














































Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Gaucher disease is a storage disorder caused by mutations in the GBA1 gene, which codes for lysosomal acid beta-glucocerebrosidase (glucocerebrosidase), resulting in accumulation of glucosylceramide (glucocerebroside). Type 2 (acute neuronopathic) and type 3 (chronic neuronopathic) Gaucher disease are in a phenotypic continuum of neurologic abnormalities with variable courses. Sudden unexpected death and unusual behavior may occur in Gaucher type 3, particularly in Egyptian patients. Enzyme replacement therapy has no effect on the neurologic complications of the disease. CSF glycoprotein nonmetastatic B (GPNMB) may be used to quantify neurologic involvement in Gaucher disease. Mutations in the GBA1 gene are the most common genetic risk factor for adult-onset, isolated Parkinson disease; multiple system atrophy; and dementia with Lewy bodies. Substrate reduction therapy, pharmacological chaperone therapy, and gene therapy for neuronopathic Gaucher disease are currently being tested or are in the advanced planning stages.
|
• Neuronopathic Gaucher disease has a very wide clinical spectrum--from congenital and early infantile Gaucher disease type 2 to very mild with horizontal supranuclear gaze palsy as the only neurologic abnormality and normal or even superior intelligence. | |
|
• Current treatment for the non-neuronopathic and for the chronic neuronopathic forms of the disease includes enzyme replacement, which targets only the non-neurologic aspects of the disease. | |
|
• Substrate synthesis reduction has shown to be effective in controlling the non-neurologic aspects of Gaucher disease, and the approach is being tried in Gaucher disease type 3 patients. | |
|
• Adult-onset, isolated Parkinson disease; multiple system atrophy; and dementia with Lewy bodies are not features of neuronopathic Gaucher disease. Rather, GBA1 mutations are a risk factor for developing these neurodegenerative diseases. |
The first example of Gaucher disease was documented in a patient with hepatosplenomegaly. The case was described in the doctoral thesis of Philippe C E Gaucher (1854-1918) (71).
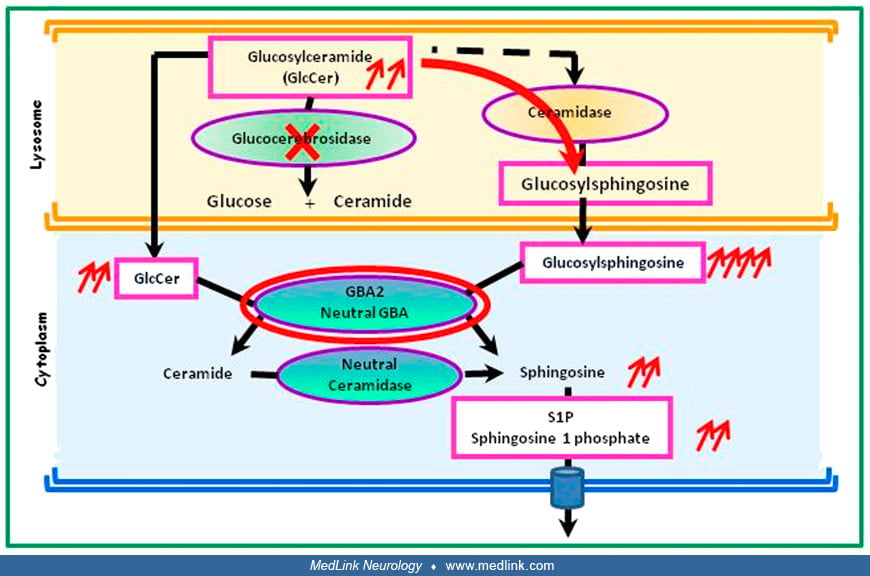
The expression of GCase varies from one cell type to another and depends on the tissue. In a mouse model of GCase deficiency (red cross), GlcCer is transformed via an alternative ceramidase pathway into glucosylsphingosine (red...
The disorder was diagnosed as an epithelioma of the spleen.
The characteristic appearance of storage in reticuloendothelial cells was noted as early as 1907 by German pathologist Felix Marchand (1846-1928) (119). The first step toward the description of the chemistry of the material accumulating in these cells evolved from the identification of its "lipoid" character by morphologists. Later, Viennese pathologist Emil Epstein (1875-1951) demonstrated that spleens from Gaucher patients yielded considerable amounts of an alcohol-soluble substance (57). (Epstein, a Jew, was dismissed from the teaching staff of the medical faculty of the University of Vienna in 1938, an effect of the annexation of Austria by the German Reich under "National Socialism.")
In 1924, Austrian medical analytical chemist Hans Lieb (1887-1979), working then as an unpaid Medical-Chemical Institute at the University of Graz (Medizinisch-Chemischen Institutes der Universität Graz), characterized this material as a cerebroside akin to the compounds described earlier by German-born British physician and biochemist Johann Ludwig Wilhelm Thudichum (known in Britain as John Louis William Thudichum; 1829-1901) (177; 112); Lieb was later promoted to full professor of applied medicinal chemistry, was appointed head of the Medical-Chemical Institute, and on three occasions was elected dean of the medical faculty.
Thudichum isolated and characterized numerous compounds of the brain, such as cephalin, sphingomyelin, galactose, lactic acid, and sphingosine. In 1884, he explained his findings in a publication titled "A Treatise on the Chemi...
In 1925, American pathologist William Bloom (1899-1972) wrote a long paper comparing the pathology of Gaucher disease and Niemann disease, for which he reviewed two cases of Gaucher disease (19; 164).
The correct identification of the sugar in the sphingolipid compound was not achieved until 1934, when French gynecologist Henriette Aghion (1906-1986) demonstrated that the lipid accumulating in the tissues of patients with Gaucher disease was a glucosyl, not a galactosyl, a derivative of ceramide (04).
The discovery of the lysosome as an organelle in 1955 by Belgian cytologist and biochemist Christian de Duve (1917-2013) and colleagues changed the definition of the storage disorders, earning de Duve a Nobel Prize in physiology or medicine in 1974. Within a short time, the first lysosomal storage disorder was described and was shown to be due to a deficiency of acid alpha-1,4-glucosidase (alpha-glucosidase) in a patient with Pompe disease (83). Other storage disorders quickly became recognized as diseases resulting from the lack of a degradative capacity—notably a lysosomal enzyme—with the expected lysosomal accumulation of substrate (84; 47). The accumulation of acid beta-glucocerebrosidase (glucocerebroside) was already well known in patients with Gaucher disease. Attention was focused on the possibility that the material accumulated because of a specific deficiency in its degradative pathway, leading to the description of the enzyme deficiency; in the mid-1960s, American biochemist Roscoe Owen Brady (1923-2016) and his colleagues at the U.S. National Institutes of Health identified the enzymatic defects in Gaucher disease (25; 26; 27; 28; 142; 24; 67).
It had been recognized that a variety of clinical disorders were related to glucocerebroside storage. Although these subtypes were originally thought to be distinguished by the relative amount of residual enzyme present (165; 196), this has been shown to be incorrect.
The discovery of the enzyme deficiency led to the development of several approaches to understand the biology of the lysosomes and to the development of enzyme replacement therapy to replace the missing gene product.
With the advent of alglucerase enzyme replenishment therapy (ERT) in 1991, the manufacturer (Genzyme Corporation) created the International Cooperative Gaucher Group (ICGG) Gaucher Registry to collect longitudinal observational "real word" information about Gaucher disease in heterogeneous patient populations to study the impact of phenotypes and genotypes on the natural history of Gaucher disease in untreated patients and to study treatment outcomes (191).
Note on nomenclature. This summary follows the current guidelines for gene and protein nomenclature and for mutation description. Please see Gene and mutation nomenclature for those guidelines. The enzyme, acid beta-glucocerebrosidase (GBA) is often referred to as glucocerebrosidase in the literature. The description of mutations in the older literature refers to the processed protein after excision of the 39 amino acid leader peptide. The current guidelines for mutation nomenclature stipulate that numbering begins with the leader peptide.
The presenting signs and symptoms of Gaucher disease vary throughout childhood (194).
(a) Dysphagia, apnea, difficulty with secretions. (b) Progressive. (c) Avascular necrosis, osteopenia, pathologic fractures. Presenting signs and symptoms of Gaucher disease are summarized Gary and colleagues; some would only b...
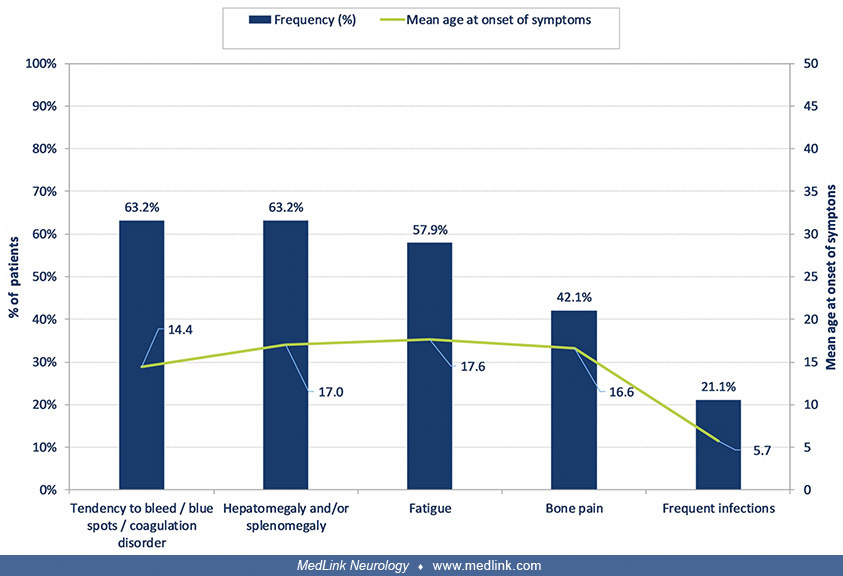
Common features include coagulopathy, hepatosplenomegaly, fatigue, bone pain, and frequent infections; frequent infections are common in childhood, whereas the others are typical of adolescence (122).
(n = 19) (Source: Mengel E, Gaedeke J, Gothe H, et al. The patient journey of patients with Fabry disease, Gaucher disease and mucopolysaccharidosis type II: a German-wide telephone survey. PLoS One 2020;15[12]:e0244279. Creati...
Based on clinical signs and symptoms, Gaucher disease has been divided into three subtypes: type 1, non-neuronopathic; type 2, acute neuronopathic; and type 3, chronic neuronopathic (106). All three types of Gaucher disease are caused by a deficiency of acid beta-glucocerebrosidase that results in the accumulation of glucocerebroside within the cells of the reticuloendothelial system. The principal differences between the subtypes are the presence of neurologic manifestations caused directly by glucocerebrosidase deficiency. Neuronopathic Gaucher disease is defined as the presence of neurologic involvement in a patient with biochemically proven Gaucher disease for which there is no explanation other than Gaucher disease (183). Because symptoms in all three types may begin in infancy, and the discrimination of subtypes depends on the evolution of clinical manifestations, especially nervous system involvement, this classification is more appropriate than one that includes reference to age of onset (ie, "adult," "infantile," and "juvenile" forms of Gaucher disease). The assignment of type, especially in children, should be made only after a careful examination for the presence and progression of related neurologic abnormalities or if a genotype study shows the presence of the N370S allele that typically is associated with Gaucher disease type 1.
Type 1: Chronic non-neuronopathic Gaucher disease. The age of onset and the severity of symptoms within this subtype vary widely and are not completely explained by the genotype. The diagnosis is frequently made later in life because of a lack of apparent clinical manifestations.
Although exceptions occur, painless splenomegaly with thrombocytopenia, anemia, and leukopenia are the usual initial signs. In many patients, these complications are not life threatening and may go unrecognized for years.
Although type 1 is characterized by the absence of primary neuronopathic involvement, the peripheral nervous system may be involved, and there may also be signs and symptoms of CNS disease according to a 2025 report of an expert panel (77). Abnormalities in saccadic eye movements may also occur in Gaucher disease type 1 (77).
Visceral abnormalities. Although hepatomegaly is often noted at the time splenomegaly is observed, the liver may not become enlarged until later in the course of the disease. Moderate hepatic dysfunction is discerned by elevated levels of liver enzymes in serum. Histologically, all patients have some degree of hepatic fibrosis (97). Hepatic failure can occur in untreated patients. Portal hypertension leading to esophageal varices is a recognized complication, occurring in patients with severe type 1 and type 3 disease.
Hematological abnormalities. Patients may have platelet counts below 50,000 without an accompanying bleeding diathesis. Conversely, some patients with Gaucher disease who have a normal prothrombin time, partial thromboplastin time, and platelet count (greater than 100,000) may have abnormal clotting times, excessive bruising, and unexpected perioperative bleeding. This variability in hematological features necessitates an individualized approach that may require the administration of platelets and fresh frozen plasma prior to and after surgical procedures.
Skeletal abnormalities. Complex degenerative skeletal changes are the leading cause of disability in patients with type 1 disease; these may include marrow infiltration, bone pain, osteopenia, fragility fractures, and recurrent avascular osteonecrosis (15). Dual-energy x-ray absorptiometry scans alone are insufficient for monitoring bone in Gaucher disease (77). Quantitative MRI techniques using Dixon quantitative chemical shift imaging have provided results that correlate with Gaucher disease severity scores, bone complications, and biomarkers for Gaucher disease bone involvement (77). Thoracic kyphosis is a common complication of Gaucher disease type 1 (77).
