelin adapter protein |
ATTCT repeat expansion in noncoding region of DAB1 gene |
Adult-onset slowly progressive ataxia, abnormal vertical eye movements |
|
SCA38 |
6p12.1 |
Very long-chain fatty acid elongase 5 |
ELOVL5 gene |
Adult-onset slowly progressive ataxia, peripheral neuropathy |
|
SCA40 |
14q32 |
Coiled-coil domain containing 88C protein |
Missense mutation in CCDC88C gene |
Adult-onset ataxia, spastic paraparesis, pyramidal features |
|
SCA41 |
4q27 |
Transient receptor potential channel 3 |
Missense mutation in TRPC3 gene |
Progressive ataxia |
|
SCA42 |
17q21.33 |
Voltage-gated calcium channel 1G |
CACNA1G gene |
Slowly progressive ataxia, impaired vibration sense |
|
SCA43 |
3q25.2 |
Neprilysin |
C143Y variant in MME gene |
Late-onset ataxia, sensorimotor neuropathy, pectus carinatum |
|
SCA44 |
6q24.3 |
Metabotropic glutamate receptor 1 |
GRM1 gene |
Adult-onset ataxia; juvenile-onset ataxia with intellectual disability |
|
SCA45 |
5q33 |
FAT2 Tumor suppressor protein |
FAT2 gene |
Late-onset, relatively pure cerebellar ataxia |
|
SCA46 |
19q13.2 |
Phospholipase D3 |
PLD3 gene |
Adult-onset ataxia, sensory axonal neuropathy |
|
SCA47 |
1p35.2 |
Pumilio 1 |
PUM1 gene |
Adult-onset ataxia; early-onset form with developmental disability and seizure |
|
SCA48 |
16p13.3 |
STUB1 |
STUB1 gene |
Late-onset ataxia, psychiatric features, cognitive dysfunction |
|
SCA49 |
7q21 |
SAMD9L |
SAMD9L gene |
Ataxia, gaze-evoked nystagmus, pyramidal features, sensory polyneuropathy |
|
SCA50 |
17q25.3 |
Neuronal pentraxin 1 |
NPTX1 gene |
Adult-onset ataxia, oculomotor apraxia, cognitive impairment |
|
SCA51 |
16q22.1 |
THAP domain containing 11 |
THAP11 gene |
Ataxia, dysarthria, nystagmus, arm tremor; myoclonic seizures and cognitive impairment reported in a severe case |
|
DRPLA |
12p13.31 |
Atrophin-1 |
CAG repeat expansion in ATN1 gene |
Ataxia, myoclonus, epilepsy, extrapyramidal signs, dementia |
|
EA1 |
12p13.32 |
Potassium channel |
Missense mutations in KCNA1 gene |
Episodic ataxia, myokymia |
|
EA2 |
19p13.13 |
Voltage-dependent P/Q type calcium channel |
Truncating and missense mutations or CAG repeat expansion in CACNA1A gene |
Episodic ataxia, nystagmus |
|
EA3 |
1q42 |
Unknown |
Unknown |
Episodic ataxia, tinnitus, vertigo |
|
EA4 |
Unknown |
Unknown |
Unknown |
Episodic ataxia, vertigo, diplopia |
|
EA5 |
2q23.3 |
Calcium channel beta 4 subunit |
Missense mutations in CACNB4 gene |
Episodic ataxia, seizures, nystagmus |
|
EA6 |
5p13.2 |
Excitatory amino acid transporter 1 (EAAT1) |
SLC1A3 gene |
Episodic ataxia, migraine, stroke-like symptoms |
|
EA7 |
19q13 |
Unknown |
Unknown |
Episodic ataxia, vertigo, migraines |
|
EA8 |
1p36.13-p34.3 |
Unknown |
Unknown |
Episodic unsteadiness, generalized weakness, slurred speech |
|
EA9 |
2q24.3 |
Voltage-gated sodium channel Nav1.2, alpha subunit |
SCN2A gene |
Episodic ataxia, infantile- or neonatal-onset seizure, developmental delay |
The autosomal dominant cerebellar ataxias are caused by expansions in either coding or noncoding regions of the genome as well as conventional mechanisms such as mutations and translocations within the DNA code. Most repeat expansions are common, benign polymorphisms. For example, the SCA1 allele commonly has 6 to 39 CAG-trinucleotide repeats. However, if the expansion becomes too large, the gene or protein product becomes dysfunctional. For example, spinocerebellar ataxia type 1 develops when the SCA1 allele has more than 39 CAG-trinucleotide repeats. In many of the cases of autosomal dominant ataxia, the clinical degeneration is due to or is associated with large repeat expansions of a trinucleotide in a coding region (SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, DRPLA, and EA2). The autosomal dominant ataxias associated with repeats in noncoding regions of the genome are SCA8, SCA10, SCA12, and SCA31. The remainder of the autosomal dominant ataxia syndromes have either a mutation (SCA5, SCA11, SCA13, SCA14, SCA15/16, SCA20, SCA27, SCA28, EA1, EA2, EA5, and EA6) or remain unknown (68).
Repeat expansions are “dynamic” mutations because they are unstable and can undergo enlargement or contraction during meiosis. SCA1, SCA2, SCA3, and SCA7 are more likely to be transmitted through the maternal lineage. A possible explanation for this discrepancy may be that the greatly expanded repeats in sperm populations are less stable than expansions within the oocyte (91). The greater expansion between generations occurs when the expansion is paternally inherited. For the most part, the severity of the disease directly correlates with the size of the repeat expansion. Therefore, in successive generations, the same disease can present at an earlier age with a more severe phenotype. This phenomenon, called anticipation, accounts for the phenotypic heterogeneity that can be seen within families affected by the same spinocerebellar ataxia syndrome. Anticipation is not readily seen in families with autosomal dominant ataxia syndromes caused by a mutation. These syndromes tend to have an earlier onset but are usually slowly progressive and not as severe as the expansion-related syndromes. The syndromes associated with expansion repeats also tend to have both cerebellar and extracerebellar features and more widespread neuronal loss. Many of the purely cerebellar syndromes are associated with mutations and have only Purkinje cell loss on autopsy. Sporadic cases occur when an allele with a repeat length in the upper limit of normal undergoes expansion or de novo mutations.
In spinocerebellar ataxia type 1, type 2, type 3, type 6, type 7, and dentatorubral-pallidoluysian atrophy, the triplet repeat expansion occurs in the polyglutamine tail of the gene. Mutations of this polyglutamine tail are believed to confer a novel neurotoxic property onto the gene (134). In most cases, the spinocerebellar ataxias have been associated with abnormal nuclear or cytoplasmic inclusions. It is unclear how these accumulations relate to the pathogenesis of disease.
Spinocerebellar ataxia type 1. This is due to a CAG trinucleotide repeat expansion within the SCA1 gene on chromosome 6p. The length of the CAG trinucleotide repeat is normally 6 to 39 with a CAT interruption (201). In affected patients, there are 40 to 81 trinucleotide repeat expansions that lack the CAT interruption. The CAT interruption must be integral to pathogenesis because normal CAG trinucleotide lengths that are not interrupted by CAT are more likely to expand. The protein ataxin-1 is encoded by the SCA1 gene. The exact function of ataxin-1 is still unknown. Transgenic mouse models with the ataxin-1 gene and SCA1 phenotype show intracellular vacuoles and intranuclear inclusion bodies.
Mastrogiacomo and Kish found markedly reduced activity of alpha-ketoglutarate dehydrogenase complex, a rate-limiting enzyme complex of the Krebs cycle, in postmortem brains of patients with the spinocerebellar ataxia type 1 (160). This suggested that these patients might experience depleted energy stores, susceptibility to excitotoxic neuronal damage, or altered levels of excitatory neurotransmitters.
One model in spinocerebellar ataxia type 1 pathogenesis is related to impaired dynamic equilibrium between two complexes, ataxin-1-CIC (transcriptional regulator capicua) and ataxin-1-RBM17 (a splicing factor RNA-binding motif protein), in the nuclei of Purkinje cells (159). The expanded polyglutamine tract impairs the balance and favors the formation of the ataxin-1-RBM17 complex. In addition, the RAS-MAPK-MSK1 pathway was found to have interactions with ataxin-1 in Drosophila and human cell lines, and suppression of multiple components of this pathway led to a reduction of ataxin-1 level (207).
Although spinocerebellar ataxia type 1 generally begins in adulthood, a direct correlation was found between the increased size of the CAG-repeat expansion and an earlier age of onset of spinocerebellar atrophy type 1 (201). This earlier-onset spinocerebellar ataxia type 1 is more often inherited from an affected father (139).
