Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Childhood absence epilepsy is the prototype idiopathic (genetic) generalized epilepsy syndrome of typical absence seizures. It is common in normal school-aged children between the ages of 4 and 10 years (peak age 6 to 7 years). It manifests with severe and frequent (pyknoleptic) typical absence seizures of abrupt onset and termination, lasting around 10 seconds each, many times per day. EEG shows bilateral, synchronous, symmetric spike-and-slow wave discharges, usually 3 Hz, on normal background activity. Females predominate. The prognosis of childhood absence epilepsy syndrome is excellent if properly diagnosed and treated. The focus is on the current understanding of its clinical and EEG phenotype, epidemiology, pathophysiology, genetics, pharmacological treatment, and outcome, along with the differential diagnosis associated with other idiopathic and genetic generalized epilepsies involving absence seizures. The author particularly emphasizes that most of the older and newer antiseizure drugs are contraindicated for the treatment of childhood absence epilepsy.
|
• Childhood absence epilepsy is the prototype idiopathic generalized epilepsy syndrome and is characterized by typical absence seizures of high daily frequency and severe impairment of consciousness in school-aged children. It is associated with generalized spike-and-wave discharges, usually 3 Hz (range 2.5 to 4 Hz). | |
|
• Typical absence seizures with different electroclinical characteristics are also part of the phenotypic expression of other idiopathic-genetic generalized epilepsies of infancy and childhood. | |
|
• The differential diagnosis of childhood absence epilepsy includes other types of epilepsies or syndromes with typical absence seizures manifesting in infancy and childhood. | |
|
• Prognosis is usually excellent for patients diagnosed on strict inclusion and exclusion criteria for the syndrome of childhood absence epilepsy. | |
|
• Ethosuximide and sodium valproate, as monotherapy or combined, are the most effective therapeutic approaches. |
According to Temkin, Poupart was the first to describe absence seizures in 1705 (215). However, Tissot's description of petits accès or petits in 1770 is better known (219). They were called "absences" by Calmeil in 1824 (31), "petit mal" by Esquirol in 1838 (81), and "epilepsia mitior" by Reynolds in 1861 (184). Gowers gave a most accurate description of the absence seizures “without conspicuous convulsions” (102). Friedman reported a long-term favorable prognosis but believed that these absences were not epileptic (90). The frequency of typical absence seizures had also been conspicuous, and Sauer coined the name “pyknolepsy” (from the Greek word pyknos, meaning closely packed, dense, or aggregated) (194). Hyperventilation as a test to introduce absences was first described by Brain in 1924 (177). In the same year, Adie reported on pyknolepsy, a form of epilepsy occurring in children with a good prognosis, and defined it as follows (02):
|
...a disease with an explosive onset, between the ages of 4 and 12 years, of short, very slight, monotonous minor epileptiform seizures of uniform severity, which recur almost daily for weeks, months, or years, are uninfluenced by antiseizure remedies, do not impede normal mental and physical development, and ultimately cease spontaneously never to return. At most, the eyeballs may roll upwards, the lids may flicker, and the arms may be raised by a feeble tonic spasm. Clonic movements, however slight, obvious vasomotor disturbances, palpitations, and lassitude or confusion after the attacks are equivocal symptoms strongly suggestive of oncoming grave epilepsy, and for the present they should be considered as foreign to the more favorable disease. I shall be well satisfied if I have made it appear probable to you that there does exist a form of epilepsy in children which is distinguishable by its clinical features and in which the prognosis is always good.” |
Gibbs, Davis, and Lennox showed that petit mal absences were associated in EEG with a rhythmic 3 Hz discharge of regular spike-and-wave complexes (94). In 1945, Lennox referred to the petit mal triad as absence, myoclonic, and akinetic seizures, and the introduction of trimethadione revolutionized the treatment of absence seizures (133). Video-EEG analysis allowed a precise clinico-EEG correlation of typical absence seizures (176).
The Commission on Classification and Terminology of the International League Against Epilepsy (ILAE) made important progress by accurately defining and differentiating typical absences of idiopathic generalized epilepsies versus atypical absences of symptomatic generalized epilepsies (48). However, all epilepsies with typical absence seizures remained for a long time clustered in the group of "petit mal" and considered as a form of "centrencephalic epilepsy."
