


Peripheral Neuropathies
Clinical evaluation of peripheral neuropathies
Jul. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Hereditary peripheral neuropathies were first described independently by Charcot and Marie in France (38; 142) and by Tooth in England (214) and have become known as Charcot-Marie-Tooth diseases. Several earlier descriptions had been published, including a 6-generation pedigree (62) and a clinicopathological study (72).
The heterogeneous nature and different forms of inheritance of the condition were soon appreciated. In 1889, Herringham recognized a family with X-linked Charcot-Marie-Tooth disease (100). Dejerine and Sottas described a more severe infancy-onset disease, which now bears their name (53), and Roussy and Levy described cases associated with tremor (184) that were defined genetically (09; 174). Different forms of inheritance were later categorized (03).
With the advent of modern neurophysiologic testing in the late 1960s, Charcot-Marie-Tooth disease was divided into two groups, one with slow nerve conduction velocities and histologic features of a hypertrophic demyelinating neuropathy (HMSN1 or CMT1) and another with relatively normal velocities and axonal and neuronal degeneration (HMSN2 or CMT2) (61; 211; 34). The features of CMT1 and CMT2 patients were outlined in two landmark publications detailing the genetic and clinical characteristics of over 200 patients (95; 96). CMT1 patients had median motor nerve conduction velocities below 38 m/sec, and CMT2 patients had velocities above 38 m/sec. As a dividing value between both forms, nerve conduction velocities of 38 m/sec are used by some and nerve conduction velocities of 42 m/sec by others (96; 115). Nerve conduction velocities in CMT1 patients typically are uniformly slowed along individual nerves and between different nerves of an individual patient, distinguishing CMT1 patients from those with acquired demyelinating neuropathies such as Guillain Barré syndrome or chronic inflammatory demyelinating polyneuropathy (133; 115).
Although the separation of neuronal and non-neuronal forms is an important etiologic and pathogenic distinction, even in CMT1, the clinical deficits correlate better with progressive axonal degeneration than with slowed nerve conduction. This is not surprising considering that demyelination secondarily disturbs axonal structure and transport. In some CMT families, patients have median motor nerve conduction velocities of 25 to 45 m/sec, and thus, are difficult to classify by electrophysiological criteria. This type of Charcot-Marie-Tooth disease was designated “intermediate CMT” (46). The distinction between demyelinating and nondemyelinating Charcot-Marie-Tooth disease was further challenged by reports of relatively normal conduction velocities suggestive of CMT2 in younger members of a family with a myelin protein zero mutation, whereas older relatives had severely slowed conduction consistent with CMT1 (49).
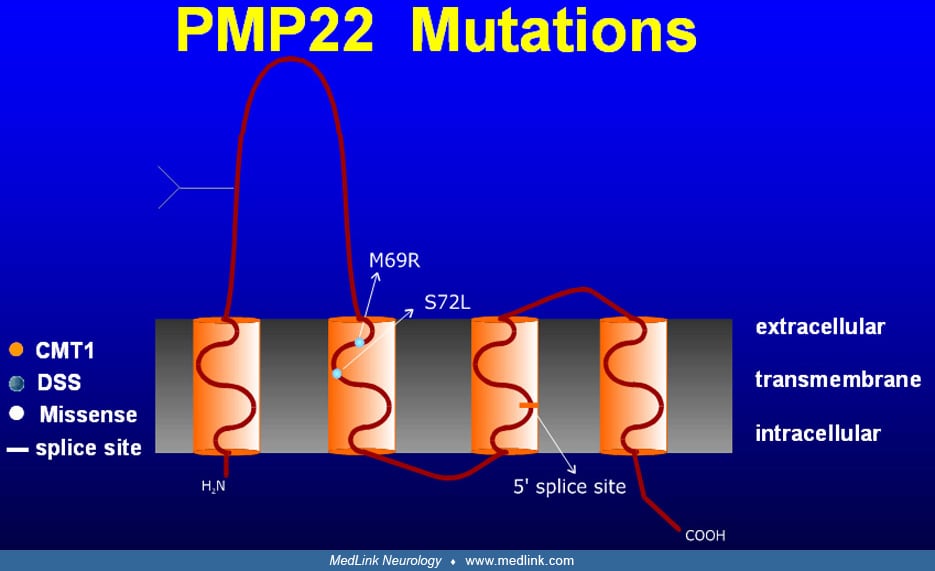
Despite clinical similarities among CMT1 patients, it was soon discovered that the group was genetically heterogeneous, as linkage studies demonstrated CMT1 loci on both chromosome 1 (27) and chromosome 17 (177; 217; 151). In 1991, two groups showed that CMT1A, the most common form of CMT1, was associated with a 1.5 mB duplication within chromosome 17p11.2 (134; 178). Some 70% of CMT1 and 90% of CMT1A cases result from this duplication (33; 93; 231; 167). Mutations in the PMP22 gene, contained within the 1.5 kB duplication on chromosome 17, have been demonstrated to cause demyelinating neuropathies in Trembler and Trembler-J mice (201; 202) as well as in some families with a CMT1 phenotype (216; 180; 159). Moreover, transgenic mice and rats that over-express PMP22 develop neuropathies resembling CMT1 (104; 139; 192); therefore, it is now believed that the extra PMP22 gene copy within the 1.5 mB duplication on chromosome 17 causes the majority of cases of CMT1A. CMT1A also occurs with partial or complete trisomy for the short arm of chromosome 17, as part of a multiorgan phenotype with developmental and growth delay, craniofacial and skeletal anomalies, and heart defects (70; 198).
The second most common subtype, X-linked recessive CMTX1, was found to result from mutations in the gap junction protein beta 1/connexin-32 on chromosome Xq13.1 (23). At least five other loci or genes for X-linked recessive Charcot-Marie-Tooth disease have been identified.
The 1990s also saw the identification of other Charcot-Marie-Tooth genes. CMT1B and some cases of Déjerine Sottas syndrome, known to be linked to chromosome 1q22-q23 (131), were found to be associated with mutations in the myelin protein zero gene (98; 123; 200). Mutations in the zinc-finger domain containing transcription factor early growth response 2 gene (EGR2 or Krox20) on chromosome 10q21.1-q22.1 were linked to CMT1D, Déjerine Sottas syndrome, and congenital hypomyelinating neuropathy (227). Homozygous EGR2 knockout mice show peripheral hypomyelination and block of Schwann cells (227). Deletion of the PMP22 gene locus was associated with hereditary neuropathy with liability to pressure palsies and several other phenotypes (36). A similar condition, hereditary brachial plexus neuropathy (or hereditary neuralgic amyotrophy with predilection for the brachial plexus) is not linked to the PMP22 locus but was mapped to chromosome 17q25 (36; 169). Mutations of all of these genes have been associated with several overlapping clinical phenotypes. For instance, Déjerine Sottas syndrome is associated with PMP22, Cx32, or myelin protein zero mutations or deletions (160; 226; 49; 179; 227).
After the initial description of the most common autosomal dominant CMT1 and CMT2 phenotypes, the quest for the most severe and rare phenotypes led to the diagnosis of autosomal recessive variants. Demyelinating autosomal recessive phenotypes are called CMT4 (or alternatively ARCMT1), whereas autosomal recessive axonal variants are called ARCMT2. In regions where consanguinity is important, up to 30% of the CMT cases are autosomal recessive (205). These forms overlap with DSS (CMT3) and CHN phenotypes. Several new disease linkages and genes have been identified.
Several loci have been identified in families with dominant intermediate Charcot-Marie-Tooth disease, ie, autosomal dominant Charcot-Marie-Tooth disease with conduction velocities between 24 and 45 m/sec. These include DI-CMTB on chromosome 19p12-p13.2 (118) and DI-CMTA, which is associated with both large fiber loss and regeneration clusters as well as onion bulbs and uncompacted enlarged myelin lamellae on chromosome 10q24.1-q25.1 (140; 220). A recessively inherited severe form of Charcot-Marie-Tooth disease with intermediate conduction velocities has been linked to chromosome 10q23 (181). Intermediate conduction velocities also occur with myelin protein zero and neurofilament light subtype gene mutations (48; 47). We reported that DI-CMTC is caused by disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase, in three unrelated families in the United States and Bulgaria (113; 112).
Since the early 1990s, over 130 genes have been found to be defective in Charcot-Marie-Tooth disease patients. Discovery has been facilitated by diagnostic tools such as target-enrichment next-generation sequencing with copy number assessment (225) and genome and RNA sequencing (141). The boundaries between CMT1 and CMT2 are less clear than originally believed and the spectrum of pathological mechanisms involved in these conditions is growing. Even when mutations reside in the nuclear genome, some CMT subtypes represent mitochondrial disorders, ie, the encoded proteins function in the mitochondria (239). Bienfait and colleagues illustrated the major challenges of diagnosing CMT2 in a large series of CMT2 patients (N=61, 18 families), where they found mutations in only three families (25). They stressed the difficulties in clinically distinguishing CMT1 from CMT2. CMT2 had later disease onset, less complete areflexia, foot deformities, and weakness of knee extensors and foot dorsal flexors (26). Rapid discovery of new genes has dramatically changed the field for several reasons. First, different mutations in a single gene can lead to CMT1, CMT2, DI-CMT, or CMT4 phenotypes, as well as milder or more severe phenotypes. Second, there is accumulating evidence for genes that can modify and complicate phenotype-genotype relations of known mutations. Third, the number of mutated genes leading to different variants has surpassed the number of letters in the alphabet; therefore, various new nomenclatures are being considered that rely on gene mutations and inheritance pattern rather than clinical phenotype (138).
An example of the current classification challenges are SORD mutations. Pathogenic biallelic variants in SORD cause autosomal-recessive CMT2. Mutations result in loss of enzyme activity and increased serum sorbitol. Phenotypic variability is large and includes axonal neuropathy, distal hereditary motor neuropathy, and protein surplus distal myopathy (43; 144; 08).
This article focuses on CMT2, intermediate CMT, and several other subtypes. Inherited neuropathies in which autonomic or sensory features predominate, conditions in which the neuropathy is part of a multiple-organ disturbance, and neuropathies with specific metabolic dysfunction are not discussed. For discussion of CMT1A, CMT1B, CMTX1, and HNPP the reader is referred to the summaries devoted to those subtypes (Charcot-Marie-Tooth disease type 1A; Charcot-Marie-Tooth disease type 1B and mutations of the myelin protein zero; Charcot-Marie-Tooth disease type X; Hereditary neuropathy with predisposition to pressure palsy).