In a patient with Gaucher disease type 1 (GBA1 genotype p.Asn409Ser/c.217delC mutations), MRI revealed diffuse bone marrow signal abnormality throughout the left femur consistent with avascular osteonecrosis. (Source: ...
A few months after initial MRI, the patient developed avascular osteonecrosis in the right femur. (Source: Basiri M, Ghaffari ME, Ruan J, et al. Osteonecrosis in Gaucher disease in the era of multiple therapies: biomarker set f...
Some degree of osteopenia and osteolysis occurs in all patients, although the extent of bone disease is variable (197; 94).
Hematoxylin and eosin stain. Necrotic bone is characterized histomorphologically by a lack of osteocytes in the lacunae. (Photomicrograph by Nephron in May 2011 via Wikimedia Commons. Creative Commons Attribution 3.0 Unported [...
Hematoxylin and eosin stain. Necrotic bone is characterized histomorphologically by a lack of osteocytes in the lacunae. (Photomicrograph by Nephron in May 2011 via Wikimedia Commons. Creative Commons Attribution 3.0 Unported [...
Hematoxylin and eosin stain. Necrotic bone is characterized histomorphologically by a lack of osteocytes in the lacunae. (Photomicrograph by Nephron in May 2011 via Wikimedia Commons. Creative Commons Attribution 3.0 Unported [...
Radiologic abnormalities are typically extensive.
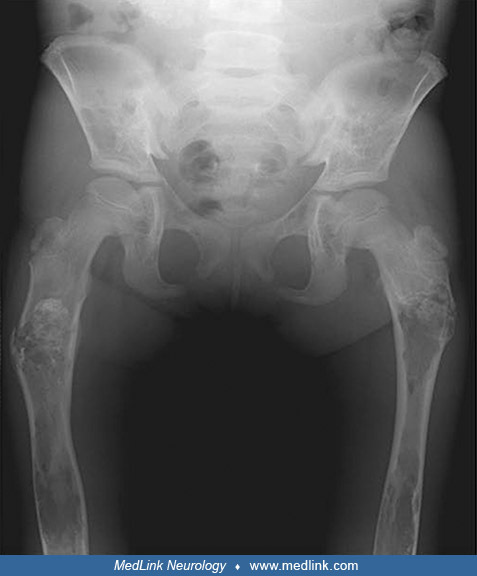
Generalized osteopenia and lytic areas in iliac wings and mid shafts of both femora. Pathological fractures both upper femora. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gau...
Generalized osteopenia and lytic areas in mid shafts of both femora and tibiae. Erlenmeyer flask deformity of both femurs, also known as metaphyseal flaring. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for...
Generalized osteopenia and lytic areas in humeri. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher disease. Orphanet J Rare Dis 2021;16[1]:431. Creative Commons Attributio...
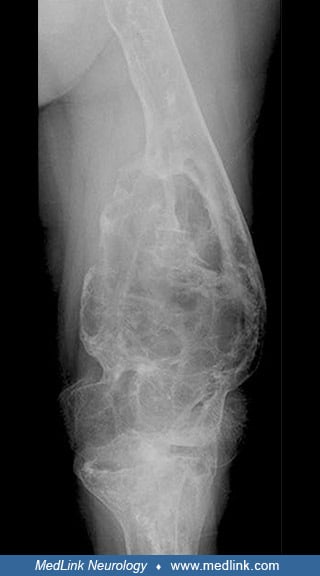
Gross expansion and coarse trabeculation of the left femur. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher disease. Orphanet J Rare Dis 2021;16[1]:431. Creative Commons ...
Extensive osteonecrosis of the right femoral head, which shows marked flattening with complete loss of joint space. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher diseas...
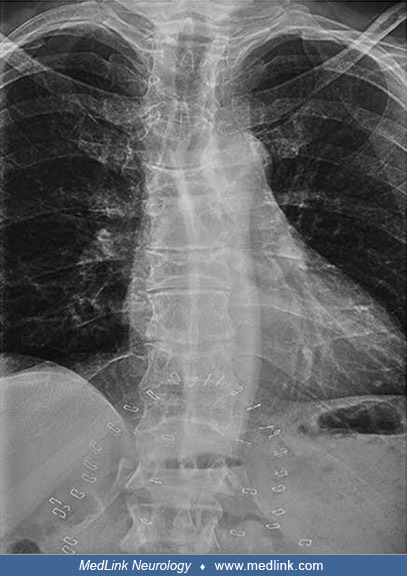
Prominent thoracic kyphosis and exaggerated lumbar lordosis with minimal lumbar curve. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher disease. Orphanet J Rare Dis 2021;1...
H-shaped vertebrae of the thoracic spine consistent with osteonecrosis. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher disease. Orphanet J Rare Dis 2021;16[1]:431. Creat...
Patients rarely have neither radiographic nor scintigraphic evidence of bone involvement. Most affected individuals have a substantial burden of bone disease that is progressive and leads to clinical presentation. Others have such severe involvement that they are confined to a wheelchair early in life because of pain, pathologic fractures, or skeletal instability. Many patients experience episodic pain in the hips, legs, back, and shoulders, referred to as "bone crises," lasting from days to months.
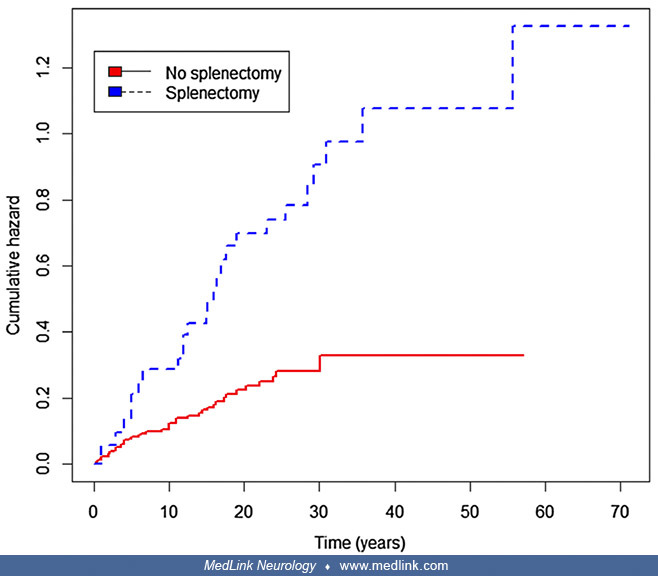
Splenectomy is associated with increased risks of osteonecrosis and fragility fractures (43). Controlling for gender, the hazard or risk of having a first osteonecrosis event after presentation of Gaucher disease is greater in patients who had splenectomy (hazard ratio of 3.32 [95% CI 1.74–5.00; p < 0.001]) (43).
Similarly, controlling for gender, the hazard or risk of having a first fragility fracture after presentation of Gaucher disease was also greater in patients who had splenectomy (hazard ratio of 2.83 [95% CI 1.33–5.99; p = 0.01]) (43).
The hazard or risk of having a first osteonecrosis event after presentation of Gaucher disease is significantly lower after starting enzyme replacement therapy (ERT) than before (hazard ratio of 0.20; 95% CI 0.11–0.38; p < 0.001) (43).
Horizontal axis shows time in years after presentation of Gaucher disease. (Source: D'Amore S, Page K, Donald A, et al. In-depth phenotyping for clinical stratification of Gaucher disease. Orphanet J Rare Dis 2021;16[1]:431. Cr...
In a study by D'Amore and colleagues, the effects of substrate reduction therapy (SRT) and bone marrow transplantation (BMT) were difficult to assess due to the small sample size (nine and two patients received substrate reduction therapy or bone marrow transplantation as first treatment, respectively) (43). Of the two patients with bone marrow transplantation, one had their first episode of osteonecrosis after the procedure.
Pulmonary features. Pulmonary complications can be identified through spirometry in up to 45% of patients with Gaucher disease type 1 (77). Pulmonary hypertension is a relatively common complication found in approximately a third of Gaucher disease patients, a direct consequence of acid beta-glucocerebrosidase storage that is worsened in splenectomized patients by the accumulation of storage cells in the lung. Fortunately, most cases are mild and subclinical with pulmonary artery pressures below 25 mmHg. The rare cases with severe pulmonary hypertension typically improve with enzyme therapy (110).
Ophthalmological abnormalities. Rare ophthalmological abnormalities may involve the vitreous, retina, cornea, uvea, and conjunctiva (52). Vitreous opacities are the most common and seen mostly in patients with Gaucher type 3 and sometimes require vitrectomy to ameliorate visual acuity.
Audiovestibular abnormalities. A retrospective analysis at a tertiary medical center identified 18 patients with Gaucher disease type 1 between January 2001 and September 2023 (139). Sensorineural hearing loss was detected in four of these patients, with bilateral involvement in two cases. The severity of hearing loss ranged from mild to moderately severe. Vestibular impairment, demonstrated by reduced vestibulo-ocular reflex (VOR) gain and catch-up saccades (CUS), was observed in one patient who also had mild bilateral sensorineural hearing loss.
Other. "Gaucheromas" may occur in patients with Gaucher disease type 1 (77); these behave like benign (ie, nonmetastasizing) neoplasms.
Type 2: Acute neuronopathic Gaucher disease. In contrast to the variability seen within type 1 Gaucher disease, type 2 is somewhat more uniform in its presentation (63). It has no ethnic predilection. The average age of onset is three months, and the presenting sign is usually massive hepatosplenomegaly.
A clinical categorization of Gaucher disease type 2 has been proposed (50). More information is available in Table 1.
|
Clinical Category |
Description |
|
Gaucher-related hydrops |
Infants stillborn or born with "collodion skin" and hepatosplenomegaly, often with contractures and evidence of hydrops fetalis. Fluid accumulations (eg, ascites, heart failure, tissue edema, effusions, polyhydramnios) compromise birth. Often born premature. |
|
Neonatal inflammatory |
Present in the first month of life (usually the first week) with hepatic involvement and a generalized inflammatory response. Typically have severe thrombocytopenia, elevated inflammatory markers (eg, ferritin), and hepatosplenomegaly. Many have "collodion skin" or ichthyosis. Likely represents an attenuated hydropic phenotype previously referred to as "lethal variant." |
|
Neonatal neurodegenerative |
Present in the first month of life with a predominant neurologic phenotype characterized by bulbar dysfunction, opisthotonos, mixed tone (typically spastic), and dystonia. |
|
Late infantile |
Present between 6 and 12 months of age with a predominant neurologic presentation (in the presence of systemic/visceral Gaucher disease) but distinguished from GD3 by the presence of global developmental delay and significant neurologic abnormalities before 2 years of age (eg, seizure disorder). Typically referred to as "GD2/3" or "2–3 intermediates." |
**Abbreviations: GD3, Gaucher disease, type 3; GD2/3 or 2–3 intermediates, intermediate between Gaucher disease, types 2 and 3.
Adapted from (50).
Neurologic abnormalities. Neurologic complications develop by 3 to 6 months of age. The presenting signs usually indicate involvement of cranial nerve nuclei and pyramidal tracts. A triad of clinical findings is very suggestive of the disease: (1) rigidity of the neck and trunk (which can progress to neck retroflexion and to frank opisthotonus); (2) bulbar signs (particularly poor suck and swallow reflexes with dysphagia and aspiration); and (3) oculomotor paresis (or bilateral fixed strabismus, particularly a convergent squint). These signs may be associated with microcephaly, trismus, hypertonia, hypokinesia, progressive spasticity, hyperreflexia, Babinski signs, and other pathologic reflexes. Seizures may occur. As neurologic deterioration proceeds, the child usually becomes apathetic and motionless. Death occurs either from apnea or aspiration pneumonia at an average age of nine months, with a range of 1 month to 2 years.
This form of Gaucher disease exhibits alterations in epidermal ultrastructure, which may provide an early and specific diagnostic tool (33).
Neonatal Gaucher disease. A rare form of Gaucher disease presents with hydrops fetalis and so-called "collodion skin" (congenital ichthyosis) (175; 168; 116).
The infant’s epidermal detachment ("collodion baby") is characteristic of perinatal Gaucher disease type 2. (Source: Sugiura T, Fujiwara A, Yo T, et al. Gaucher disease carrier with gestational thrombocytopenia and anemia: a ca...
The infant’s epidermal detachment ("collodion baby") is characteristic of perinatal Gaucher disease type 2. (Source: Sugiura T, Fujiwara A, Yo T, et al. Gaucher disease carrier with gestational thrombocytopenia and anemia: a ca...
A review of the clinical and hematological features of neonatal Gaucher disease found that this type is typically diagnosed based on postmortem pathological findings, with familial history suspected in 22% (123). Contrary to what is observed in type 2 disease, 22% of neonatal cases have dysmorphic features characterized by low-set ears, small nose with flat bridge, and anteverted nares. In a molecular analysis of 31 patients with this type of Gaucher disease, homozygosity for a recombinant allele that included the mutation p.Leu483Pro (formerly Leu444Pro) was associated with early lethality (168).
The characteristics of the clinical phenotype and the pregnancy are somewhat different between those newborns and fetuses affected by non-immune hydrops fetalis, in whom prematurity, fetal demise, and neonatal distress are prominent features. Hepatosplenomegaly is suspected prenatally in most cases with non-immune hydrops fetalis. Cardiomegaly has also been observed in some fetuses with Gaucher disease associated with non-immune hydrops fetalis (123).
Neonatal Gaucher disease should be distinguished from types 1 and 2. Type 1 cases are also diagnosed before 2 years of age and may have rapid progression in bone, liver, and spleen manifestations. CNS involvement must be established as an associated finding prior to making a diagnosis of type 2 disease.