In 1989 the Commission on Classification and Terminology of the ILAE recognized the heterogeneity of epilepsies with absence seizures and proposed to distinguish three syndromes of idiopathic generalized epilepsy (childhood absence epilepsy, juvenile absence epilepsy, and juvenile myoclonic epilepsy) (49). Further, it also recognized typical absence seizures in "other idiopathic generalized epilepsies," in "idiopathic generalized epilepsies with specific provocation," and also in a syndrome of cryptogenic generalized epilepsy (epilepsy with myoclonic absences). Panayiotopoulos and colleagues described syndrome-related characterization of absence seizures with video-EEG analysis (174).
In recognition of the diversity of typical absence seizures, the ILAE Task Force proposed four types that are probably of different pathophysiology and syndromic significance: (1) the classical typical absence seizure, (2) myoclonic absence seizures, (3) phantom typical absence seizures, and (4) eyelid myoclonia with absence (80). The task force also recognizes childhood absence epilepsy as a separate epileptic syndrome within idiopathic generalized epilepsies (79; 80). Other syndromes with predominant absence seizures identified by the ILAE Task Force are epilepsy with myoclonic absences, juvenile absence epilepsy, and juvenile myoclonic epilepsy.
All these types of seizures and electroclinical syndromes are mostly maintained in the report of the ILAE Commission on Classification and Terminology (17; 50). However, the latest ILAE position paper for the operational classification of seizure types does not consider phantom absences though “it is recognized that awareness and responsiveness can be at least partially retained during some generalized seizures, for example, with brief absence seizures, including absence seizures with eyelid myoclonias or myoclonic seizures” (86; 87). Furthermore, typical absence seizures designate a syndrome not yet recognized by the ILAE, such as facial (perioral or eyebrow) myoclonia with absences. Eyelid myoclonia with absences (previously known as Jeavons syndrome) has been recognized as a distinct genetic generalized epilepsy syndrome by the ILAE (206). Video-EEG polygraphic recordings have contributed to a better understanding of the various electroclinical characteristics and the close relationship with other types of seizures.
Nomenclature and inclusion and exclusion criteria. Childhood absence epilepsy (pyknolepsy) was defined by the 1989 Commission on Classification and Terminology of the International League Against Epilepsy as follows:
|
Pyknolepsy occurs in children of school age (peak manifestation age 6 to 7 years), with a strong genetic predisposition in otherwise normal children. It appears more frequently in girls than in boys. It is characterized by very frequent (several to many per day) absences. The EEG reveals bilateral, synchronous symmetrical spike-waves, usually 3 Hz, on a normal background activity. During adolescence, generalized tonic-clonic seizures often develop. Otherwise, absences may remit or, more rarely, persist as the only seizure type (49). |
However, this brief definition may be misinterpreted because of some ambiguity in its terminology as illustrated by the fact that many reports consider childhood absence epilepsy any type of epilepsy with onset of absences in childhood (190). Therefore, epidemiology, genetics, age at onset, clinical manifestations, other types of seizures, long-term prognosis, and treatment do not accurately reflect the syndrome of childhood absence epilepsy. A stricter definition of childhood absence epilepsy has been proposed (142; 143; 111; 168), which does not significantly differ from that of the Commission on Classification and Terminology of the International League Against Epilepsy (49):
(1) “Childhood absence epilepsy occurs in children of school age (peak manifestation age 6 to 7 years).” A simplistic but unsatisfactory practice is to label as childhood absence epilepsy any child with onset of daily absences at 10 years or earlier (as early as 2 years of age) (190; 191). The 10 years of age cut off is arbitrary and not a decisive “gold standard” to define what is and what is not childhood absence epilepsy. The arbitrary limit of 10 years is mainly based on the pioneering work of Doose and Janz who found that pyknoleptic absences usually start before 10 years of age, whereas non-pyknoleptic absences usually start after this age (76; 115). Typical absence seizures of recognized syndromes, such as myoclonic absence or juvenile myoclonic epilepsy, may start before the age of 10 years old and even in early childhood. When the onset is in early childhood, the characteristic electroclinical features of the two syndromes are recognized around 3 to 4 years later, when a successful treatment is discontinued and relapses occur. Further, “school age” does not include children younger than 3 to 4 years because absence seizures with onset at this age are heterogeneous; some may constitute discrete genetic entities; they are entirely different to those of childhood absence epilepsy (76; 106; 09; 40; 220); and they are often of bad prognosis (64; 139; 67). Childhood absence epilepsy starting before the age of 4 years and of good prognosis may be a very rare possibility that needs investigation (68; 198; 199).