Patients with Charcot-Marie-Tooth disease present with many symptoms and signs. Disease expression varies between and within kindreds and even among identical twins (15). Most patients with Charcot-Marie-Tooth disease have autosomal dominant forms involving weakness, muscle wasting, and sensory loss predominantly in the distal legs, with onset in the first 2 decades of life. Late onset in earlier generations is a possibility when a family history is apparently absent. Furthermore, de novo mutations with a truly negative family history must be considered. For instance, CMT1A has one of the highest de novo mutation rates, similar to neurofibromatosis type 1.
Enlarged and excessively firm nerves are found in more than 25% of patients with forms of Charcot-Marie-Tooth disease characterized by demyelination and remyelination. They can be visible in the superficial cervical nerves or palpable in the arms and ankles. Gait may be compromised by distal weakness, foot deformities, and poor proprioception. Ankle sprains and fractures are frequent. Reports of cold feet, hair loss, or leg edema are common. During pregnancy, some patients experience faster deterioration from which they usually, but not always, recover (185; 79). During medical procedures, prolonged immobilization of the body and limbs in particular positions can result in nerve compression.
Onset and course. Age of onset can vary with subtype, penetrance, familial phenotype, and ascertainment bias. Most symptoms start during childhood but may go unnoticed. Because of the insidious onset, some patients are unaware of their disease and seek medical attention only late in life. Diagnosis is usually not made until late adolescence or early or late adulthood. Exceptions are more severe phenotypes such as Déjerine Sottas syndrome and congenital hypomyelination neuropathy. Thomas and colleagues found that 85% of CMT1A patients developed clinical evidence of disease before 20 years of age (212).
Parents, caregivers, or teachers may notice clumsiness, frequent sprains, poor athletic performance, or toe-walking in a child. For example, one of the authors' patients noticed as a young child that her father's feet slapped the ground when he walked around the house. Later, having observed her personal series of 20 relatives, she was able to accurately diagnose Charcot-Marie-Tooth disease in her siblings’ grandchildren by watching their feet point down when sitting on the ground. Not infrequently, asymptomatic individuals are detected during screening of families after one relative has been diagnosed. Depending on the age of onset, children may have a normal clinical exam. Occasionally, the only finding is impaired heel gait.
In typical Charcot-Marie-Tooth disease, symptoms are chronic and slowly progressive, so any change in pace warrants consideration of superimposed acquired or possibly independently inherited forms of neuromuscular diseases (208). Clinical presentations may be episodic and asymmetric in patients with HNPP and inherited brachial plexus neuropathy or hereditary neuralgic amyotrophy. A distinct phenotype with early onset in the neonatal period or early infancy of weakness and wasting in the feet, with subsequent involvement of the hands, has been linked to GDAP1 mutations (CMT4A). One report described that by the late teenage years these patients develop a hoarse voice and vocal cord paresis (193).
Motor symptoms and signs. In hereditary motor and sensory neuropathy, motor symptoms usually predominate over sensory symptoms, but motor symptoms may be exclusive in hereditary motor neuropathies; these are not discussed in this article.
Patients report imbalance and tripping over objects because of foot drop; they also report impaired sensation, including proprioception. Manipulating small objects such as zippers, forks, or pencils may be difficult because of motor and sensory impairment. On examination, most patients with Charcot-Marie-Tooth disease exhibit distal dominant weakness, hypo- or areflexia, and muscle atrophy affecting the legs earlier and more severely than the arms. Wasting of distal leg muscles develops over time and may produce the classic “inverted champagne bottle” or “champagne glass” appearance.
Proximal weakness is rare except in the most severely affected, in some unusual pedigrees, and in hereditary focal neuropathies. Therefore, even patients with marked distal weakness are able to walk. Foot deformities such as pes cavus, high arches or flat feet, hammertoes, and Achilles tendon shortening result in unequal wearing of shoes and trouble finding well-fitting shoes.
Foot deformities become more prevalent with age but are variable even among same-age relatives (61). Some patients with Charcot-Marie-Tooth disease or HNPP find wider shoes a more comfortable fit for their high arches and hammertoes; wider shoes may also lessen local nerve compression.
Sensory symptoms and signs. Sensation may be normal until adulthood. Distal mild pansensory loss is common and, with semiquantitative or quantitative methods, can be documented even in children. At times, sensation is severely impaired. Sensory changes dominate in hereditary sensory neuropathy. Paresthesias, in contrast to acquired neuropathies, are typically less severe and rarely a presenting symptom, but when specifically asked about paresthesias, patients will acknowledge them. On the other hand, patients may deny sensory symptoms despite marked loss of sensation on examination. Complaints of cold feet, hair loss, or leg edema are common. Radicular pain resulting from CMT1 is rare but well described and is caused by nerve roots that are enlarged (and sometimes visible by MRI) because of ongoing demyelination and remyelination with connective tissue proliferation.
Nonneuropathic pain. Patients often have multiple non-neuropathic pain symptoms. Pain can result from pressure or strain of various structures associated with bones, joints, and tendons and abnormal posture at the knees, hips, and back, which results from foot weakness and fixed-foot deformities such as Achilles tendon shortening. Because of hammertoes and high arches, patients experience painful calluses. Scoliosis is common and leads to back pain. Patients experience leg and hand cramps that are often worse with fatigue and relieved by wearing ankle-foot orthoses.
Atypical presentations. In a large series, 34 patients fit into a "classical" Charcot-Marie-Tooth phenotype, whereas 27 patients had additional features such as CNS features, diabetes mellitus, and prominent muscle cramps (212). CNS features may be part of the disease or may indicate an independent coexisting condition and, thus, always warrant further investigation. Forty-five of 61 patients had deficits in their hands. Loss of large and small fiber sensory function, ranging from mostly mild to moderately severe, was reported in 43 patients. Tremor occurred in up to 25% of patients, and whether it was incidental or part of the syndrome is controversial (184; 174). Vucic and colleagues found two families with dominantly inherited axonal neuropathies, distal wasting, weakness, pes cavus, sensory loss, and mild pyramidal signs; no genotype was identified (224). Dyck and colleagues recognized steroid responsive forms of Charcot-Marie-Tooth disease (60). Auer-Grumbach and coworkers described an Austrian CMT1A family with slow progression and predominantly proximal upper limb weakness and wasting (10). Dematteis and colleagues reported a French CMT1A family with sleep apnea (a feature previously reported in CMT2C), which was asymptomatic in the majority and correlated with the severity of the neuropathy and sleep apnea, possibly due to pharyngeal neuropathy (57). A French family with a Thr124Met myelin protein zero mutation had respiratory, bladder, and sudomotor dysfunction, severe neuropathic pain, and an abolished pupillary light reflex; the latter allowed the discrimination of affected and unaffected relatives (199). Murakami and colleagues described two sisters with an atypical CMT1A phenotype that consisted of prominent sensory complaints, tremor, and episodes of acute paralysis (154). Studies of Déjerine Sottas syndrome (ie, CMT3 or HMSN3) demonstrated that the majority of cases may result from point mutations in PMP22, MPZ, and EGR2 (226; 49; 227). In the case of EGR2, the mutations were inherited in an autosomal dominant or recessive pattern. Nonneurologic manifestations, including endocrine disturbances, have also been described.
Other atypical presentations may possibly reflect a greater propensity of peripheral nerves in Charcot-Marie-Tooth disease to suffer axonal or demyelinating inflammatory injury. In a report, an elderly woman with no past neurologic or family history presented with severe ataxia (223). A biopsy revealed macrophage-associated demyelination with active myelin stripping consistent with a Guillain-Barré or chronic inflammatory demyelinating polyneuropathy picture. The patient responded to five courses of intravenous immunoglobulin treatment. However, a PMP22 mutation, consistent with CMT1A and clinical features of a hereditary neuropathy, including pes cavus, were also found. In another study, a probable CMT2 patient developed an acute axonal Guillain-Barré picture (164). Although this could be coincidental, the authors suggested that CMT2 could lead to an increased susceptibility to acquired axonal damage.
Disability. Disability may vary greatly between family members and can range from symptom-free with minimal findings to severe. Some adults require ankle-foot orthoses only in the sixth decade, whereas some children may already have foot drop, proximal leg weakness, and clawing of the fingers. These severely affected patients are more likely to lose their ability to ambulate independently. Pfeiffer and colleagues reported that individuals affected by CMT1 suffer emotional stress that is similar to patients with stroke and comparable disability (171). Significant disability affected 44% and depression 18% of CMT1A patients. High disability predicted attitude against childbearing, and 36% of individuals voted against childbearing. A study of 43 CMT2 patients documented slow progression of weakness and disability; most patients remained ambulatory (206).
CMT2. As with other forms of CMT disease, phenotypic variation is common between and within CMT2 families. Some reports suggest that CMT2 has a later onset in life, but this impression predates molecular studies and the recognition of different subtypes. One family with 50-year anticipation between generations has been reported (221). Patients may have greater atrophy and distal leg weakness with relatively less hand weakness. Areflexia, pes cavus, and hammertoes may be less common than in CMT1. Nerve hypertrophy is absent but is variable in CMT1 as well as CMT2 often presents a diagnostic dilemma because characteristic features such as enlarged nerves and near-pathognomonic neurophysiologic findings are absent. With later onset, this condition may be difficult to differentiate from a late-life acquired neuropathy when the family history is unclear. Thus, prevalence data are of uncertain validity, though estimates suggest that there is one case of CMT2 for every two cases of CMT1. Although mutations in several disease genes have been identified, not all can be tested commercially yet. CMT2 and CMT1 can rarely be differentiated by history and examination findings alone. Sural nerve pathology usually reveals reduced numbers of myelinated axons, especially of larger diameter. Rare myelin changes can be observed.