Type 3: Subacute or chronic neuronopathic Gaucher disease. The clinical features of Gaucher disease type 3, apart from those referable to the nervous system, are common to Gaucher disease type 1. Patients may have a combination of systemic and neurologic abnormalities, including mild systemic disease with a progressive neurologic course, or severe skeletal and hematological abnormalities with a nonprogressive neurologic course. Hepatosplenomegaly is often the presenting feature rather than the neurologic abnormalities. In well-documented and biochemically proven cases, there is marked variation in age of onset and severity of organ involvement.
A clinical classification of Gaucher disease type 3, the chronic neuronopathic variant, has been proposed (51). See Table 2 for more information.
|
Clinical Category |
Criteria | ||
|
Attenuated |
Patients meeting criteria 1 OR 2 | ||
|
1. Minimal neurologic features AND slow or no evident progression | |||
|
(a) Saccadic eye movement defect ± a further isolated asymptomatic neurologic sign on examination, eg, subtle gait ataxia or hyperreflexia | |||
|
(b) Progression, if observed, is very slow, with stability demonstrated for periods of at least 10 years | |||
|
2. Late-onset neurologic features (after age 30 years) | |||
|
Intermediate |
Patients not meeting criteria for attenuated or severe disease | ||
|
Severe |
Patients meeting criteria 1, 2, OR 3 | ||
|
1. Death due to neuronopathic Gaucher disease before adolescence (< 12 years) | |||
|
2. Seizure onset in childhood (< 12 years) | |||
|
3. Global developmental delay, evidence of spinal deformity, and strabismus before 6 years of age | |||
Adapted from (51).
Neurologic abnormalities. A neurologic hallmark of Gaucher disease type 3 (and type 2) is marked slowing of horizontal saccades, sometimes labeled saccade initiation failure or oculomotor apraxia (183; 18). Vertical saccades are also slow, but to a lesser extent, and this deficit lags the slowing of horizontal saccades. Other oculomotor abnormalities include decreased gain of smooth pursuit and of the vestibulo-ocular reflex (30).
Other neurologic manifestations may include various degrees of intellectual disability or dementia, ataxia, dystonia (especially nuchal and facial), and, more rarely, tonic-clonic or complex partial seizures, or progressive myoclonic epilepsy.
Patients who are otherwise stable medically and neurologically, and who are on long-term enzyme replacement therapy, may develop partial complex seizures of temporal lobe origin that should not be confused with progressive myoclonic encephalopathy (07; 141).
There is significant variation in neurologic pattern among different ethnicities; for example, an oppositional behavioral abnormality is seen in Egyptian patients with Gaucher disease type 3 (02). Egyptian patients may also be more susceptible to sudden unexpected death, often with epilepsy, which has otherwise been rarely observed among patients with Gaucher disease (01). Certain GBA1 mutations are associated with myoclonic encephalopathy (eg, N188S), whereas L444P/L444P rarely, if ever, is (141; 107).
A group of patients with a neurodegenerative course and cardiac valvular calcification and stenosis are homozygous for the mutation in the acid beta-glucocerebrosidase (GBA1) gene (p.Asp409His, now annotated as NM_000157.3: c.1342G > C; p.Asp448His) (37; 108). These cases have been classified as type 3c. The neurologic features occur at an older age but are no different than those of typical type 3 disease.
The neuronopathic phenotypes appear to be part of a continuum of disease severity, and some patients may have intermediate phenotypes between types 2 and 3 (20; 75). Other reported cases have novel features that may defy easy categorization (93; 108).
Audiovestibular abnormalities. A retrospective analysis at a tertiary medical center identified 24 patients with Gaucher disease type 3 between January 2001 and September 2023 (139). Sensorineural hearing loss was detected in five of these patients, with bilateral involvement in three cases. The severity of hearing loss ranged from mild to moderate. Vestibular impairment, demonstrated by reduced vestibulo-ocular reflex (VOR) gain and catch-up saccades (CUS), was observed in four patients, two of whom also had mild to moderate hearing loss.
Pulmonary features. Pulmonary complications can be identified through spirometry in up to 55% of patients with Gaucher disease type 1 (77).
Skeletal abnormalities. Dual-energy x-ray absorptiometry scans alone are insufficient for monitoring bone in Gaucher disease (77). Quantitative MRI techniques using Dixon quantitative chemical shift imaging have provided results that correlate with Gaucher disease severity scores, bone complications, and biomarkers for Gaucher disease bone involvement (77). Thoracic kyphosis is a common complication of Gaucher disease type 3 (77).
Other. "Gaucheromas" may occur in patients with Gaucher disease type 3 (77); these behave like benign (ie, nonmetastasizing) neoplasms.
The Gaucher/Parkinson disease phenotype. GBA1 mutations are a strong genetic risk factor for synucleinopathies (Parkinson disease and Lewy body dementia) (151; 161). Patients with Gaucher disease and parkinsonism have typical Lewy bodies in hippocampal regions susceptible to Gaucher-related neurodegeneration (198).
A large international collaborative study of 16 centers found significantly higher odds of 5.4 for any GBA1 mutation among 5691 patients with Parkinson disease compared with 4898 controls (161). Moreover, patients with a GBA1 mutation presented earlier with Parkinson disease, were more likely to have affected relatives, and were more likely to have atypical clinical manifestations. Although the centers varied in their ability to detect GBA1 mutations, all 16 centers could detect two particular GBA mutations: L444P and N370S. Among Ashkenazi Jewish subjects, either mutation was found in 15% of patients (n=780) and 3% of controls (n=387), whereas among non-Ashkenazi Jewish subjects, either mutation was found in 3% of patients and less than 1% of controls. GBA was fully sequenced for 1883 non-Ashkenazi Jewish patients, and mutations were identified in 7%, demonstrating that limited mutation screening may identify less than half of the mutant alleles.
A prospective cohort study of patients with Gaucher disease and GBA1-mutation carriers found that, over a 2-year period, GBA-mutation-positive individuals show deterioration in clinical markers consistent with the prodrome of Parkinson disease (16).
Even patients with Gaucher disease type 1 (ie, chronic non-neuronopathic Gaucher disease) are at a higher risk of developing Parkinson disease (31).
Although loss of function of acid β-glucocerebrosidase caused by GBA1 mutations presumably contributes to the increased risk of synucleinopathies, the pathophysiological mechanism is not clear (68). Even in the absence of GBA1 mutations, patients with Parkinson disease have lower glucocerebrosidase activity in peripheral blood (06) and in the substantia nigra (72; 36), suggesting that loss of glucocerebrosidase function, whether due to genetic or nongenetic causes, contributes to the pathogenesis of Parkinson disease. Proposed mechanisms include (1) apoptosis induced by a bidirectional pathogenic loop between glucocerebrosidase deficiency and α-synuclein (121); and (2) endoplasmic reticulum stress in susceptible neurons caused by glucocerebrosidase deficiency (118). In addition, patients with Parkinson disease carry a significant burden of other rare lysosomal storage disorder gene variants, implying that multiple genetic hits may act in combination to degrade lysosomal function, thereby enhancing Parkinson disease susceptibility (149).
Among 4190 adult Gaucher disease patients from 85 studies, 555 exhibited neurologic symptoms in adulthood (92). The median age at evaluation was 47 years, median age at neurologic symptoms onset was 44 years, and median age at Gaucher disease clinical onset was 23 years. Parkinsonism was the most reported neurologic manifestation. Other symptoms and signs included oculomotor abnormalities; peripheral neuropathy; seizures; myoclonus; and cerebellar, cognitive, and psychiatric symptoms. The genotype N370S/N370S mostly presented with parkinsonism, whereas the L444P variant presented earlier with severe neurologic symptoms.
Death in patients with Gaucher type 1 who have never received enzyme replacement therapy occurred at a median age of 66 years and was often caused by liver disease, septicemia, malignancies, and suicide (192).
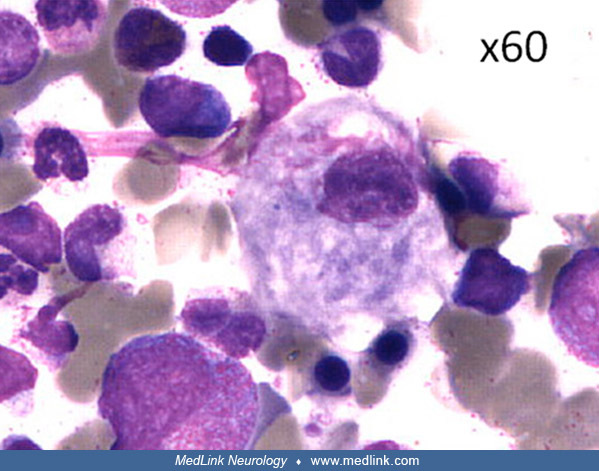
So-called "Gaucher cells" are macrophages that become full of unprocessed glucocerebroside in patients with Gaucher disease. Gaucher cells accumulate primarily in the spleen, liver, and bone marrow, causing organ inflammation and dysfunction.
The risks of monoclonal gammopathy of undetermined significance (MGUS), multiple myeloma, and non-Hodgkin lymphoma are elevated in patients with Gaucher disease compared to the general population, and strong evidence indicates that lyso-Gb1 stimulates the formation of monoclonal immunoglobulins (M-protein) in patients with Gaucher disease (77).
Visceral involvement.
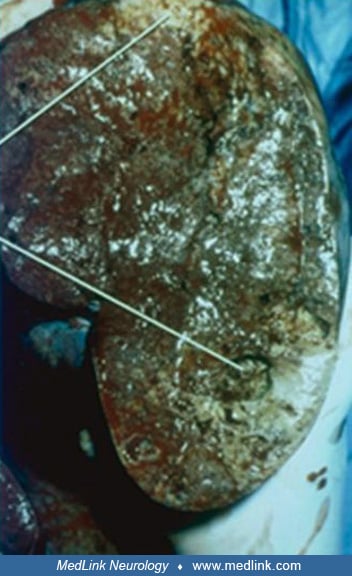
The lymphoreticular system. Painless splenomegaly is usually the earliest sign in all types of Gaucher disease. Even when the spleen is not palpable by physical examination, it usually can be demonstrated to be enlarged by diagnostic imaging techniques. The rate of enlargement of the spleen is helpful in judging the rate of progression of the disease. The preferred modality for measuring organ volume is volumetric MRI (34). The rate of splenic enlargement is often consistent for each case. Changes in that rate have been associated with malignancy or other intercurrent disease. Spontaneous rupture of the spleen is uncommon. Most cases develop hypersplenism manifested by pancytopenia and a bleeding diathesis. Red cell and platelet survival time is shortened. Splenectomy should be limited to those patients having severe bleeding diathesis, high-output cardiac failure, or mechanical interference of bowel, diaphragm, or kidney. The spleen is enlarged and firm, with pathological findings of infarcts, fibrosis, and distortion of the splenic architecture. Splenic infarction can cause abdominal pain and should be considered in the differential diagnosis of an acute abdomen in patients with Gaucher disease. The red pulp of the spleen is replaced by white collections of Gaucher cells. The surface and body of the spleen may contain dark-purple nodules that are foci of extramedullary hematopoiesis. Splenectomy is almost always followed by correction of cardiac and hematologic abnormalities. However, splenectomy poses a risk for severe, overwhelming sepsis that can be fatal within a day. For these reasons enzyme replacement therapy is the treatment of choice.
In some patients, lymph nodes may be enlarged and contain Gaucher cells. The thymus, Peyer patches in the intestine, and the pharyngeal tonsils are frequently affected.
Liver. Despite the frequent occurrence of hepatomegaly and abnormal liver function tests in Gaucher disease, hepatic failure occurs infrequently. In contrast to other lipidoses (eg, Niemann-Pick disease), hepatocytes are not involved in storage. Gaucher cells are seen within the sinusoids. In the more severely affected cases with cirrhosis, fibrosis distorts the architecture, forming small regenerating nodules that are infiltrated by Gaucher cells. Marked portal hypertension and consequent complications (eg, ascites and esophageal varices) are common in severe disease. The enlarged liver may contain sites of extramedullary hematopoiesis. Recurrent bleeding from esophageal varices has been successfully treated with a combination of aggressive medical management and sclerotherapy. Jaundice is a serious adverse prognostic sign in this disease and represents either intercurrent infectious hepatitis or hepatic failure. In cases with liver involvement, early intervention with enzyme replacement therapy is indicated. Hepatic transplantation is required in patients with end-stage liver disease.
Skeletal manifestations. In patients with Gaucher types 1 and 3, skeletal abnormalities on x-ray occur in 50% to 75% of patients and include expansion of the cortex in the distal femur (Erlenmeyer flask deformity), fractures, and other abnormalities of the acetabulum and head and neck of the femur. Prosthetic hip replacement is often necessary for patients to remain ambulatory. In some cases, the hip lesions may be confused with Legg-Calve-Perthes disease. Destruction of vertebral bodies may produce collapse, gibbus formation, and spinal cord or nerve root dysfunction.
Bone pain and bone crises. Bone infarction causes so-called "bone crises." This process affects the femoral heads and distal femur more frequently than other bones. Episodes are usually self-limited and typically last approximately 2 weeks but may be protracted over many months. During the first days of crisis, patients may require hospitalization to control pain. Bed rest is always indicated until the episode is completely resolved. Typically, a region of osteosclerosis, bone deformity, or pathologic fracture develops. Although the infarcted region can become secondarily infected, producing osteomyelitis, bone crises should be treated conservatively unless infection is highly suspected. Instrumentation of infarcted bone should be avoided unless clearly indicated because instrumentation may cause secondary infection and development of a sinus tract. Gallium scans may be helpful in distinguishing infarction from osteomyelitis, whereas x-rays and technetium scans are usually not helpful.