If diagnosis by age at onset is to be followed, the study population should be correctly defined as “epilepsy with childhood absence seizures” rather than childhood absence epilepsy, which is a subset of it. Therefore, not all typical absence seizures occurring in childhood belong to the syndrome of childhood absence epilepsy. There are idiopathic generalized epilepsies and syndromes that start in infancy, childhood, and adolescence in which typical absence seizures, myoclonic seizures, or generalized tonic-clonic seizures are the only seizure type or the predominant type, and myoclonic seizures or typical absence seizures, alone or combined, may be part of the phenotype, respectively. Table 1 is a practical phenotypical classification of idiopathic generalized epilepsies with typical absence seizures, from infancy to adolescence (54).
|
A. Absence seizures as the only type or the predominant type of seizures. Myoclonic jerks and generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Absence seizures of early onset (under 3 years old) |
|
B. Myoclonic jerks as the only type or the predominant type of seizures. Typical absence seizures and generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Early-onset myoclonic absence epilepsy (under 3 years old) |
|
C. Absence and myoclonic seizures as the predominant types of seizures. Generalized tonic-clonic seizures may be part of the phenotype. | |
|
|
• Epilepsy with myoclonic absences (previously known as Tassinari syndrome) |
|
D. Generalized tonic-clonic seizures as the only type or the predominant type of seizures. Typical absence seizures and myoclonic jerks may be part of the phenotype. | |
|
|
• Generalized tonic-clonic seizures on waking or only with or without positive response to intermittent photic stimulation and patterns |
|
| |
(2) “Childhood absence epilepsy is characterized by very frequent (several to many per day) absences.” These absences are not only “several to many per day” but also severe according to the ILAE definition:
The hallmark of the typical absence attack is a sudden onset, interruption of ongoing activities, a blank stare, possibly a brief upward rotation of the eyes. If the patient is speaking, speech is slowed or interrupted; if walking, he stands transfixed; if eating, the food will stop on his way to the mouth. Usually, the patient will be unresponsive when spoken to. In some, attacks are aborted when the patient is spoken to. The attack lasts from a few seconds to half a minute and evaporates as rapidly as it commenced (48).
(3) In childhood absence epilepsy “the EEG reveals bilateral, synchronous symmetrical spike-waves, usually 3 Hz, on a normal background activity.” This terminology presumably excludes very brief typical absence seizures and fragmented, asymmetrical, and asynchronous spike-wave discharges of 3 to 5 Hz intradischarge variations during sleep and awake EEG.
(4) In the syndrome of childhood absence epilepsy or pure childhood absence epilepsy, no seizures other than typical absence seizures are accepted in the phenotype, and, as a rule, remission occurs before or early in adolescence. Otherwise, epilepsy with absences may remit or, more rarely, persist during adolescence and thereafter.
In addition, the ILAE, by accepting epilepsy with myoclonic absences and juvenile myoclonic epilepsy as separate syndromes, probably exclude myoclonic absences and brief mild absences from childhood absence epilepsy (49). It is by this logic that eyelid myoclonia and absences (which are primarily myoclonic) is also an exclusion criterion. Further, by accepting reflex absence seizures as a distinct category, the ILAE indicates that these too may not be part of childhood absence epilepsy. That perioral myoclonia or single violent jerks occurring in the course (not at onset) of typical absence seizures is an exclusion criterion may be debatable; however, their presence indicates worse prognosis (107; 103; 111). The same applies for multiple spikes and fragmented S-PWD that also indicate a bad prognosis, coexistent myoclonic jerks, or generalized tonic-clonic seizures (174; 83; 12). Brief typical absence seizures; myoclonic seizures; generalized tonic-clonic seizures; spanioleptic typical absence seizures; fragmented, asymmetrical, and asynchronous background activity; or photosensitivity are early indicators of a less favorable outcome if included in the phenotype (62; 54). All of these additional electroclinical features may indicate early-onset idiopathic generalized epilepsy; a well-defined syndrome, eg, juvenile absence epilepsy; or an unrecognized syndrome with childhood onset. When treatment is discontinued after 3 to 4 years, children with absence seizures and aberrant features may relapse with generalized tonic-clonic seizures or may even show electroclinical features of a well-defined syndrome, eg, juvenile myoclonic epilepsy (54), which explains the heterogeneous outcome that has been reported for childhood absence epilepsy.