CMT2A. The clinical onset of CMT2A can be variable, from childhood to old age. In one family with onset typically in the second decade, conduction velocities were normal or mildly slow (22). Affected members of a large southern Italian pedigree had distal weakness, wasting, hyporeflexia, and mild panmodal sensory loss (153). Biopsies revealed axonal but no myelin abnormalities. CMT2A has been linked to chromosome 1p36.2 (21; 170; 187; 153), where a loss-of-function mutation in the KIF1B gene has been found (CMT2A1) (235). This is a motor protein in anterograde transport of mitochondria. Subsequently, mutations in the MFN2 gene (mitochondrial GTPase mitofusin 2) were found in seven families classified as CMT2A2 (129; 238). MFN2 mutations are far more common than KIF1B in CMT2A. The phenotype is indistinguishable from KIF1B-related CMT, CMT2E, and CMT2F. MFN2 mutations account for 20% to 33% of CMT2; thus, CMT2A is the most common form of CMT2 and second in frequency only to CMT1A (218). The phenotype is most commonly classical CMT, but it may be subtle. An Australian family with CMT and spasticity, previously described as HMSN V, had a MFN2 mutation (238). Other patients have optic atrophy or sensorineuronal hearing loss. MFN2 protein is a large dynamin-like GTPase that spans the outer mitochondrial membrane. There are two major forms of CMT2A: severe with early onset and mild with late onset (40; 218). Some patients develop minor CNS changes; foot deformities occurred in all patients in a large Korean series (40); these authors suggest that HMSN VI may be a variant of early-onset CMT2A. Some patients have asymptomatic, MFN2 mutations with normal nerve conduction but minor neuromuscular changes on examination. Calvo and colleagues identified 20 MFN2 gene missense mutations among 150 screened individuals with HMSN and motor conduction velocities of 25 m/sec or greater and further demonstrated the clinical diversity, including both dominant and recessive inheritance (35). Most CMT2A patients have an early onset and severe phenotype; CMT2A accounts for 91% of the severe phenotypes of CMT2 (69). Some patients also had a more prominent motor neuropathy. Optic atrophy was evidenced in one family with early-onset disease (16). An international multicenter study is underway to provide guidance for prognosis, “variant” interpretation, and clinical trial design (173).
CMT2B. This is a predominantly sensory neuropathy to the point that its classification with hereditary motor and sensory neuropathy versus hereditary sensory and autonomic neuropathy is unclear. Patients may have foot ulcerations and even amputations, but no clinical weakness. High arches, hammertoes, and hyporeflexia are also present. Although neurophysiologic findings are established early in life, clinical onset may be much later. Decreased CMAP amplitudes and denervation are electrodiagnostic features. CMT2B biopsies reveal evidence of degeneration and regeneration, with the presence of occasional onion bulbs. CMT2B was mapped to chromosome arm 3q13-q22 (125). Missense mutations (Leu129Phe and Val162Met) in the small guanosine triphosphatase (GTPase) late endosomal protein RAB7, a member of the RAS-associated GTP-binding proteins, were found (219). RAB7 is ubiquitously expressed in sensory and motor neurons. It regulates linkage of vesicles and other membranes to the cytoskeleton and plays a role in lysosomal degradation.
CMT2B1. Onset of autosomal recessive CMT2B1 in a Moroccan family was in the second decade. Features include distal and (less often) proximal weakness and pes cavus. Motor nerve conduction velocities were near normal; linkage was found to chromosome 1q21.2-q21.3 (31), but in a similar family a homozygous mutation in the LMNA gene, which encodes lamin A/C, a component of the nuclear envelope, was detected (50). In LMNA knockout mice, the authors found reduced axon density, axonal enlargement, and nonmyelinated axons. This same gene is mutated in limb-girdle muscular dystrophy type 1B, autosomal dominant Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy type 1A, mandibuloacral dysplasia, autosomal dominant partial lipodystrophy, and others.
CMT2B2. A second form of autosomal recessive axonal Charcot-Marie-Tooth disease has been mapped to a 5.5-cM on chromosome 19q13.3. CMT2B2 patients have a sensorimotor polyneuropathy with distal arm and leg weakness and wasting, hyporeflexia, and sensory loss with normal or minimally reduced nerve conduction velocities (130).
CMT2C. This subtype usually starts in the first decade of life, but onset in adulthood has been reported. Mild sensory loss is combined with weakness in the limbs (especially in the hands but proximal leg weakness is also reported), diaphragm, intercostal muscle, and vocal cords, which can lead to early death. Vocal cord dysfunction also occurs occasionally in several other CMT subtypes. The locus was mapped to chromosome 12q23-q24 (121), where Landouré and colleagues found mutations in the TRPV4 gene in two families. Worsening hand weakness in the cold, sensorineural hearing loss, and neurogenic bladder were common (127). TRPV4 gene mutations have been linked to altered calcium homeostasis, leading to allelic disorders, ie, scapuloperoneal spinal muscular atrophy or CMT2C phenotypes (56).
CMT2D. Affected CMT2D individuals may have worse hand than leg weakness and slow progression (106; 170). Onset ranges from 16 to 30 years of age. Tendon reflexes are usually absent in the arms and decreased in the legs. Progression is slow. Although initial confusion about this disorder resulted from linkage of the same region on chromosome arm 7p15 to a form of spinal muscular atrophy, both phenotypes can occur in the same family. Two CMT2D families have a 1236G-C change in the GARS (glycyl-tRNA synthetase) gene, resulting in a G240R substitution, whereas one family with distal spinal muscular atrophy type V has a 2094G-C mutation resulting in a G526R substitution (05). GARS belongs to a family of aminoacyl-tRNA synthetases that catalyze the esterification of an amino acid to its cognate tRNA.
CMT2E. CMT2E can be clinically difficult to distinguish from CMT1A, CMT1B, or CMT2A, though more severe clinical phenotypes occur (111). Onset age ranges from the first to the third decades (149; 80; 111). Multiple missense mutations and an in-frame 3 bp deletion affecting different protein domains have been reported in the neurofilament light chain gene (NF-L or NF68) on chromosome 8p21. NEFL mutations impact axonal transport, particularly that of mitochondria. Nerve conduction studies indicate axonal involvement with demyelinating features with prolonged distal latencies disproportionate to the conduction slowing, similar to findings in anti-MAG associated neuropathy (65). In some cases, conduction velocities are severely slowed (47), and for these a classification as CMT1F (sic) has been proposed. One nerve biopsy revealed axonal degenerative and regenerative features as well as onion bulbs (111). This highlights the marked variability of clinical patterns with specific CMT gene mutations. Thus, strict genotype-phenotype correlations may be an unrealistic expectation.
CMT2F. CMT2F patients exhibit slow progression and worse distal weakness. Ismailov and colleagues reported a 6-generation Russian family with autosomal dominant inheritance (108). Onset ranged from 15 to 25 years of age. Patients had early hyporeflexia and symmetric, slowly progressive weakness and leg atrophy with foot drop, whereas weakness and wasting of arm muscles ensued years later. Sensory impairment was mild-to-moderate. Linkage to a 14.5 cM interval on chromosome 7q11-q21 was established and, subsequently, mutations in the HSPB1 gene were found to be responsible (64). In an Iranian Jewish population, HSPB1 mutations caused an adult-onset, predominantly motor axonal neuropathy (87). Upper and lower motor neuron involvement were described in patients with HSPB1 mutations (117).
CMT2G. Berciano and colleagues described this form in 1986 in 10 relatives from three generations with male-to-male transmission (22). Nelis and colleagues reported on 14 affected members from the same family (157). The mean age at onset was 29 years (range 9 to 76 years). Patients presented with slowly progressive foot deformity and difficulty walking. Ankle reflexes were uniformly absent or hypoactive, knee reflexes were sometimes preserved. Mild stocking hypesthesia was present. Three nerve conduction studies were normal; others showed slight slowing. Biopsy showed regenerating fibers, fiber loss, and atrophic axons.
CMT2H. Barhoumi and colleagues described this form of autosomal recessive axonal CMT in 13 affected Tunisian patients with onset in the first decade (18). Patients had brisk upper limb and knee reflexes, Hoffman and palmomental reflexes, and severe muscle wasting. The identified locus at chromosome 8q21.3 overlaps the GDAP1 gene linked to axonal CMT4A. Specific GDAP1 mutations may cause both demyelinating and axonal forms.
CMT2I. Four different groups reported several families with CMT2 and MPZ mutations (143; 191). Several subjects were asymptomatic. Some had onset only in the seventh decade.
CMT2J. Multiple CMT2 families and two isolated CMT2 patients characterized by fourth to sixth decade onset, marked sensory abnormalities, weakness, deafness, and pupillary abnormalities have been reported (37; 48). Nerve conduction velocities ranged from below 38 m/s to normal. Biopsies showed axonal changes. MPZ mutations were found at T124M and D75V. Phenotypes include isolated Adie pupil and pupillary abnormalities with late onset axonal polyneuropathy (77).
CMT2L. Tang and colleagues described an axonal, autosomal dominant form of CMT in a Chinese family with linkage to chromosome 12q24 (204), later linked to heat shock protein 22, HSPB8 in a Korean family (107). The same mutation is also associated with distal hereditary motor neuropathy type 2A (dHMN2A). Mutations were later reported in families from Bulgaria, England, Belgium, Czech Republic, and China (156). The disease is characterized by predominant distal and lower extremity involvement and slow progression.
CMT2M. Mutations in the DNM2 gene are associated with DI-CMTB, CMT2M, and lethal contracture syndrome 5. Fabrizi and colleagues identified a family with a heterozygous mutation in the DNM2 gene associated with an axonal form of CMT and a milder phenotype (67). Patients may have congenital or early onset cataracts. The condition was also associated with centronuclear myopathy.
CMT2N. Heterozygous mutations in the AARS gene are associated with an axonal form of CMT2, with mean age of onset of 28 years and classic CMT phenotype, but with marked variability of age of onset and severity, as well as variable sensorineural deafness (128).
CMT2O. Heterozygous mutations in the DYNC1H1 gene are associated with CMT2O, a phenotype characterized by delayed milestones, classic CMT phenotype with abnormal gait, normal nerve conduction studies (but axonal changes on sural nerve biopsies), and occasional learning difficulties (229).
CMT2P. Guernsey and colleagues and Nicolaou and colleagues reported consanguineous families from Canada and Sardinia with LRSAM1 gene mutations (88; 163). Patients had episodic muscle cramps, with occasional fasciculations in the arms and legs and severe axonal neuropathy on the nerve conduction studies.
CMT2Q. Xu and colleagues reported a 5-generation Chinese family with eight affected individuals (232). Classic CMT phenotype was the usual pattern, but crane leg–like malformations with decreased motor and sensory amplitudes in the legs were seen with normal nerve conduction studies in the arms.
CMT2R. Ylikallio and colleagues described a Finnish family with autosomal recessive inheritance and early-onset axonal peripheral neuropathy (234). Hypotonia was evidenced at the age of 4 in the proband. The patient had pes cavus, and nerve biopsy revealed neurofilament accumulation within axons. Cranial nerve involvement is an important feature (137).
CMT2S. Mutations in the IGHMBP2 gene lead to spinal muscular atrophy with respiratory distress type 1 (SMARD1) and CMT2s (44). Onset occurs in late childhood with slow progression. Some patients have unusually shaped tongues.