Adequate calcium intake should be maintained. If urinary calcium is low, the diet should be fortified with calcium and vitamin D. In addition, the storage of glucocerebroside in tissue macrophages may alter the generation of competent osteoclasts and result in a failure to maintain a healthy bone matrix.
A rare occurrence in type 1 disease is the extraosseous extension of Gaucher cells to the surrounding soft tissues, a condition that may mimic a malignant process (144). Because the incidence of several malignancies, including lymphoproliferative disorders, is increased in Gaucher disease (159), a biopsy may be required for an accurate differential diagnosis.
The hypothesis that bone crises are the result of progressive vascular compromise produced directly by occlusion of vessels by Gaucher cells is not supported by scintigraphic or histologic studies. In fact, perfusion scans of bones are often enhanced. Vascular occlusion by Gaucher cells would not explain the combination of osteopenia, osteonecrosis, and osteosclerosis seen in the disorder. Other vascular complications, such as premature stroke, myocardial infarction, or renal failure, are not features of the disease, making a simple vascular occlusive process a less attractive explanation of the skeletal involvement. Metabolic and endocrinologic studies suggest an imbalance in calcium homeostasis; however, the entire skeleton is not affected uniformly. On the contrary, the lesions consist of collections of Gaucher cells scattered throughout the bone substance. Bone complications probably result from a toxic process around these foci, which then leads secondarily to edema, vascular compromise, and infarction. One study reported a correlation between the severity of type 1 disease and serum levels of macrophage derived cytokines. This correlation suggests that a cytokine imbalance may have a role in the pathophysiology of the bone lesions (87). From existing information, it is not likely that infarction is incited by vascular occlusion by Gaucher cells, but rather by the vascular compromise alluded to above.
Progressive kyphosis (sometimes with scoliosis) without any vertebral body abnormality is commonly observed in patients with Gaucher type 3. It is very rarely seen in patients with Gaucher type 1. Correction and stabilization of the kyphosis is required, and Harrington rods are often placed towards the end of the growth period (late adolescence). The mechanism of this phenomenon is unknown, but studies in a zebrafish model suggest a developmental abnormality of bone (206).
Growth retardation and delay in skeletal maturation occurs in children but is normalized by enzyme replacement therapy (100).
Other organic systems. In the kidney, Gaucher cells can be found in the cortex, medulla, or glomeruli. Renal function in Gaucher patients is usually normal. In a few cases, proteinuria or hematuria has been reported. The cause of these signs is uncertain, but they have been attributed to the infiltration of Gaucher cells.
Focal collections of Gaucher cells are found within Peyer patches and the lamina propria of the gastrointestinal tract. Patients may complain of bloating, cramps, and diarrhea. Abnormal uptake of calcium may be one of several factors affecting bone in the disorder.
The cherry-red spot that appears in the macula in some sphingolipidoses and mucolipidoses does not occur in Gaucher disease; however, white patches containing Gaucher cells have been seen in the vitreous and retina of some Gaucher type 3 patients, and they can develop while the patient is on enzyme replacement therapy (153). A case has been reported in which loss of vision due to uveitis in type 1 disease could be reversed by enzyme replacement therapy (21). There have been many reports suggesting that patients with Gaucher disease are at increased risk of developing malignancies, particularly hematopoietic tumors. A large study of 1525 patients found 2- to 3-fold risks of non-Hodgkin lymphoma, malignant melanoma, and pancreatic cancer in patients with Gaucher disease but no significant association between Gaucher disease and cancer in general or with other specific malignancies, such as multiple myeloma (109).
Because of the different clinical courses of patients with Gaucher disease, researchers have tried to develop a laboratory test that would both discriminate subtype and be useful in determining prognosis. Genotype analysis gives some guidance in this area but is not completely satisfactory. Most individuals with even one p.Asn409Ser allele do not have neurologic involvement, whereas almost all patients with the p.Leu483Pro allele have neurologic involvement. Differences in epidermal cells may provide a means to differentiate type 2 neuronopathic Gaucher disease from types 1 and 3 Gaucher disease (64; 160). Disorganized lamellar membranes in the epidermal stratum corneum are present in type 2 patients but not in type 3 patients (89; 33).
Garvey and colleagues observed that the amplitude of stretch-evoked somatosensory evoked potentials in patients with type 3 Gaucher disease positively correlated with the degree of cognitive deficit and, therefore, with neurologic disease burden (69). It is possible that measurement of somatosensory evoked potentials may be useful to monitor the response of future therapies aimed at correcting the neuropathology of Gaucher disease.
Case 1. A 5-year-old girl experienced a bladder infection that became obvious when she suddenly began to have toileting accidents. The infection was treated with sulfa drugs and cleared up initially but reoccurred 6 months later. Primary care evaluation disclosed splenomegaly and a low platelet count. She was diagnosed with a platelet disorder and treated with prednisone. Several days into the steroid treatment, her platelet count was found to be very low, and a bone marrow biopsy was performed. The biopsy showed Gaucher cells. Follow-up testing showed enzyme assay of 2 (NR 12-17) and genotype p.Asn409Ser/p.Leu483Pro.
Follow-up and discussion. The p.Asn409Ser mutation is only associated with type 1 Gaucher disease. When p.Asn409Ser occurs with a second different mutation like p.Leu483Pro, the disease sometimes presents earlier in life. This patient had two brothers, and because neither of them had symptoms of Gaucher disease, they did not have genetic testing. When a genetic condition, such as Gaucher disease, is known in a family, the brothers and sisters of a person with the condition are at higher risk for having the same condition and for being carriers. For this reason, it is important for parents to educate all of the children in an affected family about the condition. Many parents want and need help with this process. Genetic counseling is recommended for each family member when he or she becomes an adult (or before if indicated).
Case 2. At 4 months of age, the parents of an infant boy noted "something not quite right with his eye movement" and mentioned this to their pediatrician at his 6-month well baby visit. At this visit, the pediatrician noticed splenomegaly and heard a report from the parents that the boy had a choking episode during which he became cyanotic and was taken to the ER. Developmentally, the infant was meeting expected milestones and at 6 months was sitting, reaching for and grasping objects, and responding to his name. The pediatrician ordered a panel of metabolic tests including acid beta-glucocerebrosidase.
|
Physical examination | |
|
• Liver approximately two times normal size | |
|
Laboratory findings | |
|
• Platelet count 125,000 | |
Follow-up and discussion. The boy was treated with enzyme replacement therapy at a dose of 120 U/kg every 2 weeks. He did well for several months; he was walking well and saying a few words. Eventually, he failed to progress and began having more frequent choking episodes. He died during the night at age 22 months.
Case 3. A 20-month-old Caucasian boy presented with the following history: at his 12-month well-child checkup, the pediatrician noted hepatosplenomegaly. The hepatosplenomegaly was unchanged on follow-up at 15 months. At 18 months he was seen because of irritability and fever that had persisted for approximately 1 month. At that time, increased hepatosplenomegaly and cervical lymphadenopathy were noted. He was admitted to the hospital, at which time he also had hoarseness and diarrhea. He was evaluated for possible malignancy with results that included the following: chest x-ray, normal; abdominal x-ray, hepatosplenomegally; fragmented right femoral epiphysis; cervical lymph node biopsy, normal; bone marrow aspirate, Gaucher cells.
|
Physical examination | |
|
• 85 cm, 10.5 kg | |
Follow-up and discussion. Over the next few months, it became apparent from examination and parents’ observations that he had horizontal supranuclear gaze palsy with compensatory head thrusting. Treatment with enzyme replacement therapy was initiated at a dose of 60 U/kg every 2 weeks. Years later at age 11 years, he had no progression of the slow looping saccadic eye movements and no organomegaly or obvious bone disease; he played baseball and football, was an A and B student, and had the lead in the school play during the previous academic year.
Case 4. A girl was born to unrelated, healthy parents after an uneventful pregnancy and delivery (85). Bilateral esotropia was noted in early infancy, and she was referred to an ophthalmologist at her 4-month check-up. Recurrent dysphagia and cyanosis started at 11 months of age. Hypotonia, growth arrest, and psychomotor retardation were recognized by 12 months of age. At 15 months of age, she was evaluated for acutely worsening stridor and cyanosis. Bronchoscopic examination revealed laryngeal spasms, which required endotracheal intubation. She had bilateral ptosis, esotropia, and marked hepatosplenomegaly; liver and spleen were palpable 5 cm below the costal margins. Blood gas analysis excluded metabolic acidosis. Blood tests showed thrombocytopenia (103 × 109/L) and marginally elevated aspartate transaminase. NH3, alanine aminotransferase, and creatine kinase levels were unremarkable. Serum angiotensin-I converting enzyme and acid phosphatase levels were increased to 76.2I U/L (reference range 8.3–21.4) and 7150 (reference range 120–420), respectively. Echocardiography showed no abnormality. Bone marrow aspiration identified Gaucher cells. Within a few days after admission, opisthotonic postures occurred repeatedly, with or without sensory stimulation. Serial cranial MRIs found no degenerative brain lesions. Genetic testing identified compound heterozygous pathogenic variants p.L483P (formerly defined as p.L444P) and p.R502H (p.R463H) in GBA1.
Infusion of imiglucerase (60 units/kg every other week) and oral administration of ambroxol (3 mg/kg/day) was started from day 20 after admission. The dose of ambroxol was increased by 3 mg/kg every 2 to 4 weeks to reach the target doses (30 mg/kg/day) (131).
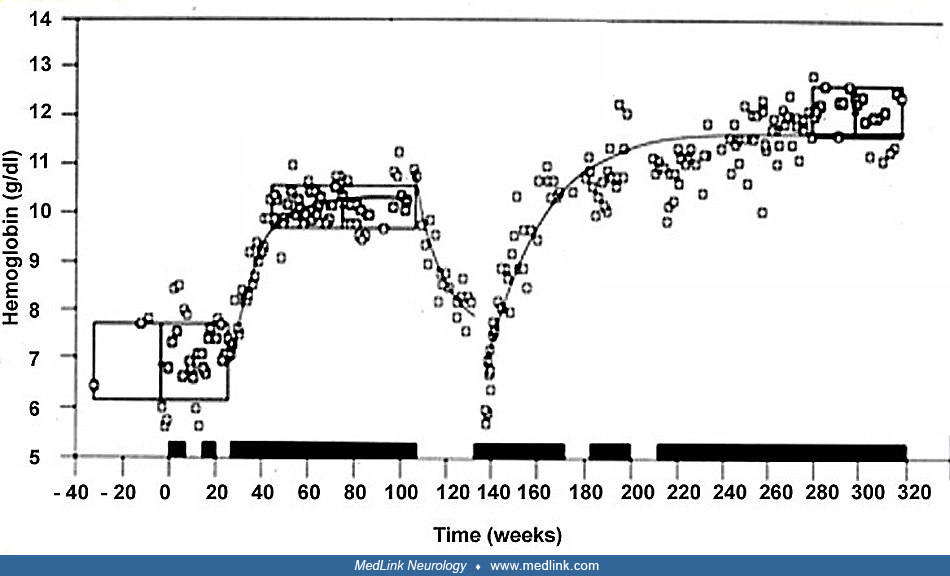
After starting imiglucerase and ambroxol therapy, she began to roll over in 1 week and to sit unaided in 2 weeks. Over this interval, spasticity and opisthotonus disappeared. Platelet counts increased to 150-200 × 109/L within 2 months of starting treatment. Because of prolonged dysphagia and laryngospasm, tracheosatomy was performed on hospitalization day 9. Given her chronic dyspepsia, the imiglucerase dose was increased to 80 units/kg every other week from 19 months of age. She then underwent laryngotracheal separation to control recurrent airway infections at 27 months of age and esophageal hiatal hernia repair at 3.5 years of age.
Serial monitoring of molecular markers supported the efficacy of treatment: (1) increased angiotensin-I converting enzyme levels declined from 76.2 U/L on admission to 50.5 U/L in 1 month and to 20.5 U/L at 2.5 years of age; (2) tartrate-resistant acid phosphatase 5b (TRACP-5b) was initially as high as 7150 mU/dL (reference range 120–420) but declined to 1770 mU/dL during treatment; and (3) serum glucosylsphingosine (Lyso-Gb1) declined from 721 ng/mL at the beginning of treatment to 130 ng/mL after 5 months of treatment.
At 3.75 years of age, she was walking independently, and her cognitive function had reached a developmental age of 12 to 16 months. Her health-related quality of life score markedly improved from before treatment to 2 years after starting ambroxol therapy.