The concise inclusion or exclusion criteria that define the syndrome of childhood absence epilepsy are summarized in Table 2 (142; 143; 144; 111; 168; 62; 54).
|
Childhood absence epilepsy |
Childhood absence epilepsy with aberrant electroclinical features | |
|
Clinical |
• Onset: 4 to 10 years of age (peak: 5 to 7 years of age) |
• Onset: before 3 to 4 years of age or after 10 years of age |
|
EEG |
• Ictal: high-amplitude rhythmic generalized spike-wave discharges and double spike-wave complexes, usually 3 Hz (range 2.5 to 4 Hz). Duration of 4 to 10 seconds, rarely above 20 seconds. |
• Brief (less than 4 seconds) generalized 3 to 4 Hz spike-wave discharges, focal spikes or slow spike-wave complexes, fragmented generalized spike-wave discharges |
|
Prognosis |
Favorable, self-limited before 12 years of age |
Less favorable |
The ILAE epilepsy manual classifies childhood absence epilepsy among epileptic syndromes of childhood (50), and the last ILAE Task Force on Nosology and Definitions classifies childhood absence epilepsy in idiopathic generalized epilepsies, a distinct subgroup of genetic generalized epilepsies (110). The term “genetic generalized epilepsies” includes other syndromes beyond the idiopathic generalized epilepsies.
The classification of childhood absence epilepsy is detailed below.
Childhood absence epilepsy. Childhood absence epilepsy is a genetic generalized epilepsy that should be considered in an otherwise normal child with multiple daily absence seizures associated with 2.5 to 4 Hz generalized spike-and-waves. Absence seizures are provoked by hyperventilation. Between 8 and 12 years of age, the distinction between the clinical syndromes of juvenile absence epilepsy and childhood absence epilepsy depends on the frequency of absence seizures.
A genetic generalized epilepsy is an epilepsy with generalized seizures associated with generalized epileptiform EEG patterns, such as generalized spike wave activity.
Clinical context. This syndrome is characterized by onset of frequent absence seizures between the ages of 2 to 12 years (peak 5 to 6 years of age). Both sexes are equally affected. Antecedent and birth history is normal. A previous history of febrile seizures may occur (seen in 15% to 20% of cases). Neurologic examination and head size are normal. Development and cognition are typically normal. Attention deficit hyperactivity disorder and learning difficulty may occur. Seizures are typically self-limiting. Self-limiting means having a high likelihood of seizures spontaneously remitting at a predictable age.
|
|
Caution. Onset of absence seizures less than 4 years, then consider glucose transporter disorders. |
Mandatory epileptic seizures. Absence seizures in childhood absence epilepsy are typically frequent (multiple daily) and brief (average 10 seconds). Awareness and responsiveness are usually severely impaired, but the child may be more responsive towards the end of the seizure.
|
|
Caution. Absence seizures longer than 45 seconds or with postictal phase should not occur. Then consider focal impaired awareness seizures (eg, due to structural brain abnormality). |
|
|
Caution. The presence of awareness during absence seizures. Then childhood absence epilepsy is unlikely, other syndromes with absences should be considered. |
|
|
Caution. If onset or offset of absences is not abrupt, or absences occur less than daily. Then consider other syndromes with absences. |
Generalized tonic-clonic seizures rarely precede or occur during periods of frequent absences but may occur later with evolution to other idiopathic generalized syndromes. Such cases may be part of childhood absence epilepsy in general, but not the syndrome childhood absence epilepsy with no aberrant electroclinical features (Table 2).
|
|
Caution. Generalized convulsive seizures before adolescence. Then consider other epilepsy syndromes. |
|
|
Exclusionary. Any other seizure type is not expected in this syndrome. |
EEG background. The background is normal. Occipital intermittent rhythmic delta activity (OIRDA) may be seen in a third of children with childhood absence epilepsy, at a frequency of 2.5 to 4 Hz and may have a notched appearance.
|
|
Caution. Focal slowing seen consistently in one area. Then consider structural brain abnormality. |
|
|
Caution. Generalized slowing is not seen. |
Interictal EEG. Generalized spike-and-wave, or fragments of generalized spike-and-wave, are seen in the interictal EEG. These are brief (usually less than 2 seconds) and occur most commonly seen in sleep.
|
|
Caution. Although focal spikes (as fragments of generalized spike-and-wave) can occur, if they consistently arise in one area, then consider structural brain abnormality. |
Activation. EEG abnormality and absence seizures are provoked by hyperventilation. If hyperventilation is poorly performed, generalized spike-and-wave may not be triggered. Intermittent photic stimulation triggers generalized spike-and-wave in a small proportion of individuals but does not induce seizures.