CMT2T. Higuchi and colleagues identified 10 patients in Japan with homozygous or compound heterozygous mutations in the MME (membrane metalloendopeptidase) gene, a gene that encodes neprilysin, one of the most prominent beta-amyloid degrading enzymes. All mutations were loss of function (nonsense, missense, splice site, and deletions) and led to a late-onset sensorimotor axonal polyneuropathy with muscle weakness, atrophy, and sensory disturbance in the legs (101). No patient had evidence of dementia or CNS disease. Both mono- and biallelic MME mutations cause late-onset axonal peripheral neuropathy (81). Biallelic mutations are more likely to be associated with rapid progression (81).
CMT2U. Mutations in the MARS gene are associated with a CMT variant characterized by late-onset, slowly progressive disease with proximal and distal involvement (84).
CMT2V. Mutations in the a-Nacetyl-glucosaminidase NAGLU gene were reported in a large French-Canadian family with CMT (207). NAGLU mutations are classically associated with mucopolysaccharidosis IIIB. Early pain was the most striking feature in this family and occasionally affected the sleep pattern.
CMT2X. Mutations in the SPG11 gene can be associated with a complex phenotype, including juvenile amyotrophic lateral sclerosis type 5; autosomal recessive spastic paraplegia 11; and CMT with foot and hand deformities, kyphoscoliosis, ankle contractures, and tremor (152)
CMT2W. Patients with mutations in the histidyl-tRNA synthetase (HARS) gene exhibit classic CMT phenotype or motor axonal neuropathy, with variable age of onset and normal or mildly decreased conduction velocities (186). Some patients have brisk patellar reflexes.
CMT2Y. Mutations in the VCP gene cause a CMT variant with variable severity. Patients may have foot deformities, and phenotype is complex, including high CK levels in the spectrum of a lower motor neuron syndrome (85).
CMT2Z. Patients with CMT and pyramidal signs were shown to have mutations in the MORC2 gene (02). In more than half of the patients, developmental delay was evidenced, and onset was in childhood or early adulthood, with late walking. Some also had dysmorphic features and seizures. A report of a patient with upper motor neuron findings, white matter changes, dystonia, and intention tremor with sensory neuropathy, and onset at age 15 extends the complex phenotype in MORC2 mutations (73).
Dejerine-Sottas syndrome (ie, HMSN3 or CMT3). Dejerine-Sottas syndrome was originally described in 1893 as a hypertrophic polyneuropathy with onset in infancy or early childhood in patients born from unaffected parents, characterized by distal sensory loss with ataxia, pes cavus, distal weakness with proximal progression, palpable hypertrophied nerves, and Argyll-Robertson pupils. Lightning pain, described in the original two cases, occurs in less than 25% of patients. Histopathologically, the hallmark was extensive nerve and root hypertrophy due to demyelination-remyelination.
In 1906, Pierre Marie reported a variant of Dejerine-Sottas syndrome with distal atrophy, nerve hypertrophy, areflexia, intention tremor, and dysarthria but without sensory ataxia or Argyll-Robertson pupils; this was later referred to as the Marie and Boveri type. Although it was noted that none of the reported cases fulfilled all criteria, Dyck and Lambert reported several common features: probable recessive inheritance, infancy onset, delayed motor milestones, severe gait disturbance, ataxia, nerve hypertrophy, increased CSF protein, extremely slow nerve conduction velocity, marked segmental demyelination, and onion bulb formation (61). The variability of neurologic manifestations is wide. Nerve hypertrophy is variable (175). Thickened spinal nerves occasionally lead to spinal cord compression (175). Although a slowly progressive length-dependent sensorimotor deficit is typical, a relapsing-remitting course has been reported (175).
Dejerine and Sottas found the muscles of their first patient to be inexcitable, whereas the responses of the less affected patient were absent in the legs only (53). Dyck and Lambert reported velocities of less than 12 m/sec in the arms with uniform slowing and marked dispersion (61). Nerve biopsies reveal decreased fiber density, segmental demyelination, onion bulbs, and sometimes giant whorls of cell processes. In young patients, teased fibers reveal segmental demyelination, thinly myelinated internodes, and irregularity of myelin sheath thickness (175).
Molecular genetics revealed that many cases are dominant. For such patients, mutations in MPZ, PMP22, and EGR2 (160; 226; 49) and linkage to chromosomes 8q23-q24 (106) must be considered. For PMP22 itself, more than 16 different mutations have been identified (175). The possible existence of milder and unrecognized hereditary neuropathies in one or both parents must be considered. Autosomal recessive forms of Dejerine Sottas syndrome are discussed in the CMT4 subgroup below.
Congenital hypomyelination neuropathy. Patients with congenital hypomyelination neuropathy present with neonatal hypotonia, areflexia, distal weakness, slow nerve conduction velocities, and at times, contractures or arthrogryposis (94). Like Dejerine Sottas syndrome, it may result from MPZ, PMP22, or EGR2 mutations, as well as from separate mutations in the same or different genes inherited from both parents or from one mutation inherited from one parent combined with a de novo mutation in the same or another gene. Histological findings include severe hypomyelination and demyelination and axonal loss. Sural nerve biopsies may differentiate congenital hypomyelination neuropathy from Dejerine Sottas syndrome. Absence of active myelin breakdown and rare onion bulbs points to congenital hypomyelination neuropathy, whereas demyelination or remyelination and abundant organized onion bulbs are seen in Dejerine Sottas syndrome. Congenital hypomyelination neuropathy results from a congenital impairment in myelin formation, whereas Dejerine Sottas syndrome results from aberrant demyelination and subsequent remyelination of the peripheral nerve (15).
Neuritis with brachial predilection or hereditary neuralgic amyotrophy or hereditary brachial plexus neuropathy (NABP or HNA). Hereditary neuralgic amyotrophy is an autosomal dominant form of recurrent focal painful neuropathy with predilection for the arms. This autosomal dominant recurrent focal neuropathy may have been first described in the case of a woman with three episodes of painful arm weakness, whose sister had experienced seven such attacks (58). Jacob and colleagues described seven patients of two unrelated families with 14 episodes of recurrent brachial neuritis with severe pain, weakness, wasting, impaired reflexes, and sensation (109). A similar condition was described in three generations of a family (91).
Individuals experience episodic brachial plexus neuropathy with weakness, atrophy, and sensory disturbances, almost always preceded by severe pain in the affected arm. Onset is between 10 and 30 years of age, rarely earlier. Near complete recovery begins within weeks to months after onset. Recurrent episodes may affect either arm. The right arm is involved more often. At times, the lower cranial nerves and sympathetic nervous system are involved as well. Isolated involvement of the long thoracic nerve with scapular winging and serratus anterior weakness has been described (172). As in the sporadic forms, attacks may follow infections or immunization (109). Phenotypic variation may occur, with some patients following the classic relapsing-remitting course and others following a chronic undulating course (150). Common dysmorphic features include hypotelorism, short stature, cleft palate, unusual skin folds, and creases in the neck or scalp referred to as cutis verticis gyrata (169; 110). Three separate studies established linkage to chromosome 17q25 in a 500 kb gene region (169; 150; 228).
CMT4. This term is applied to autosomal recessive forms of demyelinating Charcot-Marie-Tooth disease. In its more severe forms, it overlaps with Dejerine Sottas syndrome or CMT3. The classification represents distinct clinical and histologic subtypes, of which several occur only among specific small ethnic groups. Axonal forms of autosomal recessive Charcot-Marie-Tooth disease include CMT2B.
CMT4A. This condition starts in early infancy with delayed motor development, weakness, muscle atrophy, and, occasionally, scoliosis. It is the most common autosomal recessive demyelinating form. The disease may progress to severe motor weakness. It was first described in a consanguineous American pedigree (03) and later in several Tunisian and Turkish families (21; 158). Histological hallmarks include marked hypomyelination with basal lamina onion bulbs, though mixed features of demyelination and axonopathy neurophysiologically and histologically have been reported (158; 188; 193). Linkage to chromosome 8q21 has been established, where mutations in GDAP1 have been detected (19; 45; 158; 28). Mutations in the GDAP1 gene cause CMT4, ARCMT2, and intermediate conduction velocity variants. In Finland, it represents 12% of all CMT2 cases (12). This gene expressed in CNS and PNS neurons and in Schwann cells may be involved in a signal transduction pathway in neuronal development. GDAP1 is a protein in the class of glutathione transferases, which localizes within the mitochondria (168). GDAP1 mutations cause both demyelinating and axonal phenotypes, yet another example of mutations resulting in distinct classes of neuropathy. The axonal form is linked to vocal cord and diaphragmatic paralysis with midlife onset (193; 194). A Spanish report describes an autosomal dominant pattern with a milder form than the recessive cases (42). Baránkóva and colleagues found a high frequency (7.2%) of mutations of at least one GDAP1 allele in Czech families with early-onset axonal or demyelinating neuropathy and a recessive inheritance pattern (17). Among 100 Brazilian patients with axonal Charcot-Marie-Tooth, GDAP1 mutations accounted for 7.1% (71).
CMT4B1. CMT4B was first described in Japan and subsequently in European countries as a nonhypertrophic, severe, sensorimotor neuropathy during infancy with frequent cranial nerve involvement. Quattrone and colleagues and Bolino and colleagues described an autosomal recessive demyelinating neuropathy in an Italian family with onset in infancy, sometimes in the second decade (29; 176). As weakness progresses, some patients become wheelchair bound. A histologic hallmark is the presence of focally folded myelin sheaths (78). Linkage to a 4 cM interval on chromosome 11q22 was established, where several homozygous or compound heterozygous mutations in a phosphatase gene, myotubularin-related protein 2 (MTMR2), result in premature termination or frameshift (30). Such loss of function mutation could result in constitutive phosphorylation of an unknown substrate, with Schwann cell proliferation and myelin overgrowth. The autonomic nervous system can be involved, at least in some mutations (92). A multicenter retrospective study emphasized that CMT4B1 is usually more severe than CMT4B2 (166).
CMT4B2. This subtype is characterized by sensorimotor neuropathy, with onset in the first or second decade, and early-onset glaucoma has been identified in consanguineous families from Tunisia, Morocco, Brazil, and Japan. Motor nerve conduction velocities are severely reduced, and nerve biopsies showed myelin outfoldings. Early visual deficiencies and later blindness result from congenital glaucoma with buphthalmos, megalocornea, and increased intraocular pressure (13). Mutations were found in the gene for SET binding factor 2 (SBF2) on chromosome 11p15, a pseudophosphatase and myotubularin related protein that may be involved in phosphoinositide-mediated signaling events controlling myelination (13; 190).