Savoiardo and colleagues reported that neuroimaging of patients with olivopontocerebellar atrophy reveals atrophy of the cerebellum and brainstem, signal abnormalities in the basal ganglia, and supratentorial atrophy (229).
Pathology. In most cases, spinocerebellar ataxia type 1 causes olivopontocerebellar degeneration. There is Purkinje cell loss in the cerebellum, cell loss of the brainstem, and degeneration of the spinocerebellar pathways. There is usually cell loss of the dentate gyrus, red nucleus, and inferior olivary nucleus. There may also be degeneration of the third, tenth, and twelfth nuclei (202). There are no major changes in the cerebral cortex. Another study of serial tissue sections through the complete brains demonstrated more widespread involvement than previously known, including basal ganglia, thalamic sensory and motor nuclei, and brainstem dopaminergic and cholinergic systems (221).
Spinocerebellar ataxia type 2. This is due to a CAG-trinucleotide repeat expansion in the coding region of the ATXN2 gene on chromosome 12. The gene encodes for ataxin-2, a protein without a known function. The normal repeat length of 15 to 32 repeats is expanded to 33 to 200 repeats in spinocerebellar ataxia (75; 161).
Schols has shown that in spinocerebellar ataxia type 2, like other autosomal dominant spinocerebellar ataxias, longer CAG repeats are associated with earlier age of onset and more rapid worsening (235). As in spinocerebellar ataxia type 1, there is preferential anticipation with paternal transmission. Interestingly, ATXN2 alleles with 29 to 32 repeats were a risk of amyotrophic lateral sclerosis, and the risk was positively correlated with the length of repeats (255).
Ataxin-2 oligomers may play an important role in the pathogenesis of spinocerebellar ataxia type 2. In cultured fibroblasts from patients with spinocerebellar ataxia type 2, removal of ataxin-2 oligomers resulted in less apoptotic activation (289).
As in spinocerebellar ataxia type 1, type 2 is associated with olivopontocerebellar atrophy.
Spinocerebellar ataxia type 3 (Machado-Joseph disease). Type 3 is also a CAG-trinucleotide repeat disorder. The gene encodes for ataxin-3, a protein without a known function. The normal repeat length of 12 to 42 repeats is expanded to 60 to 84 repeats in spinocerebellar ataxia type 3. The length of the trinucleotide repeat is inversely related to the age of onset, pyramidal dysfunction, and dystonia (123). In addition, the length of CAG repeats is also correlated with the rate of disease progression (144). However, the length of the trinucleotide repeat does not correlate with the other neurologic findings in this disease. Therefore, it remains unclear what other factors contribute to the clinical heterogeneity that is common in spinocerebellar ataxia type 3.
Normally, ataxin-3 functions as a deubiquitinating enzyme. Mutant or expanded ataxin-3 protein with polyglutamine expansion, thought to be the toxic species, leads to misfolding, aggregate formation, and alteration of molecular interaction with other proteins (Costa and Paulson 2012).
MRI findings in type 3 show that the atrophy is widespread, including the cerebellum, vermis, pons, olives, globus pallidus, and frontal and temporal lobes (184). A study showed that on resting-state functional MRI, there were changes in functional connectivity between cerebellum, parahippocampal areas, medial prefrontal cortex, caudate nuclei, thalamus, and lateral frontoparietal cortical regions; these are correlated with ataxia severity (281). The authors called this pattern “fMRI SCA3-related pattern” (fSCA3-RP) and proposed it as a potential biomarker of the disease.
Pathology. The spinocerebellar tracts and pons undergo atrophy, but the olives may not. This may help to differentiate type 3 from type 1 and type 2.
Spinocerebellar ataxia type 4. The disease locus was mapped to chromosome 16q22.1 (79). Exonic GGC trinucleotide repeat expansions in the ZFHX3 gene were identified to be the cause by two independent groups (76; 286). ZFHX3 encodes zinc finger homeobox protein 3, a transcriptional regulator that binds to AT-rich core sequences and plays a role in neuronal differentiation. GGC repeat expansions result in polyglycine tracts and abnormal autophagy in cell models (286), supporting a toxic gain-of-function mechanism. Brain MRI reveals cerebellar atrophy.
Pathology. Gross pathology reveals cerebellar atrophy. Microscopically, there are intranuclear and intracytoplasmic p62 inclusions within neurons in the medulla oblongata, basis pontis, as well as esophageal myenteric plexuses (76; 286).
Spinocerebellar ataxia type 5. Mutations in the SPTBN2 gene cause a loss of function to the beta-III spectrin protein (114). Beta-III spectrin is expressed in Purkinje cells and possibly plays a role in stabilizing the glutamate transporter EAAT4 (114). The disease has been documented in American (216), French (260), German (23), Norwegian (36), and Japanese families (288). Descendants of Abraham Lincoln were among the first studied to discover the gene. Homozygous mutation in the same gene has been reported to cause infantile-onset autosomal recessive spinocerebellar ataxia type 14 or SCAR14 (70).
Brain MRI reveals significant cerebellar vermis and hemisphere atrophy with sparing of the pons (260).
Spinocerebellar ataxia type 6. This disease is a channelopathy due to a CAG trinucleotide repeat expansion of the CACNA1A gene, which encodes for the alpha1a subunit of the P/Q type voltage-gated calcium channel. This allele usually has 3 to 17 repeats, but in spinocerebellar ataxia type 6, there are 21 to 30 repeats (311). There is an inverse correlation between the length of the expanded allele and the age of onset, and in individuals who are homozygous for mutations, the age of onset is inversely correlated to the sum of the CAG repeats in both alleles (266). Spinocerebellar ataxia type 6, familial hemiplegic migraine, and episodic ataxia type 2 are allelic disorders. Point mutations of this same gene are associated with familial hemiplegic migraine. Truncation mutations of this gene are associated with episodic ataxia type 2. Unlike episodic ataxia type 2 and familial hemiplegic migraine, spinocerebellar ataxia type 6 is progressive.
In addition to the alpha1a subunit of the P/Q type voltage-gated calcium channel, the CACNA1A gene also encodes for alpha1ACT, a transcription factor, with a polyglutamine tract (66). The alpha1ACT protein with an expanded polyglutamine tract leading to transcriptional dysregulation has been postulated in the pathogenesis of spinocerebellar ataxia type 6.
Brain MRI demonstrates cerebellar atrophy (183). However, there is some PET evidence that the metabolic activity of the entire brain is reduced; the reduction is most prominent (63% to 66%) in the cerebellum and brainstem (253).
Pathology. There is significant cerebellar atrophy without brainstem involvement. Microscopically, there is a significant loss of Purkinje cells with less loss of granule cells, dentate nucleus cells, and inferior olive cells (226).
Spinocerebellar ataxia type 7. A CAG-trinucleotide repeat of the SCA7 gene on chromosome 3 causes spinocerebellar ataxia type 7. The normal repeat length is 7 to 17 repeats, but in the disease, it is expanded to 34 repeats or more (53). There is an inverse correlation between the length of the trinucleotide repeat expansion and the age of onset. This expansion is particularly unstable, and there is a strong anticipation. In a single generation, the expansion can become large enough to cause infant, and even embryonic, disease (12). Paternal transmission of the expansion is more likely to undergo repeat length expansion (91). However, affected individuals are more likely to inherit the mutation from their mother (greater than 80%). This dichotomy may be due to instability of the expansion within sperm.
The SCA7 gene product, ataxin-7, is a core component of SAGA complexes (Spt-Ada-Gcn5 Acetyltransferase), which has a role in transcriptional regulation. Ataxin-7 protein undergoes post-translational modification by SUMOylation. SUMOylated mutant ataxin-7 can form aggregates that lead to impaired function of SAGA complexes and transcriptional dysregulation (129).
Pathology. There is cerebellar, brainstem, globus pallidus, red nucleus, spinal cord, and occipital cortex atrophy (92). The retina shows loss of photoreceptor, ganglion, and bipolar neurons. There is also retinal pigment epithelial damage. Additional neuropathological studies revealed neuronal loss in a number of brain regions, including brainstem and thalamus, that were functional components of cerebellothalamocortical and basal ganglia-thalamocortical loops, visual, somatosensory, auditory, vestibular, and oculomotor systems as well as midbrain dopaminergic system (222).