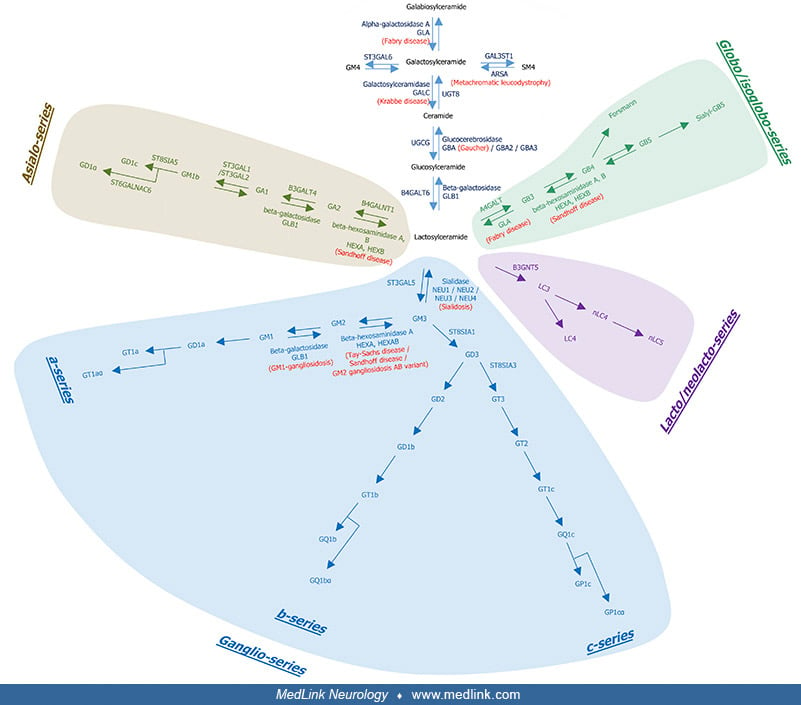
Gaucher disease in the spectrum of glycosphingolipid diseases. Glycosphingolipids are composed of a sphingosine, a fatty acid chain (these two forming a ceramide) and a carbohydrate moiety (17).
The fatty acid attached to the sphingosine may vary in chain length, degree of unsaturation, or hydroxylation (17). Glycosphingolipids include ceramide, cerebrosides (galactosylceramide and glucosylceramide), lactosylceramides, and gangliosides.
The biosynthetic and degradation pathways of glycosphingolipids in the brain are complex and incorporate metabolic loci for Fabry disease, metachromatic leukodystrophy, Krabbe disease, Gaucher disease, Sandhoff disease, Sialidosis, Tay Sachs disease/Sandhoff disease/GM2 gangliosidosis AB variant, and GM1 gangliosidosis.
This diagram shows the metabolic loci for Fabry disease, metachromatic leukodystrophy, Krabbe disease, Gaucher disease, Sandhoff disease, Sialidosis, Tay Sachs disease/Sandhoff disease/GM2 gangliosidosis AB variant, and GM1 gan...
Glucosylceramides and galactosylceramides are hydrolyzed respectively by glucocerebrosidase (also named glucosylceramidase; the GBA gene encoding lysosomal glucocerebrosidase is associated with Gaucher disease) and galactosylceramidase (encoded by the GALC gene; that deficiency causes Krabbe disease) to regenerate ceramides.
Ceramides are further deacetylated to sphingosines that can be broken down or recycled for sphingolipid synthesis by the salvage pathway.
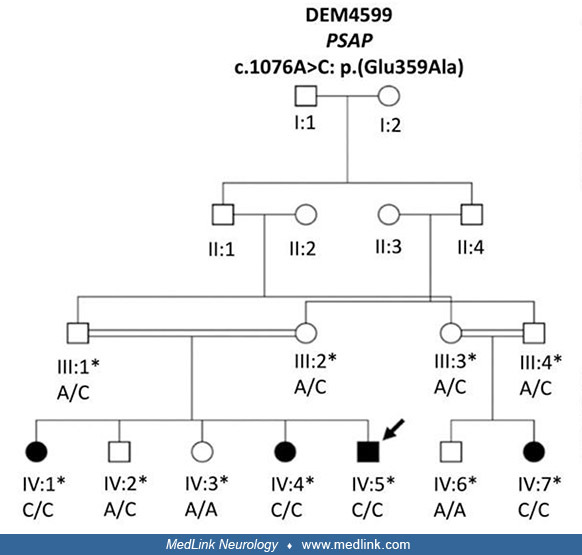
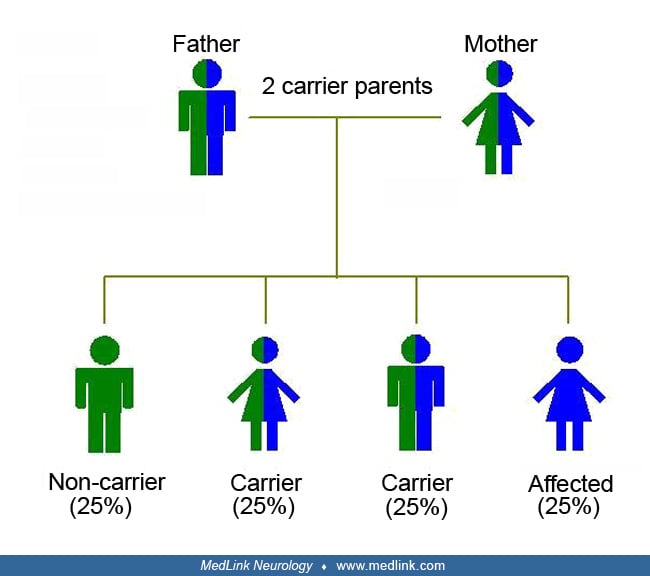
Genetics of Gaucher disease. Gaucher disease is a lysosomal storage disease that is transmitted as an autosomal recessive trait.
Circles represent females and squares males. Filled symbols indicate affected family members and clear symbols unaffected members. Double lines in the pedigree represent a consanguineous marriage. Asterisks indicate individuals...
Gaucher disease results from homozygous or compound heterozygous mutations in the gene encoding acid beta-glucosidase (GBA; OMIM 606463) on chromosome 1q22.
The wide spectrum of clinical severity in patients with Gaucher disease, particularly in Gaucher type 3 patients, is often explained by the presence of genetic modifiers. An interesting effort used animal models to identify such genes (105). The authors identify a number of variants that modify the lifespan of the affected mouse. The most interesting was Grin2b, which codes for NR2B subunit of the N-methyl-D-aspartate receptor. Pharmacological blocking of the N-methyl-D-aspartate receptor using memantine led to a significant prolongation of the Gaucher mouse model (105). Effort to identify genetic modifiers in patients with Gaucher type 3 is underway.
The precise genetic and biochemical reasons for the manifestations of the disease and the phenotypic differences among patients are not completely understood. There is little doubt that additional genetic modifiers of Gaucher disease cause marked differences in the presentation of signs and symptoms related to bone marrow, liver, spleen, lung, bone, brain, and other systems involved in the disease (73).
Acid beta-glucocerebrosidase. In addition to the 5' and 3' untranslated regions, the cDNA of the GBA gene contains 1548 base pairs encoding human acid beta-glucocerebrosidase. The molecular weight of acid beta-glucocerebrosidase calculated from the 536 amino acids deduced from the cDNA sequence is 59,716, which is in good agreement with that estimated by SDS-polyacrylamide of the product of in vitro translation of human placental mRNA (59; 58). Five potential glycosylation sites (Asp-X-Ser/Thr) were identified. Quantitative carbohydrate analysis indicates that only four are glycosylated (173). This has been confirmed by direct analysis of the amino acid sequence (B Martin, unpublished data). In addition to the amino acid sequence of the structural protein, the reading frame of the cDNA codes for 39 additional amino acids upstream of the amino terminus. This signal polypeptide contains a hydrophobic core, consisting of Gly-Leu-Leu-Leu-Leu and, in addition, has glycine at the peptidase cleavage site. These features are consistent with the properties of signal peptides of other translocated proteins. Furthermore, the cDNA sequence confirms the presence of a 2-kd signal sequence identified by pulse-labeling studies. More than 300 mutations of the GBA gene have been described as a cause of Gaucher disease, including missense or nonsense mutations as well as splice mutations, deletions, and complex gene rearrangements.
Biochemical abnormalities in Gaucher disease.
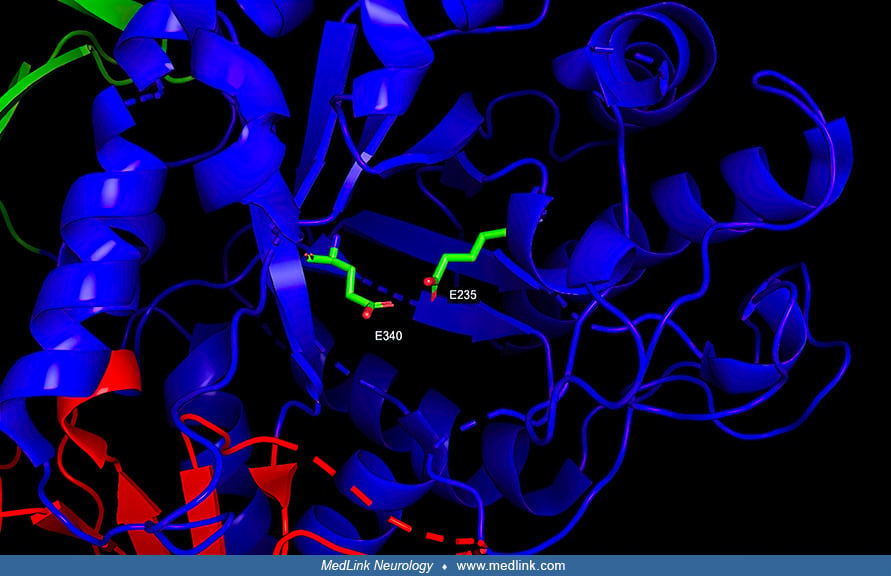
Metabolic block and storage substance. With all lysosomal storage diseases, an enzyme deficiency results in the accumulation of its substrate in the lysosome. In particular, Gaucher disease is caused by a deficiency of glucocerebrosidase (GCase; or β-glucosidase). β-Glucocerebrosidase, a member of the glycoside hydrolase family 30, has three distinct domains (I-III) (113).
Domain I forms a three-stranded anti-parallel β-sheet; this domain contains two disulfide bridges that are necessary for correct folding, and a glycosylated residue (Asn19) that is required for catalytic activity in vivo. Domain II consists of two β-sheets that resemble an immunoglobulin fold. Domain III is homologous to a TIM barrel (triose-phosphate isomerase barrel, or alpha/beta barrel), a highly conserved domain among glycoside hydrolases, consisting of 8 α-helices and 8 parallel β-strands that alternate along the peptide backbone.
Domain III harbors the active site, which binds the substrate glucocerebroside in proximity to the catalytic residues E340 and E235.
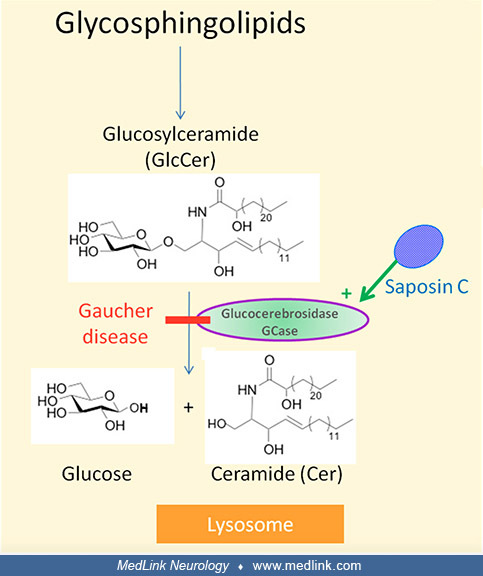
Glucocerebrosidase, which is activated by saposin C, is responsible for the lysosomal degradation of glucosylceramide (Glc-Cer; also called glucocerebroside) (62). Glucocerebrosidase maturation occurs in the Golgi apparatus from which it is delivered to lysosomes with the assistance of lysosomal integral membrane protein-2 (LIMP-2) molecule.
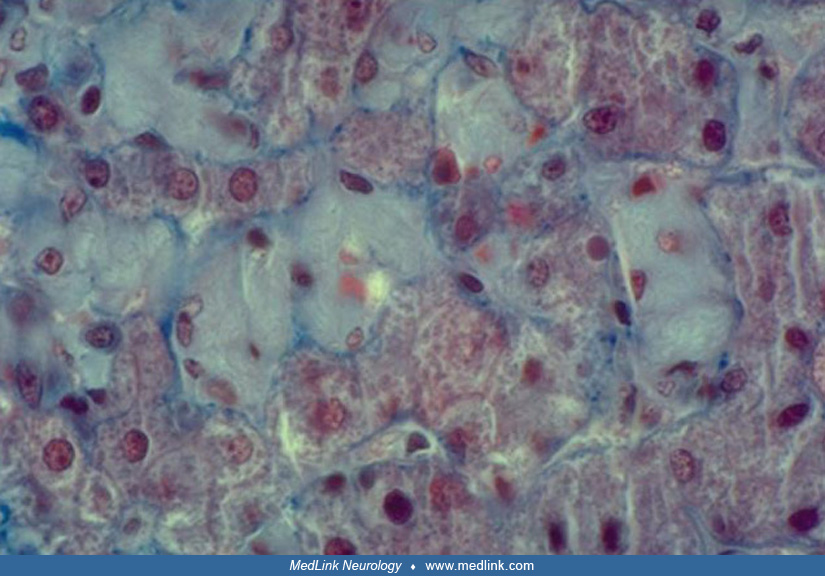
The deficiency of glucocerebrosidase in Gaucher disease leads to accumulation of glucosylceramide, as well as glucosylsphingosine (lyso-Gb1) (167; 03; 148). The accumulation of glucosylceramide causes formation of fibrillar aggregates in macrophages, giving the cytoplasm the appearance of “crumpled tissue paper” (62). Such dysfunctional "Gaucher cells" infiltrate various organs (eg, bone marrow, spleen, and liver).