CMT4B3. Only three families with CMT4B3 have been reported (155; 182). The first original Korean family had a pure sensorimotor demyelinating polyneuropathy with focally folded myelin sheaths, similar to CMT4B1 and B2. However, the second family (from Saudi Arabia) also had progressive microcephaly, intellectual disability, syndactyly, and multiple cranial nerve involvement, leading to ophthalmoparesis, absence of pupil reactivity to light, mild facial weakness, dysphagia, and dysarthria. A third family from Syria was described with a pontine-mesencephalic MRI anomaly called “fork and bracket” syndrome (182). In addition to a phenotype similar to the Saudi Arabian family, one patient developed severe oromandibular dystonia leading to impaired mouth closure, making it difficult to eat and speak.
CMT4C. This condition resembles CMT4A and CMT4B but has early onset scoliosis and is genetically distinct. LeGuern and colleagues reported two large consanguineous Algerian families with sensorimotor polyneuropathy, worse in the legs (132). Conduction velocities were 24+5.1 m/sec. Onset varies from infancy to 12 years of age. Manifestations are variable but include prominent scoliosis, early loss of ambulation, and respiratory problems (189). Nerve biopsies revealed abnormal Schwann cell processes with increased basal lamina production (189). Linkage to chromosome 5q32 was found (132; 89), where multiple truncating and missense mutations in a novel SH3/TPR domain protein of unknown function have been identified (189). In a series of 12 European and North African families CMT 4C was characterized by frequent and severe scoliosis (14). Early disease onset was confirmed, and foot and spine deformities were occasionally more pronounced than motor and sensory impairment. Hearing loss and facial paresis was seen in some. Demyelination with severe conduction slowing was the rule, but 3 of 29 patients had intermediate velocities. Giant axons were present in some. Claramunt and colleagues found a private founder p.R1109X mutation in the SH3TC2 gene in a group of Spanish gypsies with demyelinating neuropathy; the mutation probably arose 225 years ago (42). SH3TC2 mutations may be associated with predisposition to inflammatory neuropathy (103). In 29 Spanish Roma families, CMT4C was the most prevalent form (57.1%), followed by HMSN-Russe (25%) and HMSN-Lom (17.9%) (195). Of 14 French patients with CMT4C, 6 of 14 had scoliosis as the presenting sign (233). Cranial nerves (especially VII, VIII, XII, IX, and X) were involved in 10 patients. Fifty percent had proximal involvement. Electrodiagnostic studies revealed probable conduction block and temporal dispersion. In patients of Chinese background, scoliosis, cranial nerve involvement, and age of onset were variable (59).
CMT4D or HMSN-Lom. HMSN-Lom was first described in Bulgarian Roma, Lom being the town where many members of this group lives (116). This variant is characterized by gait difficulties in the first decade, skeletal deformities, deafness, and severe sensorimotor deficit, worse in the legs. Nerve conduction velocities are less than 15 m/sec in the upper extremities, with unobtainable sensory responses. In the younger patients, onion bulbs were observed, but nerve biopsies revealed chronic demyelination, severe fiber loss, and axonal inclusions. Merlini and colleagues reported a similar condition in four siblings of an Italian Roma family (148). King and coworkers documented demyelination or remyelination in five biopsies, with severe progressive axonal loss without atrophy (119). Onion bulb formations were more conspicuous in younger than older individuals. Hypomyelination and uncompacted myelin were observed together with intra-axonal accumulation of irregular curvilinear material. A mutation in the N-myc downstream-regulated gene 1 (NDRG1) on chromosome 8q24.3 was reported (116). Ubiquitously expressed, but particularly so in the peripheral nervous system, it may play a role in growth arrest and cell differentiation, possibly as a signaling protein shuttling between the cytoplasm and the nucleus; it could mediate possibly in the Schwann-cell signaling necessary for axonal maintenance.
CMT4E. Mutations in the EGR2 gene were detected in patients with phenotypes that include classical Charcot-Marie-Tooth disease, Déjerine-Sottas syndrome, and congenital hypomyelination neuropathy. More severe phenotypes with EGR2 mutations are referred to as CMT4E. Cranial nerve involvement and respiratory failure have been reported (213).
CMT4F. CMT4F was first identified in a Lebanese family (54). It presents with a disabling ataxic neuropathy with onset in the first decade, distal weakness and atrophy, pain, and prominent large fiber involvement resembling Déjerine-Sottas syndrome. Motor and sensory responses could not be evoked in most individuals. Onion bulbs, loss of axons, and hypertrophied myelin sheaths are seen in biopsies. Kabzinska and colleagues reported a boy with delayed walking (2 years) and clumsy gait noted since the age of 5 years (114). Motor or sensory responses were unrecordable since the age of 3 years. Several mutations in the periaxin (PRX) gene on chromosome 19q13.1-13.2 have been identified (90; 203). PRX codes for L and S periaxin, which are required for the maintenance of peripheral myelin. L-periaxin is a linker in a macromolecular complex consisting of laminin receptor dystroglycan and dystrophin-related protein 2 (DRP2) at the Schwann cell surface where it is required for the assembly of Cajal bands (cytoplasmic channels essential for Schwann cell growth).
CMT4G. CMT4G is caused by mutations in the HK1 gene (74). HK1 and NDRG1 mutations (CMT4D) are common causes of inherited neuropathy in Slovak and Spanish Roma (74; 195). HK1 mutations can cause retinitis pigmentosa, hemolytic anemia, and “HSMN of the Russe type.” The disease is characterized by early motor involvement in the first decade, resulting in walking difficulties, distal sensory involvement, pes cavus, and areflexia (181). Patients have important foot and hand deformities. Neurophysiological testing reveals demyelinating features and length-dependent axonal features.
CMT4H. An early-onset neuropathy with severe functional impairment and scoliosis and with eccentric folding of redundant myelin sheaths was identified in two Lebanese and Algerian families (51). Mutations in the FGD4 gene were found in the original families, three others of Lebanese and Turkish origin, one sporadic Tamil patient, and in Ireland and Japan (55; 07). Fabrizi and colleagues delineated the clinical heterogeneity of the condition, variable functional impairment, and a crucial role for frabin during myelin formation (68). Houlden and colleagues reported a less severe phenotype with slowly progressive polyneuropathy; patients remained ambulatory in middle age (102). One patient had milder motor involvement, but major sensory loss (07). In patients with milder phenotype and slow progression, cauda equina thickening has been described (06).
CMT4J. The condition is associated with mutations in the FIG4 gene on chromosome 6q21 (39). Severity is variable, with some patients having features more consistent with neuronopathy than polyneuropathy. Some patients reported trauma prior to the onset of weakness. Motor symptoms tend to be worse than sensory, and involvement of the VI and XII cranial nerves has been reported (161). Severe central nervous system involvement, including parkinsonism, cerebellar ataxia, and a continuum causing Yunis Varon syndrome, cleidocranial dysmorphism, limb malformations, and early death may be part of the spectrum of FIG4 mutations (237).
Dominant intermediate CMT (DI-CMT). Although families with conduction velocities atypical for both CMT1 and CMT2 had long been recognized, they were often considered exceptions that did not break the paradigm. Before the advent of genetic testing, Bradley, Davis, and Madrid proposed a CMT classification that included an intermediate group, characterized by median motor nerve conduction velocities of 25 to 45 m/sec and intermediate pathological changes compared to the hypertrophic neuropathy group (32; 135; 46). The degree of slowing was similar in the family members, irrespective of the degree of associated denervation. The phenotype was moderately severe, hyporeflexia developed late, nerve hypertrophy and foot deformity were uncommon, and women were less affected. Some, but not all, subjects likely had CMTX, which is also associated with intermediate nerve conduction velocities.
Molecular genetics now reveal the heterogeneity of this group: Cx32 and specific MPZ, GDAP1, or NF-L mutations can cause velocities and morphologic changes that overlap CMT1 and CMT2 and may vary between family members and change with age (162; 48; 47; 145; 19; 45; 113; 188; 193). Men with CMTX1 have nerve conduction velocities of 30 to 40 m/sec, women often greater than 40 m/sec.
DI-CMTA, DI-CMTB, and DI-CMTC were linked to chromosomes 10q24.1-q25.119, 19p12-p13.220, and 1p34-p35, respectively.
DI-CMTA. This phenotype, originally reported in 1985 (183), is clinically characterized by onset in the second decade of life, when the patients develop leg weakness, difficulty in running, and muscle cramps. Some patients had motor difficulties with exposure to cold and difficulty walking on heels since childhood. Although in the third decade progression is usually slow, in the fifth decade patients rather rapidly develop severe weakness and distal limb atrophy, steppage gait, pes cavus, areflexia, and mild distal sensory loss. Later, the course stabilizes and elderly patients are not wheelchair bound. Typical biopsy findings are chronic axonal degeneration and regeneration with large fiber loss and regeneration clusters, segmental de- and remyelination, uncompacted enlarged myelin lamellae, and onion bulbs (220). Linkage to chromosome 10q24.1-q25.1 has been established (140).
DI-CMTB. Analysis of two families permitted linkage to a 6 cM interval on chromosome 19p12-p13.2 (118; 236). A nerve biopsy in one family revealed axonal degeneration with loss of large diameter fibers and rare segmental demyelination and remyelination with onion bulb formation, similar to DI-CMTA (118). DI-CMTB is caused by mutations in dynamin 2 (238).
DI-CMTC. This form of DI-CMT has been reported in two families, one from the United States and the other from Bulgaria (209; 210; 113; 112; 165). The phenotype is discussed in the clinical vignette. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase gene was found in 2 heterozygous missense mutations (G41R and E196K) and one de novo deletion (153-156delVKQV). Homozygosity for YARS mutations is associated with a multisystem disease: poor growth, developmental delay, brain dysmyelination, sensorineural hearing loss, nystagmus, progressive cholestatic liver disease, pancreatic insufficiency, hypoglycemia, anemia, intermittent proteinuria, recurrent bloodstream infections, and chronic pulmonary disease (230).
DI-CMTD. A 4-generation Macedonian family with autosomal dominant CMT, variable distal wasting, weakness, and sensory loss (worse in the legs) was reported (145). Median and ulnar motor conduction velocities were in the range of 24 to 41 m/s and 33 to 48 m/s, respectively. Biopsies revealed axonal changes, segmental demyelination and remyelination, but no onion bulbs. An Asp6Tyr mutation in the extracellular domain of the MPZ gene was found.