Spinocerebellar ataxia type 8. This disease is caused by bidirectional transcription of CTG/CAG repeat expansion on chromosome 13 (113). The CAG DNA strand is transcribed and translated into a polyglutamine protein product, ataxin 8. The CTG DNA repeat is transcribed into a CUG noncoding RNA repeat of the ATXN8OS (Ataxin-8 Opposite Strand) gene, which is thought to have its own detrimental effects on cells. The CTG noncoding region was first implicated to be causal in SCA8; however, there was controversy regarding its role based on variable inheritance patterns and the presence of expanded repeats in asymptomatic individuals, as well as in other cases of SCA (SCA1 and SCA6). The same group that described the CTG repeat went on to discover the novel CAG expansion, thereby demonstrating that two genes spanning the repeat are expressed in opposite directions, leading to the SCA8 phenotypes. Studies in humans and animal models demonstrate the presence of polyglutamine protein-containing inclusion bodies in CTG/CAG bidirectional repeats. The presence of polyglutamine intranuclear RNA inclusion bodies in CUG repeats from ATXN8OS implicates the toxic roles of gain-of-function of protein as well as RNA in SCA8 (273). The number of alleles varies dramatically, with the normal reported range from 15 to 50 repeats and pathogenic alleles containing 71 to 1300 repeats.
The mechanism called Repeat-Associated Non-ATG (RAN) translation has been implicated in the pathogenesis of spinocerebellar ataxia type 8 (313). The RAN translation led to abnormal accumulation of three forms of RAN proteins, including polyglutamine in cerebellum and brainstem, polyalanine in cerebellum, and polyserine in white matter regions (Clearly and Ranum 2013; 06).
Neuroimaging in a few patients with spinocerebellar ataxia type 8 showed atrophy of the cerebellar hemispheres and vermis (54). Nerve conduction studies showed significantly reduced sensory amplitudes. Electromyography was normal.
Spinocerebellar ataxia type 10. Spinocerebellar ataxia type 10 is caused by a unique ATTCT-pentanucleotide repeat expansion in intron 9 of the ATXN10 gene. The normal repeat length is 10 to 22 repeats, but in affected individuals, there may be thousands of repeats (166). The ATXN10 gene is expressed throughout the brain. The ATTCT repeat may undergo intergenerational contraction as well as expansion (164). The expansion is highly unstable when paternally transmitted (164). The SCA10 gene is also expressed in the liver and hematopoietic cells. Similarly, this may explain why some patients develop systemic problems (217). The SCA10 expansions that contain the pentanucleotide (ATCCT and ATCCC) and heptanucleotide (ATATTCT and ATTTTCT) interruptions, collectively called the ATCCT interruption motif, undergo large contraction during germline transmission, especially paternal lineage (167). This does not follow the general rule of anticipation; however, individuals in younger generations still have an earlier age at onset. Furthermore, the presence of these repeat interruptions is a strong risk factor of epilepsy in patients with spinocerebellar ataxia type 10 (168).
Brain MRI shows isolated cerebellar atrophy. One neuropathological study demonstrated Purkinje cell loss (301). In one report, diffuse cerebral disturbance was demonstrated on every EEG obtained on individuals affected with spinocerebellar ataxia type 10 (217). The EEG may also show focal abnormalities in those patients with seizures.
Spinocerebellar ataxia type 11. Mutations in the TTBK2 gene that encodes for the tau tubulin kinase 2 were discovered in multiple families with a purely cerebellar form of ataxia (110). Tau tubulin kinase 2 has a crucial role in ciliogenesis (90). Primary cilia are organelles that are also present in neurons, and impairment in ciliogenesis leads to Purkinje cell death in knockout mouse models (14). The mechanisms by which tau tubulin kinase 2 regulates ciliogenesis and the exact role of primary cilia in the cerebellum, including Purkinje cells, remain unknown.
Spinocerebellar ataxia type 12. The normal SCA12 allele has 7 to 28 CAG trinucleotide repeats, but those with spinocerebellar ataxia type 12 have 55 to 78 repeats (198; 256). Srivastava and colleagues did not find a correlation between the length of the repeat expansion and the age of onset (256).
The CAG repeat expansion that causes type 12 is within the PPP2R2B gene. This gene encodes for a regulatory subunit B of serine/threonine protein phosphatase 2A, which has a ubiquitous role in cellular functions, including apoptosis. There are at least two hypotheses of the pathogenesis of spinocerebellar ataxia type 12: one is that the repeat expansion affects expression of the PPP2R2B gene, leading to impaired regulatory function of protein phosphatase 2A; the other is that the toxic species may be due to expression of the repeat itself (41) through mechanisms other than polyglutamine tracts, given that neuropathological examination did not reveal polyglutamine aggregates.
Neuroimaging of individuals affected by spinocerebellar ataxia type 12 shows mild to moderate atrophy of the cerebellum and cerebral cortex (198). Other regions of the brain are spared.
Neuropathological examination revealed marked cerebral cortical atrophy, Purkinje cell loss, and less-prominent cerebellar atrophy but no polyglutamine aggregates (199).
Spinocerebellar ataxia type 13. Spinocerebellar ataxia type 13 is caused by mutations in the KCNC3 gene, which disrupts the function of a voltage-gated potassium channel, Kv3.3 (290; 77). There are currently two main types of mutations that cause either a prolonged open state of the channel or a dominant negative effect of expressing the channel. There have not been consistent genotype/phenotype correlations yet, but seizures have been reported in a few of the cases with the KCNC3 mutations. Various mutations have different effects on the properties of Kv3.3. Nevertheless, how these physiological alterations lead to ataxia and other phenotypes remains unknown (310).
Spinocerebellar ataxia type 14. Spinocerebellar ataxia type 14 is caused by mutations or deletions in PRKCG, a gene that encodes for a protein kinase C gamma (31). The mutations in the PRKCG gene were the first described to cause a spinocerebellar ataxia. The previously known genetic causes at that time, in 2003, were all related to nucleotide repeat expansions such as the CAG repeats. A gain-of-function mechanism rather than haploinsufficiency has been speculated in the pathogenesis of spinocerebellar ataxia type 14 (03). Nevertheless, this has been debated. One study demonstrated that mutations in the C1 domain of the PRKCG gene caused cytoplasmic mislocalization and aggregation of protein kinase C gamma as well as increased protein kinase C gamma activity, supporting both gain-of-function and loss-of-function mechanisms (297). A minority of the patients in the study by Yamashita and colleagues had neuroimaging studies (305). They demonstrated slight to moderate cerebellar atrophy, especially of the vermis.
Spinocerebellar ataxia type 15 (also formerly spinocerebellar ataxia type 16). Spinocerebellar ataxia type 15 and type 16 are caused by haploinsufficiency in the ITPR1 gene that encodes for an IP3-gated calcium channel, which affects intracellular calcium signaling (102; 120). SCA16 was initially thought to be caused by a separate gene, CNTN4, which encodes for a contactin protein. A polymorphism was found in this gene in a Japanese family presenting with an autosomal dominant ataxia with tremor. Iwaki and colleagues later found a deletion in ITPR1 gene in these patients (120). The brain MRI typically demonstrates vermian atrophy (262; 272). There is generally no cerebellar hemisphere, olivary, or pontine atrophy.
Spinocerebellar ataxia type 17. This is due to a CAG/CAA repeat expansion in the TBP gene encoding for TATA box-binding protein. The normal allele contains 29 to 42 CAG/CAA repeats, and disease is associated with 47 or more repeats (188). Total CAG repeat length is inversely correlated with the age at onset (193). The TBP gene is a general transcription initiation factor and the DNA-binding subunit of RNA polymerase II transcription factor D. This complex is crucial for the expression of most genes (188). It is presumed that spinocerebellar ataxia type 17 is caused by a gain-of-function of this gene; however, the exact mechanism remains unclear. In animal models, mutant TBP was found to affect not only neurons but also glial cells, which also synergistically promoted neuronal injury (306). Unlike the other spinocerebellar ataxia syndromes caused by polyglutamine expansions, the expansion of the triplet repeat seen in the TBP gene is more stable. The TBP gene has multiple configurations for the CAG/CAA repeat. The cases in which there is only a CAG uninterrupted repeat have been shown to have more intragenerational expansion compared to CAG repeats that are interrupted with the CAA sequence (85).
Neuroimaging reveals significant cerebellar atrophy without significant brainstem degeneration (83). Putaminal rim sign seen as a hyperintense signal on T2-weighted MRI, similar to what is seen in multiple system atrophy, has also been reported (153).
Pathology. Pathological information is limited. Based on a small number of case reports, the distinctive pathology of spinocerebellar ataxia type 17 is significant cerebellar atrophy, no pontine atrophy, and only minimal cortical atrophy (83). Microscopic examination of the cerebellum revealed severe Purkinje cell loss with Bergmann gliosis, gliosis of the molecular layer, and rarefied inner granular layer. Neurons of the dentate nucleus contain perinuclear inclusions. In the brainstem, there was preservation of neuronal and glial integrity. Marinesco bodies were present in the substantia nigra.