In glucocerebrosidase deficiency, glucosylceramide can also be metabolized by an alternative metabolic pathway involving acid ceramidase, resulting in the formation of glucosylsphingosine, which is less hydrophobic (62). Glucosylsphingosine is metabolized by a cytoplasmic enzyme, the so-called "second glucocerebrosidase," which is encoded by the GBA2 gene and functions at neutral pH. This second glucocerebrosidase converts glucosylsphingosine into sphingosine, which is then phosphorylated, producing sphingosine-1-phosphate. Accumulation of glucosylsphingosine can cause neuronal dysfunction and death, whereas high levels of sphingosine can be toxic to bone. Glucosylsphingosine is absent in the brains of healthy individuals but can be detected in the brains of patients with Gaucher disease-related neurologic lesions. Glucosylsphingosine is markedly increased in neuronopathic Gaucher disease, especially Gaucher disease type 2.
Saposin C, the activator of glucocerebrosidase, is derived from the cleavage of prosaposin–precursor protein into four homologous proteins (saposins A-D) (62). These mature saposins support the activity of lysosomal hydrolases in sphingolipid degradation. Saposin C mutations can produce biochemical phenotypes mimicking Gaucher disease.
Glucosylsphingosine concentrations are markedly elevated in the brains of type 2 and 3 Gaucher disease patients, and to a lesser extent in patients with Gaucher disease type 1 (133; 140; 48).
Glucocerebroside is a compound of ceramide and glucose. The glucose moiety is esterified to the C-1 of ceramide in a beta-glucosidic linkage. The compound is similar in structure and properties to the group of sugar-containing lipids isolated from the brain by Thudichum (176; 177).
Cerebrosides are composed of ceramide esterified to a variety of different substituents at C-1. This carbon may participate in reactions with phosphorylcholine to produce the sphingomyelins, an unsubstituted monosaccharide or oligosaccharide to produce the neutral glycosphingolipids, or an oligosaccharide containing one to four molecules of sialic acid to produce the gangliosides. The common unit among these compounds is ceramide. Ceramide is derived from a long-chain base named sphingosine (D(+)-erythro-1,3-dihydroxy-2-amino-4-transoctadecene, or C18 sphingosine). This lipid is joined by an amide bond at C-2 to a long-chain fatty acid to form ceramide. The fatty acid chain length varies. In general, the neutral glycosphingolipids and sphingomyelins contain C20 and C24 fatty acids, whereas the gangliosides contain C18 fatty acids. It is from sphingosine that the group of disorders of lipid catabolism obtains its name (ie, sphingolipidoses) because the accumulating lipid compounds are derived from it.
Glucocerebroside is at the end of the glycosphingolipid catabolic pathway. The higher glycosphingolipids and gangliosides are degraded in a stepwise fashion by specific acid hydrolases, resulting in the formation of glucocerebroside, which is normally degraded to ceramide and glucose by acid beta-glucocerebrosidase. The compounds that contribute to the pool of glucocerebroside in peripheral organs are globoside, globotriose, and lactosylceramide. These are derived from the degradation of membranes, the major source of which is white blood cells (101).
The glucocerebroside found in spleen, liver, kidney, plasma, and red cells contains fatty acids with chain length of approximately C22 to C24 (63). The glucocerebroside in the brains of patients with type 2 disease is composed primarily of C18 (stearic acid) (170; 134). This conclusion has been confirmed and extended to type 3 cases (133; 39). These data have been interpreted to mean that the glucocerebroside accumulating in brain derives from gangliosides within the brain itself. This is consistent with the known fatty acid content of gangliosides. Some of the glucocerebroside, in certain type 3 cases, may be derived from sources outside the central nervous system. This may have important consequences because levels of plasma and tissue glucocerebroside increase following splenectomy in Norrbottnian cases (ie, a neuronopathic form of Gaucher disease that is relatively prevalent in Norrbotten, Sweden caused by a single mutation in exon 10 of the glucocerebrosidase gene) (134; 44; 157; 179).
Although the level of plasma glucocerebroside has been shown to increase following splenectomy in some Norrbottnian cases, this has not been reported in cases of other subtypes of the disease.
Glucosylceramide forms fibrillar aggregates that accumulate in macrophages forming so-called Gaucher cells that have a characteristic "crumpled tissue paper" appearance (23; 167). Gaucher cells result from the transformation of macrophage cells and correspond to a distinct "M2" subpopulation from an alternative differentiation pathway. The M2 subpopulation has anti-inflammatory, immunomodulatory, and tissue repair properties (167).
Alternative pathway. As was identified in a mouse model of glucocerebrosidase deficiency, glucosylceramide is also the substrate of an alternative pathway, which is favored in cases of glucocerebrosidase deficiency (ie, Gaucher disease) (126; 167).
The expression of GCase varies from one cell type to another and depends on the tissue. In a mouse model of GCase deficiency (red cross), GlcCer is transformed via an alternative ceramidase pathway into glucosylsphingosine (red...
In the alternate pathway, a ceramidase transforms glucosylceramide into glucosylsphingosine (or lyso-glucosylceramide), which then diffuses into fluids due to its reduced hydrophobicity. The enzymatic deficit of glucocerebrosidase is multifactorial, resulting from intrinsic enzymatic dysfunction as well as abnormalities during transport and delivery of the enzyme to the lysosomes. In particular, enzyme misfolding during passage through the endoplasmic reticulum can lead to premature degradation by the proteasome (152; 200; 167).
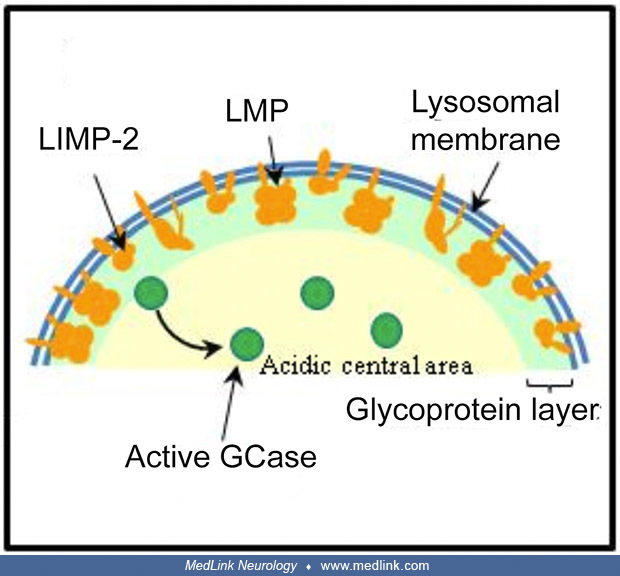
Protein maturation takes place in the Golgi apparatus; the transport and delivery of GCase to lysosomes require a particular molecule, LIMP-2, which allows GCase to reach the lysosome where the acidic pH breaks the molecular li...
LIMP-2 is a lysosomal membrane protein (LMP) whose highly glycosylated intra-lysosomal part protects the lysosome’s membrane. LIMP-2 anomalies can induce a phenotype rather than GD3 (Gonzalez A, Valeiras M, Sidransky E, Tayebi ...
Other biochemical abnormalities in Gaucher disease. Levels of acid phosphatase, angiotensin-converting enzyme, lysosomal hydrolases, lysozyme, and immunoglobulins are elevated in the plasma of patients with Gaucher disease (180; 136; 114; 189; 163; 127; 91; 150). Chitotriosidase is a particularly sensitive indicator of macrophage storage in patients with Gaucher disease (88).
The severity of neurologic symptoms is associated with glycoprotein nonmetastatic B levels (207). The same was found in a mouse model of Gaucher disease. GPNMB may prove to be a useful marker to quantify neuropathology in patients with Gaucher disease and treatment efficacy in future treatment trials (207).
Organ involvement in Gaucher disease.
Clotting cascade. Because of liver involvement, patients may have prolonged partial thromboplastin, prothrombin, and bleeding times. In addition, increased amounts of plasma glucocerebroside may interfere with the clotting cascade (22).
Reticuloendothelial system. A study of the gene expression profile in Gaucher disease showed enhanced expression of genes associated with inflammatory reactions in the affected spleen (129). In particular, the transcript abundance of the cDNAs representing cysteine proteinases (cathepsins B, K, and S) was greatly increased. These proteins are known to participate in tissue modeling, antigen presentation, and in the case of cathepsin K, bone matrix destruction. Glycolipid storage in the cells of the reticuloendothelial tissue may induce an inflammatory response characterized by the recruitment of proinflammatory cytokines and other cells of the immune system. In turn, extensive tissue damage would ensue, particularly in the liver, spleen, and bones.
Brain involvement in Gaucher disease type 1. Patients with Gaucher disease type 1 do not have clinical symptoms or signs referable to the nervous system. In this group, anatomic and biochemical examinations of the brain have been infrequent. Hippocampal gliosis has been found in brains of otherwise neurologically normal patients with Gaucher disease type 1 (198).
Pathologic features of neuronopathic phenotypes. In Gaucher disease types 1, 2, and 3, perivascular Gaucher cells have been observed within the Virchow-Robin spaces (46; 198).
In type 2 patients' brains, free Gaucher cells have been demonstrated within the parenchyma accompanied by gliosis and microglial nodules. These changes are present but much less frequently in type 3 patients' brains. Neuronal storage of lipid has been suggested in several reports, but this has not been confirmed ultrastructurally in any case of Gaucher disease. In type 2 disease, neuronophagia and neuronal cell death in the deeper layers of the cortex, thalamus, basal ganglia, brainstem nuclei, cerebellum, and spinal cord have been reported. Variable degrees of demyelination have been described in brains of type 2 patients. From the available information, one would have to conclude that the accumulation of acid beta-glucocerebrosidase in the brain produces dysfunction in surrounding cells long before discrete pathologic changes are seen (102). Extensive study of 14 brains from type 1 (including patients with parkinsonism), 2, and 3 Gaucher disease showed common neuropathologic findings in all forms of Gaucher disease (198). Unique patterns of gliosis and neuronal loss involving the hippocampal CA2-4 regions and layer 4b of the calcarine cortex were identified. Although these findings were common to all three Gaucher disease phenotypes, the extent of the changes varied depending on the severity of disease (198). Cerebral cortical layers 3 and 5, hippocampal CA2-4, and layer 4b were involved in all Gaucher disease patients. Neuronal loss predominated in both type 2 and type 3 patients with progressive myoclonic encephalopathy, whereas patients classified as having type 1 Gaucher disease had only astrogliosis. Adjacent regions and lamina, including hippocampal CA1 and calcarine lamina 4a and 4c were spared of pathology, highlighting the specificity of the vulnerability of selective neurons. Elevated acid beta-glucocerebrosidase expression by immunohistochemistry was found in CA2-4. Hippocampal 45Ca(2+) uptake autoradiography in rat brain was performed, demonstrating that hippocampal CA2-4 neurons, rather than CA1 neurons, were calcium-induced calcium-release-sensitive (CICR-sensitive). These findings match biochemical studies linking elevated glucosylceramide levels to sensitization of CA2-4 RyaR receptors and 300% potentiation of neuronal CICR sensitivity (143). In two patients with type 1 Gaucher disease and parkinsonism, numerous synuclein positive inclusions, similar to brainstem-type Lewy bodies found in Parkinson disease, were also found in hippocampal CA2-4 neurons. These findings argue for a common cytotoxic mechanism linking aberrant acid beta-glucocerebrosidase activity, neuronal cytotoxicity, and cytotoxic Lewy body formation in Gaucher disease (198). Progression of the neuropathology is very predictable in the mouse model including microglial activation and astrogliosis were spatially and temporally correlated with selective neuron loss (61). Glucocerebroside accumulates to a similar extent in most brain areas, and upon reaching a certain threshold of accumulation inflammation and neurodegeneration is initiated in susceptible brain areas (60). The same group found that receptor interacting protein (RIP) kinases-mediated necrosis plays a major role in inducing inflammation and neuronal death in neuronopathic Gaucher disease (185). Ripk3 deficiency (double knockout mice) dramatically improved the clinical course of Gaucher disease mice with increased survival and motor coordination. Therefore, Ripk3 is a new therapeutic target for neuronopathic Gaucher disease (185). Using the mouse model for the most severe form of neuronopathic Gaucher, it was shown that altered lysosomal localization and cytoskeleton disruption precede the neuroinflammatory pathways, axonal dystrophy, and neuronal loss previously characterized in neuronal forms of Gaucher disease (208).
Glucosylsphingosine has been implicated in toxic damage to neural cells. This molecule has been shown to accumulate in the cerebral and cerebellar cortex of patients with type 2 and 3 Gaucher disease (133; 133; 134) and in Gaucher mice. The highest levels of this metabolite were found in brain tissue from two fetuses with hydrops fetalis (140). Studies have found markedly elevated levels in type 1, type 2, and type 3 disease (49). Glycosphingolipids, including glucosylsphingosine, inhibit protein kinase C, mitochondrial cytochrome c oxidase, and CTP:phosphocholine cytidylyltransferase, which has been postulated to interfere with signal transduction and cellular differentiation (82).
Studies on the pathogenesis of neuropathic Gaucher disease suggest that defective calcium homeostasis is a mechanism responsible for neuropathophysiology in acute neuronopathic Gaucher disease (143). Agonist-induced calcium release via the ryanodine receptor was significantly enhanced in brain microsomes from the acute neuronopathic form of Gaucher disease (type 2) compared to the subacute (type 3) and the non-neuronopathic (type 1) forms and controls and correlated with levels of GlcCer accumulation (143). The precise mechanism by which glucocerebroside enhances calcium release via the ryanodine receptor is not known, but these findings suggest the use of certain calcium channel blockers in neuronopathic Gaucher disease. Altered expression and distribution of cathepsins in the brain of the neuronopathic Gaucher mouse suggests that inflammation plays a role in the disease and, therefore, may be a target for therapeutic intervention (184). Elevation of GBA2, a nonlysosomal glucocerebrosidase, may play a role in the disease (32).