Complex phenotypes (genes responsible for distinct disease phenotypes). Several genes have been linked to distinct clinical phenotypes, eg, amyotrophic lateral sclerosis, distal motor neuropathies, myopathies, and CMT2 or CMT4. Dominant mutations in the CHCHD10 gene were classically associated with amyotrophic lateral sclerosis or frontotemporal dementia complex. The spectrum of diseases linked to CHCHD10 mutations includes mitochondrial myopathies and late-onset spinal motor neuronopathy (spinal motor atrophy Jokela type). CHCHD10 mutations have been linked to CMT2 phenotype in four families (11). The typical phenotype included slowly progressive lower extremity weakness, later involving hand muscles. In at least 7 of the 12 patients, sensation was affected, although clinical overlap with spinal muscular atrophy Jokela type was found in some patients. HSJ1 mutations were also found to cause complex phenotypes: distal hereditary motor neuropathies and Charcot-Marie-Tooth disease (82).
Until recently, lifespan was not considered to be affected by Charcot-Marie-Tooth disease. Vaeth and colleagues reported reduced life expectancy in patients with Charcot-Marie-Tooth disease in a Danish disease registry (215). Higher standardized mortality ration and absolute excess mortality were found, especially in patients younger than 50 years and with disease duration greater than 10 years. Disability is highly variable and difficult to predict in young individuals, even among siblings. In general, Charcot-Marie-Tooth disease is slowly progressive. If progression accelerates, other causes, such as acquired neuropathies or other inherited neuromuscular conditions, should be sought (208). Often, males are affected more than females, possibly because of a greater likelihood of nerve trauma. Most patients need some kind of ankle support at some time in their life. However, a study of myelin protein zero regulation by androgens and progesterone derivatives suggests a possible genetic course of this gender difference (136; 146). Weakness sometimes spreads to proximal leg or arm muscles, and then ambulation can be impaired even with ankle braces. One study reported significant disability in 44% and depression in 18% of patients with CMT1A. In addition, 36% of patients opted against childbearing (171).
Rarely, patients with Charcot-Marie-Tooth disease may have laryngeal dysfunction with aspiration and voice problems.
Patients with Déjerine-Sottas syndrome are more likely to lose their ability to ambulate independently, and congenital hypomyelination neuropathy can lead to early death.
Since the age of 12 years, school friends of a 56-year-old woman had noted that she walked funny, eg, on the tips of her toes. She had been aware of clumsiness and had experienced multiple falls. During her adolescence, she was never able to take part in sports because of her clumsiness. Also, she had recognized the sound her father made while walking as “different” since she was 5 years old. Her symptoms progressed slowly. She had recently noticed intermittent left foot drop and decreased sensation in the legs, but she had no autonomic complaints. Otherwise, she was healthy and had no other medical problems.
Her general exam was normal. Her mental status and cranial nerves were intact. The dorsal interossei and wrist movement were impaired. Weakness was worse in the legs, and she had moderate foot drop. Deep tendon reflexes were attenuated, and the ankle jerks were absent. Plantar responses were flexor. Proprioception, vibration, and pinprick sensation were diminished, more so in the distal legs. She could not stand on one foot or heel- or toe-walk. She had pes cavus and hammertoes. No skin ulcers were noticed in the feet.
Nerve conduction studies revealed absent peroneal and tibial motor responses (recorded at the EDB and abductor hallucis longus, respectively). Ulnar and median motor velocities were approximately 40 m/sec. The sural and median sensory responses were absent. Electromyography revealed sparse fibrillations and increased duration and amplitude motor unit potentials in the legs. A sural biopsy showed thinly myelinated fibers and clusters of regenerating fibers but no onion bulbs.
Multiple family members from four generations had similar clinical, neurophysiologic, and histologic findings and slow progression starting in childhood. None of the relatives were wheelchair-bound, even after decades of disease.
Genetic testing for the commercially available CMT mutations was unrevealing. Linkage analysis excluded the known loci for intermediate conduction velocity Charcot-Marie-Tooth disease. A genome screen revealed linkage to chromosome 1p34-35, which was confirmed in a second family from Bulgaria. We have classified this condition as DI-CMTC (209; 210; 113; 112).
Table 1 provides a summary of known Charcot-Marie-Tooth disease genes and loci. Somewhat arbitrarily, genes involved in the CMT disorders can be clustered in groups based on function of their proteins:
|
(1) One group involves mutations in structural myelin proteins that cause mostly demyelinating phenotypes, though as discussed, axonal presentations occur. This group of proteins consists of PMP22, P0 (MPZ), and periaxin. | |
|
(2) Proteins that are either part of or associated with the cytoskeleton are implicated in several subtypes. The second group consists of mutations in the structural and motor proteins in nerve axons, which lead to axonal neuropathies. This group includes KIF1B beta, LMNA, gigaxonin, neurofilament light subtype, Rab7, and periaxin. | |
|
(3) Mutations in the gap junction protein connexin-32 lead to demyelinating or mixed phenotypes. As discussed at length in the CMTX1 article, connexin-32 forms gap junctions in regions of non-compact myelin, ie, the paranodes and Schmitt-Lantermann incisures, where it may provide a 1000x faster path for diffusion of ions and small molecules across the myelin sheath compared to the circumferential path along the Schwann cell cytoplasm. That such a dramatic delay in intracellular trafficking would interfere with cellular function, stands to reason. | |
|
(4) Mutations in the transcription factors EGR2 and SOX10 (central and peripheral demyelination) constitute another group. These gene products appear to regulate expression of other myelin genes, eg, MPZ, Cx32, myelin basic protein, proteolipid protein, and periaxin. | |
|
(5) Signal transduction and cell cycle associated proteins include GDAP1, MTMR2, SBF2, NDRG1, the neurotrophin receptor gene TrkA (NTRK1), SPTLC1. | |
|
(6) Genes and their proteins found to be responsible for other inherited neuropathies include enzymes. |
|
Disorder |
Gene |
Chromosome |
|
CMT1A |
PMP22 |
17p11 |
|
CMT1B |
P0 or MPZ |
1q22 |
|
CMT1C |
LITAF/SIMPLE |
16p13.1-12.3 |
|
CMT1D |
EGR2 |
10q21 |
|
CMT1F |
NF-L (NF-68) |
8p21 |
|
HNPP |
PMP22 |
17p11 |
|
NABP/HNA |
Unknown |
17q25 |
|
CMTX1 |
Cx32 |
Xq13 |
|
CMTX2 |
Unknown |
Xp22.2 |
|
CMTX3 |
Unknown |
Xq26 |
|
DI-CMTA |
Unknown |
10q24.1-q25.1 |
|
DI-CMTB |
DNM2 |
19p13.2-p12 |
|
DI-CMTC |
YARS |
1p34-P35 |
|
DI-CMTD and others |
PMP22, P0/MPZ, Cx32 | |
|
CMT2A1 |
KIF1Bb |
1p36.2 |
|
CMT2A2 |
MFN2 |
1p36.2 |
|
CMT2B |
RAB7 |
3q13-q22 |
|
CMT2B1 |
LMNA |
1q21 |
|
CMT2B2 |
Unknown |
19q13.3 |
|
CMT2C |
TRPV4 gene |
12q24.13 |
|
CMT2D |
GARS |
7p15 |
|
CMT2E |
NF-L (NF68) |
8p21 |
|
CMT2F |
HSPB1 |
7q11-q21 |
|
CMT2G |
Unknown |
12q12.q13.3 |
|
CMT2H |
GDAP1 |
8q21.3 |
|
CMT2I |
P0/MPZ |
1q22 |
|
CMT2J |
P0/MPZ |
1q22 |
|
CMT2L |
HSPB8 |
12q24 |
|
CMT2M |
DNM2 |
19p13.2 |
|
CMT2N |
Unknown |
16q21 |
|
CMT2O |
DYNC1H1 |
14q32.31 |
|
CMT2P |
LRSAM1 |
9q33.3-q34.1 |
|
CMT2Q |
DHTKD1 |
10p14 |
|
CMT2R |
TRIM2 |
4q31.3 |
|
CMT2S |
IGHMBP2 |
11q13.3 |
|
ARCMT-2T |
MME |
3q25.2 |
|
CMT2U |
MARS |
12q.13.3 |
|
CMT2V |
NAGLU |
17q21.2 |
|
CMT2X |
SPG11 |
15q21.1 |
|
CMT2W |
HARS |
5q31.3 |
|
CMT2Y |
VCP |
9p13.3 |
|
CMT2Z |
MORC2 |
22q12.2 |
|
DSS/HMSN3/CMT3 |
PMP22, EGR2, P0/MPZ, PRX | |
|
CHN |
PMP22, EGR2, P0/MPZ | |
|
CMT4A |
GDAP1 |
8q13-21.1 |
|
CMT4B1 |
MTMR2 |
11q23 |
|
CMT4B2 |
SBF2/MTMR13 |
11p15 |
|
CMT4B3 |
SBF1 |
22q13.33 |
|
CMT4C |
SH3/TPR domain protein of unknown function |
5q32 |
|
CMT4D (Lom) |
N-myc downstream-regulated gene 1 (NDRG1) |
8q24.3 |
|
CMT4E |
EGR2 |
10q21 |
|
CMT4F |
PRX |
19q13.1-q13.3 |
|
CMT4G |
HK1 |
10q23.2 |
|
CMT4J |
FIG4 |
6q21 |
|
CMT4H |
Frabin (FGD4) |
12p11.21-q13.11 |
|
HMN VII |
Unknown |
2q14 |
|
HSN IA |
SPTLC1 |
9q22.1-q22.3 |
|
HSNII |
HSN2 |
12p13 |
|
HSANIII |
IKBKAP |
9q31 |
|
HSAN IV/CIPA |
NTRK1, TrkA |
1q21-q22 |
|
HMSN-P |
Unknown |
3q13.1 |
|
HMSN-R |
Unknown |
10q23.2 |
|
Giant axonal neuropathy |
Gigaxonin |
16q21.4 |
|
CMT with pyramidal signs |
MORC2 |
22q12.1–q12.3 |
Traditionally, Charcot-Marie-Tooth disease has been divided into two categories of pathophysiology: a predominantly demyelinating process resulting in low conduction velocities (CMT1) and a predominantly axonal process resulting in low potential amplitudes (CMT2). However, even for CMT1, axonal damage may be more relevant to the disease manifestations and progression than demyelination and remyelination, and because axonal and myelin maintenance are inextricably related, pure forms are actually counterintuitive.