Spinocerebellar ataxia type 18. Spinocerebellar ataxia type 18 is also known as autosomal dominant sensorimotor neuropathy with ataxia. Brkanac and colleagues described a multigenerational family with this phenotype, and in 2009, identified a candidate gene, IFRD1, which encodes for interferon-related developmental regulator 1 (17). More studies need to be performed in individuals with similar phenotypes to confirm causality.
Spinocerebellar ataxia type 19/22. Mutations in the KCND3 gene encoding for the voltage-gated potassium channel (Kv4.3) are responsible for both spinocerebellar ataxia types 19 and 22, merging these two types of spinocerebellar ataxias (67; 141). Thus, the disorder is considered a channelopathy.
Brain MRI reveals marked atrophy of the cerebellar hemispheres and moderate vermian atrophy (230; 231). Neuropathological examination demonstrates not only cerebellar but also brainstem involvement (239). In addition to Purkinje cell loss, there is evidence of neurodegeneration in the brainstem, including substantia nigra pars compacta and omnipause neurons of the nucleus raphe interpositus, and inferior olivary nuclei.
Spinocerebellar ataxia type 20. The locus was mapped to the pericentromeric region of chromosome 11, but the gene has not been identified. Dentate calcification is evident on the CT and MRI of the brain (135).
Spinocerebellar ataxia type 21. This is caused by mutations in the TMEM240 gene encoding for a transmembrane protein of unknown function (56). Lysosomal dysfunction and activation of microglia and astrocytes were found after TMEM240 overexpression in cell culture and mouse model, but their implications to the pathogenesis require further studies (241). Individuals with this form of spinocerebellar degeneration show cerebellar atrophy, especially of the superior vermis, without atrophic changes in the brainstem (61; 275).
Spinocerebellar ataxia type 23. Bakalkin and colleagues identified missense mutations in the PDYN gene in four Dutch families with progressive ataxia (07). This gene encodes for prodynorphin, a precursor protein for opioid neuropeptides, alpha-neoendorphin, and dynorphins A and B (07). It has been postulated that elevated mutant dynorphin A may be critical in the pathogenesis (250). The neurotoxic effects of mutant dynorphin A may be through NMDA-mediated excitotoxicity and loss of opioid signaling (251).
Spinocerebellar ataxia type 25. The PNPT1 gene was identified from whole-exome and whole-genome sequencing in individuals from the originally reported French family and an Australian family (09). This gene encodes polyribonucleotide nucleotidyltransferase PNPase 1, which is located in the mitochondria and intermembranous space. Mutations in this gene result in the accumulation of double-stranded mitochondrial RNAs, leakage into the cytoplasm, and subsequent activation of type 1 interferon. Brain MRI shows significant global cerebellar atrophy without brainstem involvement (258).
Spinocerebellar ataxia type 26. This is caused by a heterozygous P596H mutation in the eEF2 gene on chromosome 19p13.3, encoding for eukaryotic elongation factor 2 (105). This region is adjacent to the gene location for spinocerebellar ataxia type 6. The brain MRI of the affected patients demonstrated atrophy confined to the cerebellum (Yu t al 2005).
Spinocerebellar ataxia type 27A. The disease is caused by point mutations, deletions, or translocations that disrupt the FGF14 gene on chromosome 13q34, encoding for fibroblast growth factor 14 (20). Patients from a Dutch pedigree had an F145S mutation in this gene. Other mutations or variants have also been reported (172).
Spinocerebellar ataxia type 27B. The disease is caused by intronic GAA repeat expansions in the FGF14 gene encoding fibroblast growth factor 14 (211; 214; 215). Current data indicate that pathogenicity with full penetrance is associated with more than 300 GAA repeats, whereas 250 to 300 repeats show incomplete penetrance; however, these thresholds may need to be refined (210; 116).
Fibroblast growth factor 14 is involved in the function of voltage-gated sodium channels, affecting the rhythmic spontaneous firing of Purkinje cells. It has been proposed that the loss-of-function pathomechanism of the FGF14 gene involves repeat expansions forming a “sticky” DNA secondary structure, which, in turn, inhibits transcription of the gene, similar to the mechanism of GAA repeat expansion in Friedreich ataxia (210).
Brain MRI demonstrates pure cerebellar atrophy, most prominently affecting the vermis, typically of mild-to-moderate severity. Longitudinal progression of cerebellar atrophy is typically mild, corresponding to the characteristically gradual clinical course of SCA27B compared with many other spinocerebellar ataxias (295).
Hyperintense signals along the superior cerebellar peduncle tract on T2-weighted images have been reported in 50% to 63% of patients (33). Quantitative imaging analyses also show involvement of lobule X of the cerebellum, which correlates with downbeat nystagmus, a frequently observed eye movement abnormality in SCA27B (32).
Neuropathological examination revealed cerebellar atrophy, which was more prominent in the cerebellar vermis than the cerebellar hemispheres (211). Microscopic examination showed widespread Purkinje cell loss, gliosis in the molecular layer, and mild cell loss in the granule-cell layer of the cerebellum. There were no intranuclear or intracytoplasmic inclusions observed on p62 staining, indicating an absence of protein aggregates. Postmortem cerebellum specimens and induced pluripotent stem cell (iPSC)-derived motor neuron cell lines from patients showed a reduction in both FGF14 RNA expression and FGF14 protein, suggesting FGF14 haploinsufficiency as a pathogenic mechanism.
Spinocerebellar ataxia type 28. The locus is 18p11.22-q11.2 (27). Di Bella and colleagues described the mutations in the AFG3L2 gene (63). This gene encodes for an ATP-dependent mitochondrial matrix-AAA (m-AAA) protease (ATPase associated with diverse cellular activities) (137). Di Bella hypothesized that either AFG3L2 or a substrate of AFG3L2 is involved in protecting the cerebellum from neurodegeneration. Loss-of-function mechanism was previously postulated; however, additional data supported gain-of-function or dominant negative mechanism (143).
Spinocerebellar ataxia type 29. Heterozygous mutations in the ITPR1 gene were identified by exome sequencing (111). This disorder may be an allelic variant of spinocerebellar ataxia type 15. The gene encodes inositol 1,4,5-trisphosphate receptor type 1, a ligand-gated calcium channel that regulates calcium release from endoplasmic reticulum and is highly expressed in Purkinje cells.
Brain MRI reveals cerebellar atrophy, especially of the superior cerebellar hemispheres and vermis (309).
Spinocerebellar ataxia type 30. Storey and colleagues published a case of a family in Australia (261). In the paper, his team identified a possible region at chromosome 4q34.3-q35.1 through linkage analysis based on this family.
Spinocerebellar ataxia type 31. Sato and colleagues identified large, 2.5-3.8-kb, insertions of a pentanucleotide repeat (TGGAA) in the noncoding intronic regions around the genes for BEAN and TK2 on opposite strands at chromosome 16q22.1 (227). Intergenerational instability is not prominent (307). The same chromosomal region is thought to be involved in spinocerebellar ataxia type 4. The affected families described with spinocerebellar ataxia type 31 did not have any sensory neuropathy symptoms.
Neuropathological examination reveals Purkinje cell loss, especially in the anterior lobe, and characteristic abnormal dendritic processes of surviving Purkinje cells with halo-like amorphous materials (01).
Spinocerebellar ataxia type 32. The disease locus was mapped by a genome-wide linkage scan to chromosome 7q32-q33 (127). Brain MRI reveals cerebellar atrophy.
Spinocerebellar ataxia type 34. This disorder is caused by mutations in the ELOVL4 gene, which encodes for very long-chain fatty acid elongase 4, which has a function in the elongation of very long-chain fatty acid (26). A different mutation was reported in two Japanese families (204).
Spinocerebellar ataxia type 35. This is caused by mutations in the TGM6 gene encoding for transglutaminase 6 (287). The pathogenesis of neurodegeneration in this disorder remains unknown, but it has been postulated that mutant transglutaminase 6 can lead to the activation of unfolded protein responses, loss of enzymatic function, change in subcellular distribution of the enzyme, and sensitization to apoptosis (96; 276). Interestingly, an antibody targeting transglutaminase 6 can also lead to an autoimmune disorder, celiac disease, or gluten ataxia. However, the association between these autoimmune and genetic disorders as well as the role of gluten-free diet as a treatment of the genetic form is to be further elucidated (148).
Spinocerebellar ataxia type 36. This is caused by a GGCCTG hexanucleotide repeat expansion in the first intron of the NOP56 gene (115). NOP56 encodes for Nop56p, a component of small nucleolar ribonucleoprotein that has a role in 60s ribosomal subunit assembly and pre-rRNA processing.