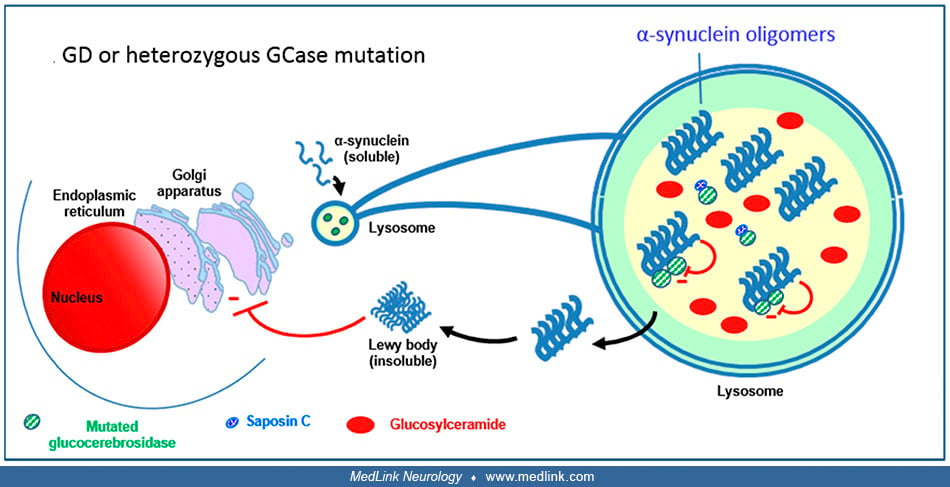
Relationship between glucocerebrosidase and synucleinopathies. Normally, glucocerebrosidase interacts with its substrate glucosylceramide as well as monomers of α-synuclein in lysosomes, facilitating the breakdown of both at acidic pH (167).
Normally, GCase interacts with its substrate glucosylceramiide (GlcCer) as well as monomers of alpha-synuclein in lysosomes, facilitating the breakdown of both at acidic pH. (Source: Stirnemann J, Belmatoug N, Camou F, et al. A...
A decrease in the activity of glucocerebrosidase (eg, due to mutations) causes a gradual buildup of glucosylceramide and also causes a slowdown of α-synuclein degradation with the resulting formation of α-synuclein oligomers and fibrils (42; 201; 130).
Mutated GCase or decreased levels of GCase cause a slowdown of alpha-synuclein degradation with the resulting formation of alpha-synuclein oligomers and fibrils (Cullen et al 2011; Yap et al 2011; Murphy et al 2014). Glucosylce...
Glucosylceramide stabilizes the α-synuclein oligomers (121), which are able to bind to the mutated glucocerebrosidase molecules and inhibit the enzymatic activity of glucocerebrosidase, further decreasing enzyme activity (121; 204; 203). These impaired lysosomes show impaired chaperone-mediated autophagy and autophagosome fusion. This results in an increased accumulation of α-synuclein in the cytoplasm, forming insoluble aggregates that ultimately form Lewy bodies. These aggregates block trafficking of glucocerebrosidase from the endoplasmic reticulum (ER) to the Golgi (162). Mutant glucocerebrosidase is retained in the endoplasmic reticulum (ER), which causes ER stress and evokes the ER stress response ("unfolded protein response") (90). Saposin C can have a modulating effect on this by binding to glucocerebrosidase (202; 79).
Mouse models of Gaucher disease. The transgenic acid beta-glucocerebrosidase-deficient mouse model obtained by targeted knockout of the acid beta-glucocerebrosidase gene exhibits a severe phenotype with prenatal or perinatal death (181). A number of other mouse models were of limited use and did not represent a genuine model for the neuronopathic form of the disease (199). Two true models of the acute neuronopathic form of Gaucher disease were produced. One of the models had normal enzyme activity in hematopoietic cells but no enzyme activity in neuronal and astrocytic cells. There was no significant mitigation of the neuronal disease in this model, confirming the observation in patients that bone marrow transplantation is not effective in neuronopathic Gaucher disease (56).
All types of Gaucher disease are inherited as an autosomal recessive trait. The collective subtypes of Gaucher disease constitute one of the most prevalent forms of the sphingolipidoses and the most common lysosomal storage disorder. Gaucher disease type 1, the non-neuronopathic form, occurs with an incidence of about 1 in 40,000 to 60,000 in the general population and 1 in 500 to 1000 among Ashkenazi Jews. Patients with Gaucher type 3 disease (chronic neuronopathic Gaucher disease) constitute about 5% of the population of Gaucher patients in the United States and in Europe, with an estimated incidence of about 1 out of 100,000. However, reports suggest the neuronopathic form of Gaucher disease predominates in countries like China, Japan, Korea, Taiwan, Egypt, and Syria (187; 38; 172; 98; 53; 05; 78; 74). The reason is that the N370S mutation is common in European countries but almost absent in Northern Africa and Asia, whereas the L444P mutation is particularly common in the East (187; 38; 172; 98; 53; 05; 78).
An epidemiological study in France utilized the French Gaucher disease registry (FGDR), which includes all known patients with Gaucher disease in France (132). Between 1980 and 2024, there were 706 confirmed Gaucher disease cases. In 2024, 447 patients were alive (413 type 1; 34 type 3). Gaucher disease incidence was 0.21 per 1,000,000 person-years, and Gaucher disease prevalence was 0.61 and 0.05 per 100,000 inhabitants for type 1 and 3, respectively. The standardized mortality ratio was 0.70 for type 1 Gaucher disease and 16.23 for type 3 Gaucher disease. Over this period, the delay between first symptoms and diagnosis decreased significantly; it was 5.4 years before 2000 and 0.8 years after 2020. The decreased diagnostic delay was facilitated by enzyme assays becoming the primary diagnostic method. This led to a reduction in splenectomies and a gradual increase in the use of substrate reduction therapy in type 1 Gaucher disease. The incidences of bone events, malignancies, and Parkinson disease were 23, 2.7, and 1.07 per 1000 person-years, respectively.
Although it is more frequent among Eastern European Jews, Gaucher disease type 1 is usually less severe in this group. It is more severe among blacks. This generalization is helpful in counseling, but there are exceptions. Therefore, it is important to know the types of complications of the disease that have occurred in a family. In general, the severity of the disorder tends to be similar among siblings. A single exceptional family has been reported in which one type 1 and one type 2 case occurred in full siblings.
Implications for genetic counseling. Testing and genetic counseling of immediate family members of affected patients are recommended. The differences in clinical subtypes and variability of some forms of the disease should be pointed out. Because of the high carrier frequency among the Ashkenazi, wide-scale carrier testing has been suggested. Because the disease among this group is not uniformly catastrophic (in fact, many cases do not come to medical attention until late in life), this kind of testing, if desired, should be done within a system providing careful counseling that includes adequate information about the disorder and an explanation of the limitations of carrier detection. More than 300 mutations have been described in the GBA1 gene, four account for the majority of the nucleotide alterations. In the Ashkenazi Jewish population, the p.Asn409Ser mutant allele accounts for about 75% of the abnormal genes (78). In this ethnic group, four alleles (p.Asn409Ser [formerly described as Asn370Ser], c.84insG [formerly described as 84GG], p.Leu483Pro [formerly described as Leu444Pro], and IVS2+1G> A) account for more than 95% of the chromosomes, thus, permitting population screening (12). In the general population, many "private" alleles or mutations confined to a single kindred are found. Testing for the four mutations listed above results in identification of fewer than 50% of the cases. Thus, except for the Eastern European Jewish group, population screening is not appropriate for this disease with currently available technology.
Prenatal diagnosis. Affected fetuses with any form of Gaucher disease can be diagnosed prenatally by enzymatic assay of cultured amniocytes or chorionic villi. This is particularly important in families in whom the neuronopathic subtypes have occurred. Because of the difficulty of the assay for carriers (45) and the lack of appropriate control data, prenatal carrier detection is unreliable by enzyme assay. However, molecular genetic methods permit this identification without difficulty in families where the mutations are known.
Other childhood diseases with signs and symptoms overlapping with Gaucher disease include the following (194):
|
• Cancers, particularly lymphoproliferative malignancies | |
|
• Hematologic disorders (eg, idiopathic thrombocytopenia, von Willebrand disease) | |
|
• Sarcoidosis | |
|
• Legg-Calvé-Perthes disease | |
|
• Metabolic bone disease (eg, rickets, vitamin C deficiency, copper deficiency sickle cell disease, Paget disease) | |
|
• Bacterial osteomyelitis | |
|
• Other lysosomal storage diseases (eg, GM1 gangliosidosis, lysosomal acid lipase deficiency, and Niemann-Pick disease types A and C). |
Painless splenomegaly is the most common initial presenting sign in Gaucher disease type 1. Lymphoproliferative malignancies are the chief concern in the diagnostic evaluation of patients presenting in this manner. The results of bone marrow biopsies may suggest the presence of Gaucher cells; however, the diagnosis should be made by enzymatic assay of acid beta-glucocerebrosidase in leukocytes or fibroblast cultures. If a patient is Jewish, Gaucher disease should be considered and an enzyme assay performed. Results can usually be obtained within several days. A disease virtually identical to Gaucher can rarely be caused by a deficiency in saposin c, the protein that activates glucocerebrosidase (182; 99). In this case, glucocerebrosidase activity is normal but the patient has clinical features resembling Gaucher disease. In infants with hepatosplenomegaly and a neurodegenerative course, Niemann-Pick disease type A and GM1 gangliosidosis are sometimes similar in presentation but usually can be distinguished by clinical presentation.
The diagnosis of Gaucher disease should be considered in any case of unexplained splenomegaly with or without a bleeding diathesis or other manifestations of the disease in the skeleton or liver. The disorder should be considered likely in any infant with hepatosplenomegaly and a neurodegenerative course. Elevation of tartrate-resistant acid phosphatase in serum is suggestive of the disease. Demonstration of characteristic Gaucher cells in bone marrow biopsies narrows the diagnostic possibilities. The definitive diagnosis is made by assay of acid beta-glucocerebrosidase in leukocytes, fibroblasts, chorionic villi, or urine.
A diagnostic algorithm has been proposed for pediatric patients without a family history of Gaucher disease (104; 194).
(Source: Weinreb NJ, Goker-Alpan O, Kishnani PS, et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol Genet Metab 2022;136[1]:4-21. Adapted from: Kishnani PS, Al-Hertani W,...
In a cross-sectional study of 44 patients, dry blood spot samples were used to assess levels of the deacylated form of glucocerebroside, glucosylsphingosine (lyso-Gb1) for Gaucher disease diagnosis (49). Of 444 screened subjects, 99 (22%) were diagnosed with Gaucher disease at a median age of 21 years (range 1 to 78). Lyso-Gb levels for genetically confirmed patients with Gaucher disease (median 252 ng/mL [range 9 to 1340]) were markedly higher than for patients without Gaucher disease (median 5.4 ng/mL [range 1.5 to 16]). Patients diagnosed with Gaucher disease type 1 and mild GBA1 variants had significantly lower lyso-Gb1 levels (median 194 ng/mL [range 9 to 1050]) compared to Gaucher disease type 1 and severe GBA1 variants (median 447 ng/mL [range 38 to 1340]), and neuronopathic Gaucher disease (median 325 ng/mL [range 116 to 1270]).
Subjects with heterozygous GBA1 variants (carrier) had significantly higher lyso-Gb1 levels (5.8 ng/mL [2.5 to 15.3]) compared to wild-type GBA1 (4.9 ng/mL [1.5 to 16]). Based on these results, the authors called for a change in the diagnostic approach to Gaucher disease, based on lyso-Gb1 measurements and confirmatory GBA1 mutation analyses in dry blood spot samples (49).
After a diagnosis of Gaucher disease has been established, the burden of the disease should be assessed. Evaluation of the characteristic features of the disease should include hematological, visceral, skeletal, and quality of life parameters, as suggested by the Gaucher Disease Registry.
Minimum assessment recommendations in children with Gaucher disease type 1 have been proposed (100; 194).
(a) MRI should include sagittal T1-weighted scan of the spine and T1-weighted scan of the head of the femur. (b) DXA should include spine and total body Z-scores, which require comparison with age- and gender-specific norms. (c...
Additional recommended tests for children with Gaucher disease type 3 include neurologic examination, neuro-ophthalmological investigations, testing of hearing, brain imaging, electroencephalography, periodic pulmonary evaluation for interstitial lung disease, and neuropsychological evaluation. In addition, for those with Gaucher disease type 3c, characterized mainly by cardiovascular and neuro-ophthalmological findings (108), periodic chest x-ray, echocardiography, cardiac computed tomography, angiography, or coronary artery catheterization have been recommended (194), but with little detailed guidance of when and how such studies will be useful in impacting the course of the disease or quality of life.
Hematologic workup. Hemoglobin, platelet count, tartrate resistant acid phosphatase, angiotensin converting enzyme, and chitotriosidase should be measured when the diagnosis is established; follow up measurement is recommended every 3 months for patients on enzyme replacement therapy and at least every 12 months for patients who choose not to receive enzyme replacement therapy.
Visceral studies. Spleen and liver volume should also be assessed at the time of diagnosis. The recommended technique is volumetric CT scan or MRI. These tests are recommended every 12 months in patients receiving enzyme replacement therapy and every 24 months in those patients who achieve a stable state.