In some hereditary neuropathies (eg, HNPP and IBPN/HNA), focal asymmetric features predominate; in others (eg, certain PMP22 gene mutations in CMT1A, HMSNP, and IBPN/HNA), proximal weakness predominates. But typically, a predilection exists for distal limbs as the site of disease onset and for more severe symptoms and signs. Furthermore, although significant variation in nerve conduction velocities exists between and within families, velocities do not predict severity, with the exception of the very low velocities (ie, less than 5 m/sec) observed in Déjerine-Sottas syndrome and congenital hypomyelination neuropathy.
Axonal degeneration predicts disability. This suggests that, in most cases, axonal damage, not demyelination, is the root cause of the neuropathy (122). However, the gene mutations responsible for the different forms of CMT1 are clearly myelin-associated genes. Although the mechanisms are currently speculative, myelin disturbances result in axonal damage. This is not surprising given the strong evidence for interaction between myelin and axon gene expression in development and after experimental nerve injury. On the other hand, axonal damage can result in secondary demyelination.
Myelinating Schwann cells form a myelin sheath around a single axon and express high levels of myelin-related proteins and messenger RNA (mRNA). Axonal degeneration leads to Wallerian degeneration, in which myelin sheaths are phagocytosed, previously myelinating Schwann cells dedifferentiate, and mRNA expression is downregulated. When Schwann cells re-ensheathe axons, levels of proteins or mRNA, or both, increase. Myelin genes and products affected in this manner include myelin protein zero, PMP22, connexin-32, myelin-associated glycoprotein, myelin basic protein, EGR2, periaxin, and others.
Whether Schwann cells differentiate into myelinating or nonmyelinating (a misnomer because such Schwann cells myelinate more than one axon) phenotypes depends on axonal characteristics, which are determined, at least in part, by transcription factors such as Oct-6 (POU domain family) and EGR2.
Charcot-Marie-Tooth disease is the most common inherited neurologic disorder worldwide. In the United States, it affects approximately 150,000 people. An exhaustive study from Norway indicated a prevalence of 3.6 cases per 10,000 people (196). A worldwide meta-analysis estimated a prevalence of 1 case in 10,000 people (63). A Japanese epidemiologic study demonstrated a prevalence of 10.8 cases per 100,000 people (124).
Estimates of the frequency of CMT subtypes vary (63). CMT2 accounts for about 22% of autosomal dominant neuropathies, CMT1A accounts for some 60%, CMTX for about 16%, and CMT1B for approximately 1.6%. A Finnish study of 435,000 individuals found a prevalence of 16 cases per 100,000 people for HNPP and 20 cases per 100,000 people for Charcot-Marie-Tooth disease in general (147). The other forms are rarer (105).
Charcot-Marie-Tooth disease is found worldwide in people of all races and ethnic groups. Some rare CMT subtypes, particularly autosomal recessive forms, are restricted to particular racial groups, eg, CMT4D (Lom) (see Table 1). In the United States, Charcot-Marie-Tooth disease may be less common among African Americans. Whether this represents a lower frequency of the specific mutations or protection from disease manifestation through unknown disease-modifying genes remains unclear.
CMT subtypes may be inherited in an autosomal dominant, autosomal recessive, or X-linked pattern. Importantly, although CMTX (similar to other X-linked diseases) is usually more severe in men, it often produces overt disease in women. Woman can be severely affected, most likely because of unequal inactivation of the X chromosome (lyonization), which results in predominant expression of the abnormal connexin-32 allele in nerves. Charcot-Marie-Tooth disease may have a more severe phenotype in men, possibly because of environmental (nerve trauma) or X-linked neuroprotective factors, but, in practice, this impression is of little value because of the great phenotypic variability between and within families.
Not surprisingly, the autosomal recessive forms are rare; several only have been described in 1 to 3 families, often in small ethnic groups. However, as previously stated, recessive or compound heterozygous forms of Charcot-Marie-Tooth disease may also result in offspring of two parents with known or unknown Charcot-Marie-Tooth disease of any subtype.
Inherited neuropathies cannot be prevented at present unless affected parents choose not to have children. In addition, CMT1A occurs relatively frequently as a new mutation; thus, even if patients had no children, CMT1A would remain a prevalent disorder. As Charcot-Marie-Tooth disease does not usually affect life span, intellect, or independent living, most patients have children. Prenatal detection and screening may become easily available (52; 131; 24).
Secondary prevention focuses on awareness and avoidance of intercurrent medical problems or interventions that can lead to systemic or focal neuropathies, such as diabetes mellitus, hypothyroidism, vitamin deficiencies, neurotoxic drugs, carpal tunnel syndrome, and prolonged immobilization of limbs during surgery. Disease awareness on the part of the patient and health care providers is essential: For example, development of cancer may lead to the consideration of neurotoxic drugs such as platinum compounds or vincristine (86). At times, other treatment protocols may be equally effective, at other times not. This obviously also applies to many other conditions in which a potentially neurotoxic drug might be replaceable by one without this risk.
Patients should maintain a well-balanced diet and avoid obesity, which can contribute to back pain, spinal root disease, and certain entrapment neuropathies (meralgia paresthetica); naturally, carrying an overweight body is more of a strain on weakened muscles. Obesity and other causes of glucose intolerance are also particularly undesirable because of the risk of diabetic neuropathies.
Avoiding excessive alcohol use is important. Whether alcohol abuse alone without associated nutritional deficits leads to neuropathy is unclear; however, intoxication with alcohol or other drugs can result in nerve or other trauma. Although to our knowledge neurotoxic alcohol intake in patients with preexisting neuropathy has not been studied, common sense suggests that patients with Charcot-Marie-Tooth disease should consume less alcohol than unaffected individuals. One of the authors was confronted with the prospect of bariatric surgery for one of his DI-CMTC patients. Given the growing popularity of this approach, it is likely that a CMT patient (whose diagnosis may be known or unknown), will undergo one of the various forms of this surgery. One can hope only that extreme attention to nutritional issues will then be given, as is required regardless of the presurgical presence of a neuropathy.
Patients with Charcot-Marie-Tooth disease should lead as much of a full lifestyle as they can manage. As long as patients feel capable of performing activities, no clear reason exists for them not to do so. However, avoidance of exhaustion is important because of evidence that in Charcot-Marie-Tooth disease the intrinsic hand muscles of the dominant and, thus, more active hand may be weaker than those of the nondominant hand (221). Another caveat concerns HNPP patients, who, as much as possible should avoid work and recreational activities that can compress or otherwise injure nerves.
The differential diagnoses for Charcot-Marie-Tooth diseases include the following:
|
• Acute inflammatory demyelinating polyradiculoneuropathy |
Other problems to be considered. The first challenge for the clinician is to demonstrate that a patient's weakness and sensory loss result from peripheral nerve disease and not from abnormalities elsewhere in the nervous system. This can usually be accomplished by a clinical examination revealing distal weakness and muscle wasting, stocking-glove type sensory loss, and hyporeflexia. Pes cavus and hammertoes are stigmata of the disease and although nonspecific (as they occasionally occur in other forms of chronic acquired neuropathies), should raise the suspicion of Charcot-Marie-Tooth disease if the clinical context is appropriate. If the patient has a neuropathy and a positive family history, Charcot-Marie-Tooth disease is likely.
Nerve conduction velocities distinguish CMT1 from DI-CMT and CMT2, though significant variation may exist within families. Although exceptions exist, uniform conduction slowing distinguishes most type 1 cases from acquired disorders such as chronic inflammatory demyelinating polyneuropathies, in which conduction slowing typically varies along the same nerve and between nerves. Guillain-Barré syndrome also has asymmetric slowing and a more rapid onset. Dispersion and conduction block are rarely described in Charcot-Marie-Tooth disease and are more compatible with acquired neuropathies.
Finally, innumerable acquired disorders may cause neuropathy, including diabetes mellitus; alcohol abuse; monoclonal gammopathy; infections such as HIV, hepatitis C, leprosy, and Lyme disease; and renal disease. Medications should be evaluated when considering the possibility of inherited neuropathies in patients.
Laboratory studies. When considering an inherited neuropathy, the goal is to prove or refute this diagnosis and possibly to discover coexisting treatable conditions such as nerve entrapment and acquired neuropathy. Thus, the workup must address causes of neuropathies such as endocrine, infectious, and immunologic disorders, as well as vitamin and nutritional abnormalities or deficiencies, and nerve compression. Required screening tests include rapid plasma reagin, vitamin B12, folate, antinuclear antibodies, erythrocyte sedimentation rate, thyroid-stimulating hormone, and serum and urine protein electrophoresis.
Standard serum protein electrophoresis is too insensitive to identify small quantities of monoclonal proteins that may well be pathogenic; therefore, immunofixation electrophoreses is preferable. Similarly, clinically relevant vitamin B12 deficiency is not excluded by a reference range serum B12 of 170 ng/L or more (111 pM/L) for the radioimmunoassay and 250 ng/L or more (184 pM/L) for the chemiluminescent assay. When suspected, this test must be supplemented by methylmalonic acid and homocysteine levels, which are elevated in up to 1% of patients with B12 levels above 300 (197).
The cerebrospinal fluid is usually normal in Charcot-Marie-Tooth disease, but the protein may be elevated above 100 mg/dL. It is often high in Déjerine-Sottas syndrome, rarely in PMP22 deletion neuropathy. In a comparison of CMT1A, CMT1B, and CMTX, CSF protein (and CK) elevations were more common with myelin protein zero mutations (97). Cerebrospinal fluid analysis may be helpful in unclear clinical situations in which an acquired immune neuropathy is being considered.
Pedigree analysis. Clarifying inheritance patterns can narrow the differential diagnosis and eliminate the need for some genetic tests. Specifically, it is important to determine if there is male-to-male transmission, which essentially rules out X-linked disorders. On the other hand, the absence of a family history cannot be used to rule out a hereditary disorder because of the possibility of difficult ascertainment, variable penetrance, and expressivity, nonpaternity, and de novo mutations.
Genetic testing. When the clinical and neurophysiologic phenotype and the family history suggest Charcot-Marie-Tooth disease, the patient should undergo genotyping. This is important because clinical examination and electrodiagnostic study findings often cannot definitively establish a precise diagnosis due to the overlap between clinical syndromes and the significant variability between family members with an identical genotype. Genotyping permits sound genetic and prognostic counseling and advances the scientific understanding of phenotypes. The importance of genetic testing is exemplified by the report of two sisters with severe CMT1 and healthy parents, for whom autosomal recessive inheritance had been presumed until genetic testing identified low-level somatic and germline mosaicism of a myelin protein zero extracellular domain Gly74Glu mutation in the healthy mother, which she transmitted to her affected daughters (66).