Brain MRI reveals cerebellar atrophy, which begins in the superior vermis and progresses to involve the cerebellar hemispheres, as well as pontine and cortical atrophy in later stages (115).
Spinocerebellar ataxia type 37. The disease locus was initially mapped to chromosome 1p32 (242). The gene was later found in 2017 (240). This disorder is caused by an ATTCT pentanucleotide repeat mutation in the non-coding region of the DAB1 gene. This gene encodes for DAB1, the reelin adaptor protein. Upregulation of reelin-DAB1 and PI3K/AKT signaling due to mutant DAB1 has been postulated in the pathophysiology of this disorder (45).
Brain MRI reveals cerebellar but no brainstem atrophy (242). Neuropathological examination demonstrates extensive Purkinje cell loss and empty basket cells in the Purkinje cell layer (45).
Spinocerebellar ataxia type 38. This is caused by mutations in the ELOVL5 gene encoding for very long-chain fatty acid elongase 5 (64).
Brain MRI reveals vermian atrophy with brainstem sparing.
Spinocerebellar ataxia type 40. A missense mutation, R464H, in the CCDC88C gene was found in one reported family from Hong Kong (277). This gene encodes for a coiled-coil domain containing 88C protein, which is involved in the c-Jun N-terminal kinase (JNK) pathway.
Brain MRI shows pontocerebellar atrophy.
Spinocerebellar ataxia type 41. A missense R762H mutation in the TRPC3 gene was found in the reported 40-year-old patient (80). This gene encodes for transient receptor potential channel 3, a nonselective cation channel linking to mGluR1 and other ataxia-associated pathways (11).
Brain MRI of the reported patient revealed very mild vermian atrophy (80).
Spinocerebellar ataxia type 42. This is caused by mutations in the CACNA1G gene encoding for voltage-gated calcium channel 1G (47).
Brain MRI demonstrates cerebellar atrophy, especially of the vermis (47; 133).
Spinocerebellar ataxia type 43. This is caused by a C143Y variant in the MME gene encoding for neprilysin, a zinc-dependent metalloprotease that has a role in peptide cleavage at the amino side of hydrophobic residues (58).
Brain MRI reveals vermian atrophy.
Spinocerebellar ataxia type 44. This is caused by mutations in the GRM1 gene encoding for metabotropic glutamate receptor 1 (291). There are genotype-phenotype correlations. Affected individuals in two families presenting with adult-onset cerebellar ataxia were found to have gain-of-function missense mutations, whereas the affected individual in the other family with juvenile-onset ataxia had a de novo truncation mutation.
Brain MRI reveals pontocerebellar atrophy.
Spinocerebellar ataxia type 45. This is caused by mutations in the FAT2 gene (194). This gene is a human homolog of the Drosophila fat gene encoding for a tumor suppressor important for cell proliferation control during development in Drosophila.
Brain MRI reveals vermian atrophy. Hemosiderin deposition in the mesencephalon was found in one patient.
Spinocerebellar ataxia type 46. This is caused by a missense mutation in the PLD3 gene encoding phospholipase D3 (194). Brain MRI reveals absent or mild cerebellar atrophy.
Spinocerebellar ataxia type 47. This is caused by mutations in the PUM1 gene encoding for Pumilio1, an RNA-binding protein that has a role in ataxin-1 regulation (87; 88). There are genotype-phenotype correlations. The mutations causing about 25% reduction of the Pumilio1 protein level were associated with the adult-onset form, whereas those causing 50% reduction of the protein level (or haploinsufficiency) were associated with the early-onset form with more severe phenotype.
Brain MRI in the adult-onset form reveals vermian atrophy. Shortening of the vermis and enlarged forth ventricle are seen in the early-onset form.
Spinocerebellar ataxia type 48. This is caused by a mutation in the STUB1 gene encoding for STIP1 homologous and U-box-containing protein 1, which is involved in protein quality control (86). This gene is also responsible for autosomal recessive spinocerebellar ataxia type 16 (SCAR16) (244). However, mutations in this gene are typically biallelic in SCAR16, and heterozygous (monoallelic) in SCA48. In SCAR16, patients present with early-onset spastic ataxia, cognitive impairment, hyperkinetic movement disorders, peripheral neuropathy, and hypogonadism. Overlapping clinical features between SCAR16 and SCA48 represent a phenotypic spectrum or continuum of autosomal recessive and autosomal dominant forms of the same gene (60; 219). Some studies have suggested an interaction between STUB1 mutations and TBP allele with intermediate or high-normal (40) CAG/CAA repeats, indicating possible digenic inheritance in SCA48 (156; 10). Of note, CAG/CAA repeat expansions in the TBP gene are responsible for spinocerebellar ataxia type 17, which has overlapping phenotypes with SCA48. However, one case series of 21 patients with ataxia carrying heterozygous STUB1 mutations showed that 71% of these patients had normal TBP allele (< 40 repeats) (283). Intermediate (41-42 repeats) and high-normal (40 repeats) TBP expansions were found in 29% of patients, who also exhibited more pronounced cognitive impairments. This study suggests that SCA48 is a monogenic disorder, and the expansion in the TBP gene likely has a modifying effect on phenotypic expression in some cases. This is also supported by two additional reported families with SCA48 with STUB1 mutations but normal CAG/CAA repeat ranges in the TBP gene (205).
Spinocerebellar ataxia type 49. This is caused by a mutation in the SAMD9L gene encoding for SAMD9L protein. All nine affected individuals in the same family in the original report had the same mutation, c.1877C>T (p.Ser626Leu) (44). Mutations in this gene are also associated with ataxia-pancytopenia syndrome (30). In addition, mutations in the paralogue SAMD9 gene are associated with MIRAGE syndrome (Myelodysplasia, Infection, Restriction of growth, Adrenal hypoplasia, Genital phenotypes, and Enteropathy) (192). Interestingly, patients with spinocerebellar ataxia type 49 have not been found to have hematological abnormalities. It has been hypothesized that early-onset myelodysplasia requires secondary genetic hits, such as monosomy of chromosome 7 or somatic mutations, in addition to primary mutations in the SAMD9L or SAMD9 gene (187). The SAMD9L protein localizes in the mitochondria, and its roles in mobility, as well as vestibular and sensory functions, have been implicated from a study in zebrafish (44).
Electrophysiologic studies show reduced sensory nerve action potential amplitudes predominantly involving the lower extremities, indicating sensory axonal polyneuropathy. There can also be abnormal sympathetic skin responses in the lower extremities, indicating autonomic dysfunction. Brain MRI shows cerebellar atrophy and evidence of demyelination at subcortical cerebral white matter.
Spinocerebellar ataxia type 50. This is caused by missense mutations in the NPTX1 gene encoding for the neuronal pentraxin 1 protein. Overexpression of the NPTX1 variants results in a change in endoplasmic reticulum morphology and activating transcription factor 6 (ATF6)-mediated endoplasmic reticulum stress (48). This mechanism has been proposed to cause cerebellar ataxia.
Brain MRI demonstrates cerebellar atrophy, predominantly in the vermian region (48; 59).
Spinocerebellar ataxia type 51. This is caused by CAG repeat expansions in exon 1 of the THAP11 gene, which encodes the THAP domain-containing protein 11. A repeat length of 45 or greater is pathogenic, whereas the significance of 40 to 44 repeats remains unclear. Healthy controls have 19 to 39 CAG repeats in the THAP11 gene (74; 269). Both affected and healthy individuals may carry CAA repeat interruptions, but their role and potential disease-modifying effects remain unclear.
The THAP11 protein is a transcription factor that regulates ATXN1, the gene responsible for spinocerebellar ataxia type 1. Overexpression of THAP11 (Ronin) in mouse cerebellar Purkinje cells leads to neurodegeneration and severe ataxia, with increased ataxin-1 expression in the cerebellum (314). Ataxin-1 expression was decreased in patient skin fibroblasts and transfected neuro-2a cells expressing mutant THAP11-(CAG)100 (269). THAP11, in complex with host cell factor 1 (HCF-1), plays a role in cell cycle, proliferation, and transcription (208), and it regulates cobalamin metabolism via methylmalonic aciduria and homocystinuria cblC type (MMACHC) expression (35).
Brain MRI typically reveals cerebellar atrophy. Nerve conduction studies and electromyography are usually unremarkable (269).
Pathology. Data on neuropathology in patients with THAP11-related spinocerebellar ataxia are not available. However, increased intracellular aggregations of p62 and autophagic vacuoles have been observed in the cytoplasm of patients’ skin fibroblasts (269). This suggests that polyglutamine expansions within the THAP11 protein may lead to abnormal autophagy and protein quality-control pathway dysfunction.