EEG abnormality is enhanced by sleep deprivation and by sleep. Generalized spike-and-wave often becomes fragmented with sleep deprivation or in sleep. Fragmented generalized spike-and-wave can appear focal or multi-focal but usually is not consistently seen in one area. The morphology of the focal spike-and-wave typically appears similar to the generalized spike-and-wave.
|
|
Caution. Where hyperventilation is performed well for 3 minutes, and no generalized spike-and-wave is seen in an untreated patient, childhood absence epilepsy is unlikely. |
Ictal EEG. Regular 3 Hz generalized spike-and-wave occurs associated with absence seizures. Polyspike-and-wave can occur in the ictal EEG.
|
|
Caution. Slow spike-and-wave (less than 2.5 Hz) is exclusionary. |
Imaging. Neuroimaging is normal. If the electroclinical diagnosis of childhood absence epilepsy is established, and there are no atypical features, neuroimaging is not required.
Genetics. It has a complex pattern of inheritance. In children who have absences with onset before 4 years of age, 10% have GLUT1 deficiency, which is caused by a mutation in SLC2A1 (208; 08). Other genes linked to this syndrome include GABRG2 and CACNA1A.
Family history of seizures/epilepsy. Up to 20% of patients may have a first-degree relative with seizures. When a family history is present, the affected family members typically have childhood absence epilepsy or a related genetic or idiopathic generalized epilepsy, eg, juvenile absence epilepsy, juvenile myoclonic epilepsy, or, less commonly, genetic epilepsy with febrile seizures plus.
A genetic generalized epilepsy is an epilepsy with generalized seizures associated with generalized epileptiform EEG patterns, such as generalized spike wave activity.
|
Differential diagnoses. | |
|
• Juvenile absence epilepsy: Consider if there are infrequent (eg, once daily) absence seizures in a child who is 8 years of age or older. | |
|
• Epilepsy with eyelid myoclonias: Consider if there is repetitive, rhythmic, fast greater than 4 Hz jerks of the eyelids, with upward deviation of the eyeballs, and with head extension; seizures are often very frequent and induced by eye closure (voluntary or on command) and photic stimulation. | |
|
• Epilepsy with myoclonic absences: Consider if there are 3 Hz myoclonic jerks of upper limbs with tonic abduction. | |
|
• Nonepileptic disorders such as daydreaming and inattention | |
|
| |
According to the position paper of the ILAE Commission for Classification and Terminology, childhood absence epilepsy is an idiopathic or genetic self-limited epilepsy (195). The 2017 ILAE classification suggested that the term “genetic generalized epilepsies” be used for the broad group of epilepsies with generalized seizure types and generalized spike-waves based on a presumed genetic etiology arising from twin and family research study data. It was suggested that the term “idiopathic generalized epilepsy” be reserved for the four syndromes: childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and generalized tonic-clonic seizures alone.
The four idiopathic generalized epilepsy syndromes continue to be regarded as a special group under the umbrella term “idiopathic generalized epilepsy syndromes” in the 2022 ILAE classification and are regarded as a distinct subgroup of genetic generalized epilepsies (110). The term “genetic generalized epilepsies” includes other syndromes beyond the idiopathic generalized epilepsy syndromes, such as epilepsy with myoclonic absences and epilepsy with eyelid myoclonia (previously known as Jeavons syndrome). The recognition of the idiopathic generalized epilepsy syndromes as a distinct subgroup of the genetic generalized epilepsies remains helpful as they carry prognostic and therapeutic implications.
In summary, childhood absence epilepsy occurs in an otherwise normal child, with daily absence seizures associated with 2.5 to 4 Hz generalized spike-waves at seizure onset. Age at onset is typically 4 to 10 years (range 2 to 13 years).