Although fresh blood samples are routinely required for DNA analysis, a report documented that chromosomal changes of the PMP22 gene can be diagnosed in highly degraded DNA from sural nerve biopsy specimens that are up to 12 years old (20).
The limitations of genotyping must be recognized because they do not exclude mutations with 100% certainty. It should be emphasized that one only tests for known mutations; thus, negative results only tend to rule out the mutations one has tested for. Laboratory errors such as mislabeling occur. DNA can degrade in transit, and submission of another sample is sometimes required. If the result is counterintuitive, the test should be repeated with a new blood sample; typically, the testing laboratory does not charge for a second test. Until recently, the PMP22 gene was only tested for deletion and duplication; now the gene can be tested for point mutation, but this test must be requested separately. Point mutation analysis is limited to the open reading frame, ie, the protein coding sequences. It does not include a search for changes in promoter, enhancer, silencer, or other nontranslated sequences, which could result in too much or too little RNA.
De novo mutations are particularly common with PMP22, but they can occur with any gene. In other words, a general tenet holds for the family history: absence of evidence is not evidence of absence. Testing is possible only for mutations in known genes that are sufficiently common to make commercialization feasible, usually after three independent pedigrees have been identified. As indicated by the ever-increasing number of mutations, which makes a publication instantaneously obsolete, many additional genes exist that will be discovered in the future. Updated information is readily available at several web sites, including National Center for Biotechnology Information, GeneTests, and the Inherited Peripheral Neuropathies Mutation Database.
Electrodiagnostic studies. Slowed nerve conduction velocities in patients with peroneal muscular atrophy have been described independently by several authors (99; 126; 83). The concept of uniform slowing in inherited neuropathies (to differentiate them from acquired neuropathies) was later established by Lewis and Sumner (133). “Uniform slowing” suggests that all myelinated fibers are affected in the entire nerve length, in contrast to disorders such as CIDP, characterized by patchy involvement of different nerve segments. Conduction block and temporal dispersion, for example, are characteristic of acquired disorders and, therefore, are not ordinarily observed in inherited neuropathies.
Median motor nerve conduction velocities are below 38 m/sec in CMT1 and above 38 m/sec in CMT2, although some studies have proposed a cut-off of 42 m/sec. Nerve conduction studies in CMT2 typically reveal mild slowing, with median nerve velocities above 38 m/sec, reduced or absent CMAP amplitudes (< 4 mV), and reduced or absent sural nerve sensory action potential (SNAP) amplitudes (< 10 µV). Phrenic CMAP also shows reduced amplitudes. EMG reveals signs of chronic denervation.
The distinction between demyelinating and nondemyelinating CMT is not always clear. Relatively normal nerve conduction velocities have been reported in younger members of a family with a particular myelin protein zero mutation, whereas older relatives had severely slowed nerve conduction velocities (48). In studies of CMT1A, nerve conduction velocities have ranged from 10 to 42 m/sec, again illustrating that the distinction of CMT1 and CMT2 cannot rest on nerve conduction velocity findings alone. Conduction values are symmetric in CMT1, and few differences exist between proximal and distal nerve segments. Nerves often are refractory to stimulation or require higher amplitude and prolonged stimulation. Sensory nerve conduction velocities in all forms of CMT1 are reduced and often unrecordable. Sensory loss correlates with median sensory nerve conduction velocities and CMAP amplitudes.
EMG findings may be normal in proximal muscles but show distal changes with increased duration and amplitude motor unit potentials. Signs of active denervation such as increased insertional activity and fibrillation potentials are not prominent in muscles unaffected by weakness. Diffusely slow sensory nerve conduction velocities independent of nerve entrapment were found in another study, consistent with a background demyelinative polyneuropathy. Slowed motor conduction was less common in HNPP, although DML were frequently prolonged, indicating that a distal motor polyneuropathy is present, similar to that observed in IgM monoclonal gammopathy against myelin-associated glycoprotein or sulfated glucuronyl paragloboside.
Imaging studies. In demyelinating forms of Charcot-Marie-Tooth disease, MRI can demonstrate enlarged nerves, not only at the level of the spinal roots but also in the limbs. Occasionally, areas of demyelination are identified in the CNS, which should always be imaged in cases of symptoms or signs that cannot be attributed to the PNS. MRI of leg muscles may be a promising tool for the evaluation of the disease progression in CMT1A patients (76). In CMT2A, MRI demonstrated fatty infiltration of superficial posterior compartment muscles, whereas CMT1A was characterized by peroneal nerve innervated muscle involvement (41). MRI in CMT2 patients with dynamin 2 mutations showed calf muscle infiltration, often in a length-dependent fashion (75).
Conservative care. Currently, no medical therapy is capable of preventing the progression of Charcot-Marie-Tooth disease. Therefore, therapy should be focused on the management and prevention of the development of physical disability related to Charcot-Marie-Tooth disease.
Experimental approaches that may benefit humans in the future include the introduction of recombinant DNA encoding nl wild-type versions of mutated CMT genes into the nerves of knockout mice. Another approach explores neurotrophin gene transfer into the spinal cord to prevent secondary axonal changes in models of Charcot-Marie-Tooth disease.
Pain may result from joint deformities or compensatory overuse of certain muscle groups. Abnormal gait and scoliosis lead to back pain. Some types of pain may respond to nonsteroidal anti-inflammatory drugs. Dysesthetic pain may occur but is typically mild except in rare subtypes; it responds to antidepressants such as amitriptyline, desipramine, or paroxetine and to anticonvulsants such as gabapentin or carbamazepine. Patients suffer from leg and hand cramps. For a discussion of neuropathic or musculoskeletal cramping and other pain, see the article titled Treatment of neuropathic pain.
Surgical care. Depending on the degree of foot deformities, patients may benefit from Achilles tendon lengthening, tendon transfers, hammertoe correction, and release of the plantar fascia. However, such surgeries can often be prevented by conservative measures and lifelong follow-up with physical therapists. Patients should only be referred to orthopedic surgeons or podiatrists with specific training for foot surgery and experience with Charcot-Marie-Tooth disease. Similarly, because of concerns that the median and ulnar nerves may be more sensitive to manipulation in CMT patients, special caution must be exercised during entrapment surgery. The authors routinely refer patients for separate opinions from more than one surgeon. Orthopedic surgeons also play a role in the treatment of secondary joint problems at more proximal sites and in the evaluation and treatment of scoliosis.
Consultations. Referrals to physical therapists and prosthetics or orthotics specialists are often required to prevent and treat joint deformities. Orthotics and ankle-foot orthoses frequently enable patients to continue performing activities they enjoy while preventing falls that might result in broken ankles and other injuries that can severely limit future independence for the patients. In addition, orthotics and ankle-foot orthoses can prevent Achilles tendon shortening and extend near-normal ambulation. At times, boots can delay the need for such ankle braces. Multiple types of ankle-foot orthoses of different weights and sturdiness exist. With moderate foot drop, lighter ankle-foot orthoses may be more appropriate than with severe foot drop. Thick-handle tools and cutlery can render certain activities of daily living easier. For details, the reader is referred to the article titled Rehabilitation of peripheral nerve diseases.
Dealing with a life of initially mild, but typically worsening, disability can increase the risk of depression and lead to maladjustment. This should be borne in mind and addressed in patients of all ages, including teenagers. Referrals to mental health professionals may be indicated.
Genetic counseling is often indicated, particularly when affected individuals or unaffected individuals with an affected child contemplate procreation.
Numerous associated conditions can compromise pulmonary and upper airway function and sleep, some of which, including restless leg syndrome, sleep apneas, and vocal cord dysfunction, may respond to treatment (01).
Patient education. Patient education is an important aspect of the long-term management of patients with Charcot-Marie-Tooth disease. Education helps them cope with the progression of disability and leads to prevention of further nerve damage (eg, avoiding exposure to drugs or toxins with known deleterious effect on the peripheral nerves). Internet resources include sites such as the Charcot-Marie-Tooth Association in North America, which has patient support groups in most major cities, and the National Organization for Rare Diseases.
American patients should acquaint themselves with the Americans with Disabilities Act, and if necessary, seek advice on their rights in the workplace or mediation between employees and employers.
Parents should address issues of disability in their affected children with teachers and school counselors. A fine line exists between expecting too much from a child and being overprotective, for well-intentioned parents and teachers alike.
Special concerns. Patients, family members, and physicians must be aware of drugs that can affect the peripheral nervous system. Drugs with various degrees of nerve toxicity include the following:
|
• Adriamycin |
• Alcohol |
Some patients suffer exacerbations or rapid progression during pregnancy, usually, but not always, with recovery (185; 79). As with surgical procedures, prolonged positioning of the body and limbs in particular positions during delivery can result in nerve compression, which could make any underlying neuropathy worse. Furthermore, due to the variability of clinical manifestations, couples who both have symptomatic or asymptomatic Charcot-Marie-Tooth disease might have homozygous or compound heterozygous offspring with Déjerine-Sottas syndrome or congenital hypomyelination neuropathy.
In a series of 161 surgical procedures performed on 86 patients with Charcot-Marie-Tooth disease, the patients had no difficulty tolerating anesthetics, even with succinylcholine (04). However, in patients with progressive Charcot-Marie-Tooth disease, using succinylcholine may be inadvisable; nitrous oxide may prove neurotoxic by inactivating the cobalamin-dependent enzyme methionine synthase and causing B12 deficiency (120). Measuring B12 levels and treating a deficiency before nitrous oxide anesthesia may be indicated. Prolonged body and limb positions can result in nerve compression. Regional anesthesia is relatively contraindicated in Charcot-Marie-Tooth disease.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Florian P Thomas MD MA PhD MS
Dr. Thomas of Hackensack Meridian School of Medicine has no relevant financial relationships to disclose.
See ProfileFrancisco de Assis Aquino Gondim MD MSc PhD
Dr. Gondim of Universidade Federal Ceará, Fortaleza, Brazil, received consulting fees and travel grants from Daiichi Sankyo Brasil.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University received a consultant honorarium from Roche and Ovid Therapeutics.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jul. 16, 2026
Peripheral Neuropathies
Jul. 14, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
May. 12, 2026
Neuromuscular Disorders
Apr. 23, 2026
Peripheral Neuropathies
Apr. 09, 2026