Dentatorubral-pallidoluysian atrophy. Dentatorubral-pallidoluysian atrophy is caused by an expanded CAG repeat in the ATN1 gene on chromosome 12. The normal allele has 7 to 23 repeats, but individuals affected with dentatorubral-pallidoluysian atrophy have 49 to 79 repeats. The allele is more likely to undergo expansion if inherited from the paternal lineage. The gene product, atrophin-1, has an unknown function. Truncated fragments of atrophin-1 accumulate in the neuronal nuclei of a transgenic mouse model of dentatorubral-pallidoluysian atrophy (232). Evidence suggests that atrophin-1 is a component of a transcription corepressor complex (298). At this time, it is unclear how repression of transcription contributes to the pathogenesis of dentatorubral-pallidoluysian atrophy.
Pathology. There is variable atrophy of the dentate nucleus, lateral corticospinal tract, red nucleus, putamen, caudate, globus pallidus, thalamus, inferior olives, pons, posterior columns, spinocerebellar tracts, and cerebral cortex (267). There are neuronal intranuclear inclusions (179). Corneal endothelial degeneration has been reported in patients with dentatorubral-pallidoluysian atrophy, but it is not always associated with impairment in visual acuity (119).
Episodic ataxia type 1. Episodic ataxia type 1 is due to missense mutations in the KCNA1 gene on chromosome 12p13, encoding for the voltage-gated potassium channel, Kv1.1 (18).
Episodic ataxia type 2. Episodic ataxia type 2 is due to a mutation in the CACNA1A gene on chromosome 19, encoding for the voltage-dependent P/Q type calcium channel. The mutation can be a CAG trinucleotide repeat expansion or missense mutation causing truncation of the gene product (98). These mutations are likely to prevent the assembly of functional calcium channels. Dysfunction of these channels may affect the control of neurotransmitter release and aberrant activity of Purkinje cells (98; 270). Episodic ataxia type 2 may be associated with cerebellar atrophy, especially of the anterior vermis (263). This calcium channel is linked to other neurologic disorders such as spinocerebellar ataxia type 6, familial hemiplegic migraine, and epilepsy.
Episodic ataxia type 3. The disease locus was mapped to a 4-cM region on chromosome 1q42 (25).
Episodic ataxia type 5. This is caused by missense mutations in the gene encoding calcium channel beta 4 subunit, CACNB4 on chromosome 2q, and the clinical features are similar to those of episodic ataxia type 2 (72).
Episodic ataxia type 6. This is caused by a heterozygous mutation in a glutamate transporter gene, SLC1A3. The functional studies from in vitro models show the mutations caused decreased glutamate uptake (125; 62).
Episodic ataxia type 7. This has been linked to chromosome 19q13. There are no mutations identified in the KCNC3 or SLC17A7 genes (131).
Episodic ataxia type 8. The disease locus was mapped to chromosome 1p36.13-p34.3 (42). Exome sequencing identified two candidate variants in the HSPG2 and UBR4 genes. The authors postulated that the latter gene was more likely, due to its role in calcium control within neurons. Mutations in the UBR4 gene were then also identified in a series of Korean patients with episodic ataxia (37).
Episodic ataxia type 9. This is due to mutations in the SCN2A gene encoding the alpha subunit of voltage-gated sodium channel, Nav1.2. Mutations in this gene are associated with marked phenotypic variability, such as benign familial neonatal seizures, Ohtahara syndrome, epilepsy of infancy with migrating focal seizures, infantile spasms or West syndrome, Lennox-Gastaut syndrome, and myoclonic-astatic epilepsy, among others (296). Mutations associated with the episodic ataxia phenotype include p.A263V resulting in toxic gain-of-function, as well as mutations affecting the S4 segment or the cytoplasmic loop linking the S4 and S5 segments of the sodium channel (238). Some of these mutations have also been found in patients with epileptic encephalopathies.
|
• SCA3 is the most common spinocerebellar ataxia worldwide, except in some specific populations. For example, SCA2 is the most common subtype of spinocerebellar ataxias in Cuba and India. | |
|
• SCA27B is one of the most common causes of adult-onset or sporadic late-onset cerebellar ataxia. |
Most of the spinocerebellar degenerations have been documented only as case reports or as family studies. However, some epidemiology data have been published. European studies report between one and three per 100,000 Europeans have autosomal dominant cerebellar ataxia. The polyglutamine expansion spinocerebellar ataxias are currently the most prevalent. SCA3 is the most prevalent, ranging from 50% of cases in Norway (235) to 20% of cases in a predominantly French population (68). Durr provides a pie chart in her paper based on 826 patients with spinocerebellar ataxias. She reported that 48% of cases were unknown and did not correlate with any of the known causative genes. SCA2 comes in second with 10% of cases, SCA1 with 8%, SCA7 with 6%, SCA15/16 with 3%, and SCA11, SCA13, SCA17, and SCA28 with 1% each. Durr also reports founder effects with a higher number of cases of SCA2 in Cuba and SCA3 in Brazil (69%), Portugal (58%), China (49%), and Japan (26% to 63%); a lower number of cases in Canada (24%) and the United States (21%); and a rare number of cases in India (3%). The prevalence of type 1 in a Spanish population has been reported to be 1.2 per 100,000, but there are regional differences due to the founder effect (213).
SCA2 and SCA10 are common in Mexicans (218), and type 10 is rare in other populations (165). SCA10 is also the most common spinocerebellar ataxia in Peru (43). In India, SCA12 is prevalent, but SCA2 remains the most common subtype (185). Spinocerebellar ataxia type 31 is one of the common autosomal dominant ataxias in Japan. Dentatorubral-pallidoluysian atrophy also has high prevalence in Japan but has also been reported much less commonly in non-Asian populations (28).
SCA27B has been identified as one of the most common causes of adult or sporadic late-onset cerebellar ataxias in various populations, including Australian, German, Italian, Indian, Spanish, and Greek, with a prevalence ranging from approximately 10% to 30% (117; 211; 214; 215; 130; 228). Notably, there is a particularly high prevalence of 61% of SCA27B in the French-Canadian population, suggesting a founder effect (211). However, the prevalence is relatively low (2.2%) in Jewish populations (100).
The prevalence of SCA48 may be higher than previously thought. In a study cohort of 235 unrelated patients with adult-onset cerebellar ataxia who had negative testing for common spinocerebellar ataxias, an estimated frequency of SCA48 (STUB1) mutations was 3.4% (147).
There is no method of preventing autosomal dominant inherited ataxias.
This section will discuss the differential diagnosis of all forms of inherited ataxia, including those that are inherited in an autosomal dominant, autosomal recessive, X-linked, or mitochondrial pattern. Because this article is categorized in the pediatric neurology section, the focus will be on pediatric ataxic disorders, but adult-onset disorders will also be mentioned.
The unifying factor among these various diseases is ataxia, most often progressively worsening in nature. The differential diagnosis for ataxia is extensive. Grouping the various ataxic disorders by their associated features can help differentiate them. The associated features include the inheritance pattern, temporal course, signs of pyramidal tract involvement, ophthalmologic abnormalities, eye movement abnormalities, peripheral nervous system involvement, the occurrence of seizures, and other features (eg, acanthocytes).
Inheritance pattern. If the patient with ataxia does not have a family history of ataxia, genetic, nongenetic, and sporadic causes should be considered. The sporadic causes of spinocerebellar syndromes include multiple system atrophy, autoimmune and paraneoplastic cerebellar ataxia, infectious or postinfectious etiologies, nutritional deficiency (eg, vitamin E deficiency), structural lesions (eg, stroke, posterior fossa tumors), and superficial siderosis, among others (82).
If a child with progressive ataxia, pes cavus, kyphoscoliosis, and signs of spinocerebellar dysfunction has a sibling with ataxia, Friedreich ataxia is a likely diagnosis. Because Friedreich ataxia is autosomal recessive, the parents usually are asymptomatic.
If either parent is affected with ataxia, then the disorder is more likely to be one of the autosomal dominant spinocerebellar ataxias. However, ataxia in a parent does not necessarily exclude Friedreich ataxia (140). In Friedreich ataxia, the patient is usually areflexic. The spinocerebellar ataxias usually cause hyperreflexia.