Enzymatic diagnosis. A variety of natural and artificial substrates provide accurate assays for homozygotes, but they all have the same limitations when used for heterozygote detection (196; 45). The best substrate employed for the assay is the 4-methylumbelliferyl-beta-D-glucopyranoside. Modifications and improvements in the original description of the assay involving the addition of taurocholate have made it useful as a diagnostic tool and equivalent to assays employing the natural substrate. The assay is easy to use, and the substrate is widely available, making it the method of choice for a diagnostic laboratory. Conduritol B-epoxide is a specific inhibitor of mammalian acid beta-glucocerebrosidase and permits confirmation of the enzyme deficiency in systems where nonspecific beta-glucosidase may be interfering. Leukocytes and fibroblasts may be prepared and shipped to laboratories for assay as whole cells or extracts. High throughput method for newborn screening has been developed (137).
From the time of the introduction of the assay of acid beta-glucocerebrosidase, it has been observed that the values of heterozygotes overlap those of control subjects for the activity of acid beta-glucocerebrosidase in leukocytes and leukocyte subpopulations. This subject has been reviewed extensively. A variety of methods have been developed, but all have the same problem in the detection of heterozygotes. In studies of known heterozygotes, approximately 20% of the carriers fall into the normal range. Fibroblasts have a higher specific activity of acid beta-glucocerebrosidase than leukocytes; however, a wide range of activity in control cells, which varies with time in culture and conditions, makes these cells no more useful in heterozygote detection. In those for whom the genotype is known, carrier detection by molecular methods is completely reliable.
Genetic studies. Whole GBA1 sequencing is superior to exome sequencing because of potential effects of deep intronic variants on gene expression and because it has the capacity to detect variant alleles that might be missed with gene panels (77).
Skeletal assessment. Computerized tomography, radionuclide scan, and magnetic resonance imaging have been useful in assessing the extent of bone abnormalities. The recommended method for routine assessment of bone disease is T1- and T2-weighted MRI of the entire femora (34).
Bone density should also be assessed in patients with Gaucher disease. DEXA studies are useful to determine and assess the risk of pathological fractures. Spine, femur, forearm, and whole-body densitometries can be performed in these patients. These studies should be performed at baseline and every 12 months in patients receiving enzyme replacement therapy.
Unfortunately, there is a lack of treatments capable of reaching the CNS to slow or halt the progression of neuronopathic disease in patients with Gaucher disease type 2 or 3, or to potentially reduce the risk of Parkinson disease in Gaucher disease type 1 patients and heterozygotes for GBA1 variants (77).
Gaucher disease type 1. Consensus management goals for Gaucher disease type 1 have been developed (178; 115)
Eliglustat tartrate. Eliglustat tartrate is a P-glycoprotein substrate, possibly accounting for its poor distribution into the brain (158). Eliglustat tartrate is an orally active specific inhibitor of glucosylceramide synthase (GCS), the enzyme that creates glucosylceramide (GL-1); therefore, it is a substrate synthesis inhibition therapy (SSIT) agent (77). By partially blocking the production of glucosylceramide, eliglustat reduces the amount of substrate that needs to be broken down; this balances the rate of glucosylceramide production with the limited ability of the deficient enzyme to break it down, preventing harmful buildup. Eliglustat has shown efficacy that is similar to that of enzyme replacement therapy at the 60 IU/kg/2 weeks dose (40). Eliglustat can significantly improve common manifestations of Gaucher disease type 1, including: (1) reducing spleen and liver volume; and (2) improving hematological parameters (eg, increased hemoglobin and platelet counts). Eliglustat was approved by the FDA in 2014 for adults with Gaucher disease type 1 who are "extensive," "intermediate," or "poor" metabolizers of the liver enzyme CYP2D6 (145); because the metabolism of eliglustat is susceptible to the genotype of cytochrome P450 CYP2D6a, a medical provider must order a test to determine a patient's CYP2D6 metabolizer status before starting this medication. Studies of switching from enzyme replacement therapy to eliglustat, or between different ERT products, have indicated that changing treatment is safe, although efficacy outcomes vary.
Gaucher disease type 2. This severe form of Gaucher disease poses significant diagnostic, management and ethical challenges (195). None of the specific therapeutic approaches described above have been shown to be efficacious in patients with the most severe form of Gaucher disease. The overall management of patients with Gaucher disease type 2 is very difficult and has been reviewed (195).
Cell and organ transplantation. Splenic and renal transplantations have had little or no effect on the disease process in patients with Gaucher disease. Bone marrow transplantation has been reported in both Gaucher disease type 1 and type 3 patients. Rapid resolution of the enzyme deficiency in circulating white cells was achieved, indicating successful engraftment.
The indications for hematopoietic cell transplantation vary according to the subtype of Gaucher disease. In type 1 disease, allogeneic bone marrow transplantation leads to resolution of organomegaly within several months to 2 years. The bone disease is also ameliorated, with disappearance of Gaucher cells from the marrow, regression of bone lesions, improvement in linear growth, and disappearance of bone pain (171; 147; 86). Bone marrow transplantation may, therefore, be curative for type 1 disease, even though this therapeutic option is limited by donor availability and high short-term risks, which include a high percentage of procedure associated morbidity and mortality of about 15%. In type 2 and 3 disease, bone marrow transplantation has no effect on the neurologic disease and should be avoided (183).
Enzyme replacement. The ultimate development of enzyme replacement therapy was a fortunate intersection of scientific disciplines that provided key findings:
• Discovery of receptors for glycoproteins (lectins) (09) |
The mannose moieties in the oligosaccharides are the key to successful enzyme replacement therapy. Mannose, rather than mannose-6-phosphate, binds to the mannose receptor on Kupffer cells. There are also mannose receptors on hepatocytes, although some of these are non-endocytic. Acid beta-glucocerebrosidase can, therefore, gain entry into both hepatocytes and Kupffer cells, but the delivery to Kupffer cells is much better for mannose-terminated (modified) acid beta-glucocerebrosidase.
Early studies of enzyme replacement produced great hope and excitement that Gaucher disease might be amenable to therapy by infusions of enzyme (29). The initial encouraging biochemical and clinical reports were not confirmed. Even though amounts of enzyme in subsequent trials were increased significantly, no changes could be measured in response to enzyme administration. These trials were unsuccessful because they failed to target the enzyme to the proper cells in sufficient quantity.
A method was devised to improve that aspect of the problem (66). Using enzymatic degradation of the oligosaccharide side chains of acid beta-glucocerebrosidase to expose mannose residues, the enzyme molecule was tailored to bind to the naturally occurring mannose receptors on macrophage plasma membranes. In animal model studies, this engineered acid beta-glucocerebrosidase was delivered to reticuloendothelial cells in quantities 10 times greater than the unmodified native enzyme (66). The function of the oligosaccharide side chains is not fully understood, but they are probably important in maintaining the tertiary structure of the protein during its synthesis and translocation to the lysosome. If addition of the sugar side chains is prevented completely, the protein is not properly folded and has no enzyme activity.
A clinical study in one patient demonstrated that this mannose-terminated acid beta-glucocerebrosidase was therapeutic (11). A formal clinical trial confirmed these results (14; 13). The commercial preparation is now widely used in patients and successfully reverses the visceral, hematologic, and skeletal manifestations of the disease.
After several clinical trials of placental derived acid beta-glucocerebrosidase (alglucerase) for the treatment of Gaucher disease, alglucerase was approved in 1991 (13). Production of the same protein using recombinant DNA technology was achieved, and the resultant drug, imiglucerase, received regulatory approval in 1994.
A review of the hepatic, splenic, and hematologic outcome from 175 patients was reported by Weinreb and colleagues (193). The authors concluded that the following four responses could be expected after 12 to 24 months of enzyme replacement therapy:
|
(1) Patients with hepatomegaly will have approximately a 20% decrease in hepatic volume. |
The skeletal complications of Gaucher disease respond well within the same time frame as the other signs and symptoms of the disease. Bone pain is relieved, and the incidence of bone infarct is reduced dramatically to near zero in patients treated with 60 U/kg of recombinant human acid beta-glucocerebrosidase every 2 weeks prior to the development of a significant structural abnormality. The radiographic assessment of the skeletal complications, however, remains inadequate as a tool to measure the baseline and improvements in the bone in response to therapy.
Enzyme replacement has the same salutary effect on patients with type 3 Gaucher disease as it has in type 1 patients (155; 07). Treatment is indicated in these patients at a dose of 60 IU/kg of body weight every 2 weeks and should be closely monitored clinically and with laboratory studies to establish the response to the therapy (183). When initiated early in the course of the disease, enzyme replacement normalizes growth, corrects the hematologic abnormalities, and prevents the skeletal complications that occur in patients with type 3 Gaucher disease. This therapy has markedly ameliorated the course of the disease in this group of patients who used to die in their teens prior to the advent of enzyme replacement. However, in type 2 disease, enzyme replacement therapy may lengthen the course of the illness, but the neurologic deterioration progresses, and the poor prognosis of the disease is unaltered (77); therefore, enzyme replacement therapy should not be initiated in this group (146).
The adverse effects of enzyme replacement therapy are few and rarely a reason for discontinuing the therapy (128). Antibodies to enzyme replacement therapy have been reported in approximately 15% of patients. The incidence of serious complications is very low.
Effects of enzyme therapy cessation and withdrawal have been evaluated in patients who completely discontinued their therapies. These studies suggest that a complete cessation of therapy will inevitably endanger the patient by deteriorating hematological, biochemical, and skeletal parameters (186; 190). However, successful experiences of enzyme withdrawal for short periods of time have been reported (156).
Early initiation of enzyme replacement therapy is very important in children because it allows prevention of serious, irreversible skeletal complications (77). Prompt initiation of treatment in pediatric patients is important to ameliorate or prevent impaired growth, lower peak bone mass, and delayed puberty (100; 08; 77).
The COVID-19 pandemic ushered in a new era of next-generation mRNA vaccines. Studies have explored the use of mRNA as a potential tool for protein replacement therapy for a wide range of genetic disorders. Introduction of synthetic mRNA into a patient’s cells leads to production of therapeutic proteins that are otherwise missing or defective due to genetic mutations (62). The mRNA serves as a template to direct the synthesis of a therapeutic protein. Another type of RNA molecule, so-called self-amplifying RNA (saRNA), can replicate itself, allowing for sustained production of therapeutic proteins within cells. Both approaches have the potential to improve the delivery of protein replacement therapy in various genetic diseases, including Gaucher disease.
(Left) When self-amplifying RNA (saRNA) is injected and delivered into cells, the translation of replicase takes place. The replicase uses the saRNA as a template and makes a negative saRNA strand, which in turn serves as a tem...
Other therapeutic approaches. The success of enzyme replacement therapy has lessened motivation to develop a gene therapy approach for the nonneurologic aspects of Gaucher disease. Because there is an unmet need to reverse or prevent the primary neurologic complications of Gaucher disease, research is ongoing to develop approaches that will enable delivery of acid beta-glucocerebrosidase or the normal copy of the GBA1 gene across the blood-brain barrier and into brain cells (166).
Substrate deprivation. N-butyldeoxynojirimycin (OGT-918), an inhibitor of the glucocerebroside synthase, initiates the glycosphingolipid pathway and catalyzes the formation of glucocerebroside (41). The rationale of this therapy is to decrease the formation of glucocerebroside to rates at which the residual acid beta-glucocerebrosidase activity of the patients would be sufficient to catabolize the substrate. Most patients experienced diarrhea, the most frequent side-effect, and two patients withdrew from the study because of this complaint. Some patients developed peripheral neuropathy. These results do not compare favorably to those obtained with enzyme replacement therapy, which normalizes or greatly impacts the clinical parameters with negligible side-effects (10; 125).
Newer inhibitors of ceramide glycosyltransferase but with good brain penetration are being developed and are being tested in human patients (120).
In a randomized controlled trial, miglustat was found not to be effective for the treatment of type 3 Gaucher disease (154).
Pharmacological chaperones. A proposed approach for the treatment of Gaucher disease and other lysosomal storage diseases relies on the administration of active site competitive inhibitors that promote the correct folding and trafficking of the mutant protein (and even wild-type molecules). This process leads to increased levels of the mutant enzyme that can then function during the drug washout period (205; 188). The only pharmacological chaperone that is currently being clinically tested is ambroxol in high dose (117; 103).
Ceramide, glucosylceramide shift between the Golgi apparatus and lysosomes. Ceramides (Cer), generated in the endoplasmic reticulum (ER), are transported to the Golgi apparatus, where UDP-glucosylceramide synthase (UGCG synthas...
It shows some promise in myoclonic forms of Gaucher disease type 3 (103). However, no controlled studies have been performed to confirm the clinical benefit of ambroxol.
Gene therapy. Gene therapy using an adeno-associated virus vector injected intrathecally is in the advanced planning stages.
Patients with Gaucher disease can become pregnant and deliver normal healthy infants. Precautions should be taken for women who are anemic or thrombocytopenic during the pregnancy. Excessive bleeding is a frequent complication following delivery. The teratogenic potential of acid beta-glucocerebrosidase has not been evaluated, and patients contemplating pregnancy should be advised of this fact. Prudence suggests that pregnant women should not be treated with enzyme replacement during the first trimester. However, pregnant patients who were treated with at least two forms of enzyme replacement have delivered normal babies (54; 55).
Patients with anemia, thrombocytopenia, or pulmonary involvement are at an increased risk for complications related to anesthesia and surgery. A particularly instructive case is a patient who was not diagnosed prior to cardiac surgery (124).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Peripheral Neuropathies
May. 12, 2026
Neurogenetic Disorders
May. 08, 2026