Features that cast doubt on the diagnosis of Friedreich ataxia include the following (177):
|
• intellectual disability |
|
Autosomal dominant | |
|
• Spinocerebellar ataxias (SCA1-48) | |
|
Autosomal recessive | |
|
• Friedreich ataxia | |
|
Metabolic (inborn errors of metabolism) | |
|
X-linked | |
|
• Adrenoleukodystrophy: Spinocerebellar ataxia variant | |
|
Mitochondrial | |
|
• Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes | |
Temporal course. Periodic episodes of ataxia associated with vomiting are observed in urea cycle defects. Other causes of episodic ataxia include pyruvate dehydrogenase deficiency; propionic acidemia; methylmalonic acidemia; isovaleric acidemia; maple syrup urine disease; myoclonic epilepsy with ragged-red fibers; mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes syndrome; and Hartnup disease. The remainder of the inherited ataxias usually causes progressive ataxia. When patients present with an acute or subacute time course of ataxia, acquired etiologies need to be sought and excluded.
Spasticity. Hereditary spastic paraplegia causes slowly progressive spastic weakness of both lower extremities. If it occurs in the “pure” or “uncomplicated” form, the patient has lower extremity stiffness and weakness, urinary dysfunction, paresthesias, diminished vibratory sensation, and hyperreflexia of the lower extremities. This symptom complex is rarely confused with spinocerebellar degeneration. However, there are “complicated” forms that cause other neurologic problems such as ataxia, pes cavus, optic neuropathy, retinopathy, extrapyramidal disturbance, dementia, ichthyosis, intellectual disability, and deafness (78). The complicated forms may cause confusion with Friedreich ataxia.
The ataxia of juvenile GM2 gangliosidosis, unlike Friedreich ataxia, is associated with spasticity and progressive mental deterioration in the absence of neuropathy. A deficiency of hexosaminidase A confirms the diagnosis. Areflexia with amyotrophy and spasticity of the lower limbs also occurs in xeroderma pigmentosum and Chediak-Higashi syndrome.
Ophthalmologic abnormalities. Ophthalmologic findings have been associated with several ataxic disorders. Corneal clouding is characteristic of adult beta-galactosidase deficiency and multiple sulfatase deficiency. Cataracts have been seen in Marinesco-Sjogren disease, Usher syndrome, and cerebrotendinous xanthomatosis (cholestanolosis) as well as in some patients with Friedreich ataxia and spinocerebellar ataxia. Retinal disease, in particular retinitis pigmentosa, has been recognized in abetalipoproteinemia, hypobetalipoproteinemia, Refsum disease, the spinocerebellar ataxias, and Kearns-Sayre syndrome. Spinocerebellar atrophy type 7 is particularly likely to cause impaired vision and retinal disease (128). Disordered macular pigmentation or cherry red spots of the macula occur in neuronal ceroid lipofuscinosis, Niemann-Pick disease, multiple sulfatase deficiency, adult-onset beta-galactosidase deficiency, and hexosaminidase B deficiency. The myoclonus of the cherry red spot-myoclonus syndrome or sialidosis is occasionally confused with ataxia. Optic atrophy occurs in Marinesco-Sjogren syndrome, Friedreich ataxia, ataxia-telangiectasia, Leigh disease, and some forms of spinocerebellar ataxia.
Eye movement abnormalities. Eye movement disorders may occur in syndromes that cause ataxia. Vertical nystagmus has been observed in multiple sulfatase deficiency. Primary-position upbeat nystagmus has been seen in episodic ataxia or in syndromes that cause cerebellar vermian atrophy. Spontaneous downbeat nystagmus has been noted in late-onset cerebellar ataxia and in other lesions of the craniocervical junction, including Chiari malformations, multiple sclerosis, and neoplasms. Anticonvulsant toxicity may also present with downbeat nystagmus.
When saccades are slow or when there is a combined loss of pursuit and vestibular function, one should consider an autosomal dominant spinocerebellar ataxia (182). Slow saccade, especially in the horizontal direction, is a characteristic eye movement abnormality in spinocerebellar ataxia type 2 but can also be seen in other types of spinocerebellar ataxia. Oculomotor apraxia can be seen ataxia-telangiectasia and ataxia with oculomotor apraxia types 1 and 2 (271).
Peripheral nervous system abnormalities. Spinocerebellar ataxia type 1 and spinocerebellar ataxia type 2 can cause a neuronopathy, whereas spinocerebellar ataxia type 3 and spinocerebellar ataxia type 7 can cause both a neuronopathy and axonopathy (279). Yet a neuropathy has been associated with several disorders that cause ataxia, including Friedreich ataxia, xeroderma pigmentosum, Behr syndrome, Refsum disease, adrenomyeloneuropathy, cerebrotendinous xanthomatosis (cholestanolosis), and Dejerine-Thomas ataxia. Diagnosis of a spinocerebellar ataxia may be particularly difficult early in the course of the disease when only impaired vibratory sensation, ataxia, and areflexia in the legs are present. In these cases, hereditary motor and sensory neuropathy should be considered. Motor nerve conduction velocity is decreased in hereditary motor and sensory neuropathy type I but is usually normal in Friedreich ataxia and in hereditary motor and sensory neuropathy type II. DNA testing may be necessary to differentiate Friedreich ataxia from hereditary motor and sensory neuropathy type II.
Weakness and distal amyotrophy are seen in Friedreich ataxia, ataxia-telangiectasia, the spinocerebellar ataxias, and Joseph syndrome.
The differential diagnosis of childhood-onset areflexia combined with extensor plantar responses includes Friedreich ataxia and neurodegenerative disorders such as metachromatic leukodystrophy, globoid cell leukodystrophy, neuroaxonal dystrophy, and the rare syndrome of congenital absence of intrinsic factor with B12 deficiency. Areflexia may be seen late in the course of ataxia-telangiectasia, cerebellar ataxia with hypogonadism, some forms of spinocerebellar ataxia, and juvenile metachromatic leukodystrophy. Cockayne syndrome and mitochondrial disorders, which may cause areflexia and ataxia, is typically associated with short stature and cerebral leukodystrophy.
Seizures. Seizures can be a prominent feature in xeroderma pigmentosum, some congenital ataxias, the progressive myoclonic ataxias, and the progressive myoclonic epilepsies. For example, Baltic myoclonus (Unverricht-Lundborg disease), reported predominantly from Scandinavia, presents with myoclonus or generalized seizures followed by ataxia and dysarthria (49). Many inborn errors of metabolism can cause ataxia and seizure, including pyruvate dehydrogenase deficiency, L-2-hydroxyglutaric aciduria, biotinidase deficiency, urea cycle defects, Gaucher disease type 2, galactosialidosis, and neuronal ceroid lipofuscinosis. Dentatorubral-pallidoluysian atrophy can cause epilepsy, but epilepsy rarely occurs in the autosomal dominant spinocerebellar ataxias (except spinocerebellar ataxia type 10).
Other abnormalities. Neuronal intranuclear hyaline inclusion disease is similar to Friedreich ataxia with seizures but without cardiac abnormalities (252).
Acquired vitamin E deficiency can cause symptoms that mimic Friedreich ataxia. There are multiple etiologies for acquired vitamin E deficiency, including inflammatory diseases (celiac disease, inflammatory bowel disease, chronic pancreatitis, chronic cholestatic liver disease), infections (Whipple disease, tropical sprue), malnutrition, postgastrectomy, short bowel syndrome, and cystic fibrosis (121). It is an important deficiency to screen for because it can be treated.
The association of tendon xanthomas with mental deterioration differentiates cerebrotendinous xanthomatosis from the spinocerebellar ataxias.
In sporadic cases where no skeletal deformities are found, the neurologic signs of Friedreich ataxia may be similar to those of multiple sclerosis. The younger age distribution and absence of deep tendon reflexes in Friedreich ataxia differentiate it from multiple sclerosis. In addition, the history of optic neuritis helps to establish the diagnosis of multiple sclerosis.
Adrenomyeloneuropathy, a phenotypic variant of X-linked adrenoleukodystrophy, may present with a spinocerebellar syndrome in males typically between the ages of 10 and 35 years (220).
|
• Whole exome sequencing has limitations in detecting repeat expansions, which are the underlying genetic bases in the most common spinocerebellar ataxias. | |
|
• Long-read sequencing, a relatively new next-generation sequencing technology, has emerged as a crucial tool for diagnosing spinocerebellar ataxias due to repeat expansion. |
Identifying the repeat expansion with molecular testing is diagnostic. Most of the spinocerebellar ataxias have commercially available DNA tests. The GeneTests website provides an up-to-date list of available DNA tests. Genetic counseling should be involved in the work-up. Even though there is clinical heterogeneity between the syndromes, one needs to be thoughtful in their approach to using the clinical information about the patient and other affected individuals in the family, if possible. Neuroradiologic evaluation may demonstrate abnormalities. Although these abnormalities are not diagnostic for a specific spinocerebellar atrophy syndrome, they may be suggestive.
With advances in genetics, especially the development of next-generation sequencing, the list of spinocerebellar ataxias has been rapidly growing after the identification of new genes. The questions on how to appropriately apply these new technologies to clinical practice and how to select between single gene testing, gene panel, and whole exome sequencing remain a moving target. Potential results of having variants of unknown significance and cost need to be taken into consideration. It is important to note that the whole exome sequencing usually does not detect repeat expansions, which are underlying causes of the most common types of spinocerebellar ataxia (types 1, 2, 3 and 6). Thus, screening for these common repeat expansion disorders before pursuing larger gene panels or whole exome sequencing may be one reasonable approach (81; 280). Long-read sequencing, a relatively new next-generation sequencing technology, has emerged as a crucial tool for the diagnosis of repeat expansion disorders, which are frequent causes of spinocerebellar ataxias (71). This approach can detect unusually long repeats that are difficult to size using polymerase chain reaction or short-read sequencing, which is particularly important in disorders such as spinocerebellar ataxia type 27B. Additionally, long-read sequencing provides accurate sizing and detection of repeat interruptions. Ultimately, phenotypic characterization remains crucial, and genetic results should not be separately interpreted without clinical correlation.
Schols and colleagues reported the electrophysiology characteristics of the most prevalent spinocerebellar ataxias (236). The goals of the electrophysiology testing are to diagnose associated neuropathies, even when clinical symptoms may not be present. These studies may help distinguish between an Autosomal dominant cerebellar ataxia types I/II and type III, the purely cerebellar ataxia type. The clinical presentation of the various spinocerebellar ataxias is heterogeneous.
Spinocerebellar ataxia type 2. One should consider performing trinucleotide repeat testing in individuals with levodopa-responsive familial parkinsonism, especially if the family history shows anticipation of parkinsonism symptomatology (209).
Spinocerebellar ataxia type 10. Polymerase chain reaction usually cannot amplify large expansions, such as those seen in some affected individuals with spinocerebellar ataxia type 10. However, Matsuura and Ashizawa report a polymerase chain reaction technique that can distinguish those who are homozygous for normal allele length from those who are heterozygous carriers or homozygous affected individuals (163). Nevertheless, positive results should be confirmed with Southern blot analysis because the technique is newer and cannot be used to determine the expansion size.
|
• There are currently no disease-modifying therapies for autosomal dominant hereditary ataxias. | |
|
• Management includes supportive and symptomatic therapies, as well as genetic counseling. |
There are no approved disease-modifying therapies for any of the autosomal dominant ataxias at this time. The management focuses on symptomatic relief of comorbid conditions, such as associated movement disorders, sleep disturbances, and rehabilitative measures. The past two decades have focused on the genetic characterization of the hereditary ataxia syndromes. Researchers are now looking to better characterize the disorders from a molecular perspective to try to identify biomarkers for each disorder. Serum neurofilament light chain has been found to be elevated in patients with autosomal dominant spinocerebellar ataxias (245) as well as the pre-ataxic stage of spinocerebellar ataxia type 1 (294). This could potentially serve as a biomarker. Biomarkers may be useful in assessing disease burden as well as possible targets for therapy. Rehabilitative measures, which include speech, physical, and occupational therapy, play a major role in the management of all patients with ataxia. Early rehabilitation goals are safe ambulation and independence in daily living. As the disorders progress, patients require training with assistive devices such as walkers, lower-extremity orthotic devices, and wheelchairs. It is also necessary to maintain an adequate range of motion of all joints. Orthopedic procedures for pes cavus or scoliosis may improve function. General supportive and rehabilitative measures are of paramount importance. Genetic counseling needs to be a part of all evaluations of possible hereditary disorders.
Gene therapies, such as antisense oligonucleotides, have been under active investigation (170; 169). Some of these may target either toxic downstream effects or polyglutamine expansions within the spinocerebellar ataxia genes (21). Stem cell therapies have also been studied. These experimental therapeutic modalities are still in early phases, and clinical trials in humans are required before clinical application. Challenges in the development of disease-modifying therapies in spinocerebellar ataxia include heterogeneity of disorders in this group and identification of biomarkers to monitor disease progression in clinical trials.
Spinocerebellar ataxia type 2. Type 2 can cause familial parkinsonism. In those cases, levodopa may be indicated (243).
Spinocerebellar ataxia type 3 (Machado-Joseph). A double-blind, crossover trial of trimethoprim-sulfamethoxazole in patients with spinocerebellar ataxia type 3 showed that this medication is not an effective treatment for the movement disorders and visual disturbances that can occur (237). Fluoxetine, a serotonin-selective reuptake inhibitor, has also been shown to be ineffective (176). Dopaminergic agents may be helpful for the parkinsonian features. In a patient with dystonia as the sole presentation of spinocerebellar ataxia type 3, significant clinical improvement occurred with levodopa therapy (190). Dopaminergic agents may decrease the severity of restless legs syndrome if it is present with any of the ataxia syndromes. Transcranial direct current stimulation (tDCS) had no benefits in improvement of ataxia severity and restoration of cerebellar-motor cortex connectivity in a study of 2-week treatment in mildly to moderately affected patients with spinocerebellar ataxia type 3 (155). Troriluzole, an oral prodrug of riluzole, is a glutamate modulator under investigation as a potential disease-modifying therapy for several spinocerebellar ataxias, including types 1, 2, 3, 6, 7, 8, and 10. In a matched-cohort analysis comparing treated patients with untreated natural history cohorts, the troriluzole group was associated with significantly slower disease progression, by 50% to 70% over 3 years, corresponding to a 1.5- to 2.2-year delay in disability (151). The U.S. Food and Drug Administration has not yet approved troriluzole, but its new drug application has been accepted and granted priority review, with a Prescription Drug User Fee Act decision expected in the fourth quarter of 2025.
Spinocerebellar ataxia type 6. Acetazolamide has been shown to help diminish the ataxia in individuals affected with type 6 (126; 304). In a double-blind, crossover study, branched-chain amino acid therapy significantly decreased ataxia severity in individuals with spinocerebellar degeneration, including spinocerebellar ataxia 6, spinocerebellar ataxia 7, and olivopontocerebellar atrophy (178). The improvement was most notable in subjects with spinocerebellar ataxia type 6. D-cycloserine, a partial NMDA receptor agonist, has been shown to significantly improve posture and gait in patients with spinocerebellar degeneration, including spinocerebellar ataxia type 6 (196). Two phase 3 randomized double-blind placebo-controlled studies of patients with SCA6, SCA31, or cortical cerebellar atrophy showed no significant difference in Scale for the Assessment and Rating of Ataxia total scores between the treatment and the placebo group (195). Most recently, a multicenter, randomized, double-blind, placebo-controlled phase 2 clinical trial of L-arginine in genetically confirmed spinocerebellar ataxia type 6 demonstrated a trend toward benefit on the Scale for the Assessment and Rating of Ataxia total score over 48 weeks, but statistical significance was not reached (118). Larger clinical trials with adequate statistical power are warranted to confirm the therapeutic potential of L-arginine in this population.
Spinocerebellar ataxia type 10. Those individuals with seizures should be treated with appropriate anticonvulsant therapy.
Spinocerebellar ataxia type 27B. In a cohort study, 86% of patients treated with 4-aminopyridine reported a good response that positively impacted everyday living (295). It was shown to significantly reduce daily symptomatic time and symptom severity in a series of three prospective n-of-1 treatment experiences. A study of 170 patients with idiopathic downbeat nystagmus found that 48% had 250 or more GAA repeats in the FGF14 gene, and 12% had 200 to 249 repeats (212). Patients with either 250 or more repeats or 200 to 249 repeats showed significant clinician-reported and self-reported improvements with 4-aminopyridine, compared to those with fewer than 200 GAA repeats.
Spinocerebellar ataxia type 38. Given mutations in the ELOVL5 gene encoding for a very long-chain fatty acid elongase 5 important for polyunsaturated fatty acid synthesis, docosahexaenoic acid was studied and found to improve both clinical symptoms and cerebellar hypometabolism (157).
Episodic ataxias. The attacks of ataxia in the episodic ataxias may be responsive to acetazolamide. Phenytoin may be helpful in controlling the myokymia associated with episodic ataxia type 1 (19; 154).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Pichet Termsarasab MD
Dr. Termsarasab of Ramathibodi Hospital, Mahidol University has no relevant financial relationships to disclose.
See Profile
Alcy R Torres MD FAAP
Dr. Torres of Boston Medical Center and Boston University Chobanian and Avedisian School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Neurology
May. 13, 2026