Neurologic examination is normal. Development and cognition are typically normal, but subtle specific learning difficulties and attention deficit hyperactivity disorder may be present. Epilepsy remits in 60% of children, often within 2 years of onset or by early adolescence.
Background EEG is normal. Fragmented generalized spike-waves can appear focal or multifocal but are not consistently seen in one area. The morphology of the focal spike-wave is similar to the generalized spike-wave. Polyspike-waves may be seen in drowsiness and sleep only, but not during wakefulness. Ictal EEG is characterized by regular 3 Hz (range 2.5 to 4 Hz) generalized spike-waves in the first second of seizure onset with absence seizures. Disorganized discharges, defined by brief (less than 1 second) or transient interruptions in the ictal rhythm or waveforms of different frequency or morphology, are significantly less common than in juvenile absence epilepsy.
Absence seizures are brief but may occur in clusters and are provoked by hyperventilation (110). Incontinence and loss of postural control can be seen. Seizures typically occur multiple times a day but are often under-recognized. Generalized tonic-clonic seizures rarely precede or occur during the period of frequent absence seizures in childhood. More commonly, they begin in adolescence, often after the resolution of absence seizures, and may herald evolution to another idiopathic generalized epilepsy syndrome.
Childhood absence epilepsy is the most common form of pediatric epilepsy, representing 10% to 17% of children with epilepsy (132). In general, childhood absence epilepsy is characterized by frequent (many per day) typical absence seizures. Severe impairment of consciousness is the essential feature of absence seizures in childhood absence epilepsy. Any other type of seizure at this early stage is likely to be part of the clinical expression but is incompatible with the diagnosis of the syndrome of childhood absence epilepsy.
Childhood absence epilepsy. Typical absence seizures are characterized by an abrupt onset and termination of complete impairment of consciousness, responsiveness, and memory with or without other signs. Within 1 or 2 seconds of the discharge, the eyes open spontaneously, and all activity stops (54). Spontaneous eye opening (when not caused by motor events of the seizure), like all other automatisms, occurs only when consciousness is severely impaired (173; 168). Impairment of consciousness may be the only clinical symptom (10%) or may be associated with other ictal clinical manifestations, such as mild clonic, tonic, atonic, automatisms, and autonomic features (90%). Mild clonic-myoclonic and tonic components of the eyelids and eyebrows may be observed at onset. However, marked eyelid, facial (perioral and or eyebrow), or limb and trunk myoclonic jerks are excluded by definition, particularly if they continue in the course of the absence seizure. Transient impairment of postural tone may lead to unsteadiness but rarely to a fall. Atonic falls do not occur (168; 54). The automatisms occur in two thirds of cases; are not stereotyped, even in the same patient; and may be de novo or perseverative. De novo automatisms are mostly simple, ie, lip licking, chewing, swallowing, scratching, rubbing, and fumbling with one’s clothes. Perseverative automatisms are more complex, ie, the child continues his or her previous activity in an unorganized way and is usually surprised by his or her actions or position at the end of the absence (54). Variability of the semiology exists not only among individuals but also within the same individual. Automatisms are related to the duration of the attack, ranging from 22% in a seizure lasting less than 3 seconds to 95% in a seizure lasting more than 16 seconds (176). The associated clinical phenomena usually appear in combination and are of no prognostic significance. Pallor is common. Incontinence of urine or genital and sexual manifestations represent rare clinical phenomena in typical absence seizures. Absence seizures occur many times a day in a predisposed individual, either spontaneously or influenced by various factors, such as emotional, physical, and mental inactivity; lack of sleep; reading difficulties; and metabolic factors (hypoglycemia, hyperventilation). Of these, the main precipitating factor and the easiest way to provoke absence seizure is hyperventilation. In contrast, absence seizures do not usually occur during periods of mental or physical activity (54). The duration of absence seizures is around 10 seconds (range 4 to 20 seconds, exceptionally longer), and there are many per day.
Untreated absences with mild impairment of consciousness may be an exclusion criterion (167; 169). The hallmark of the absence attack is a sudden onset and interruption of ongoing activities, often with a blank stare. Lennox and Lennox stated: “If warning occurs, the diagnosis of petit mal may be questioned” (134). The attack ends as abruptly as it commenced. The child resumes his or her ongoing activity as if nothing happened and is usually unaware of the seizure.
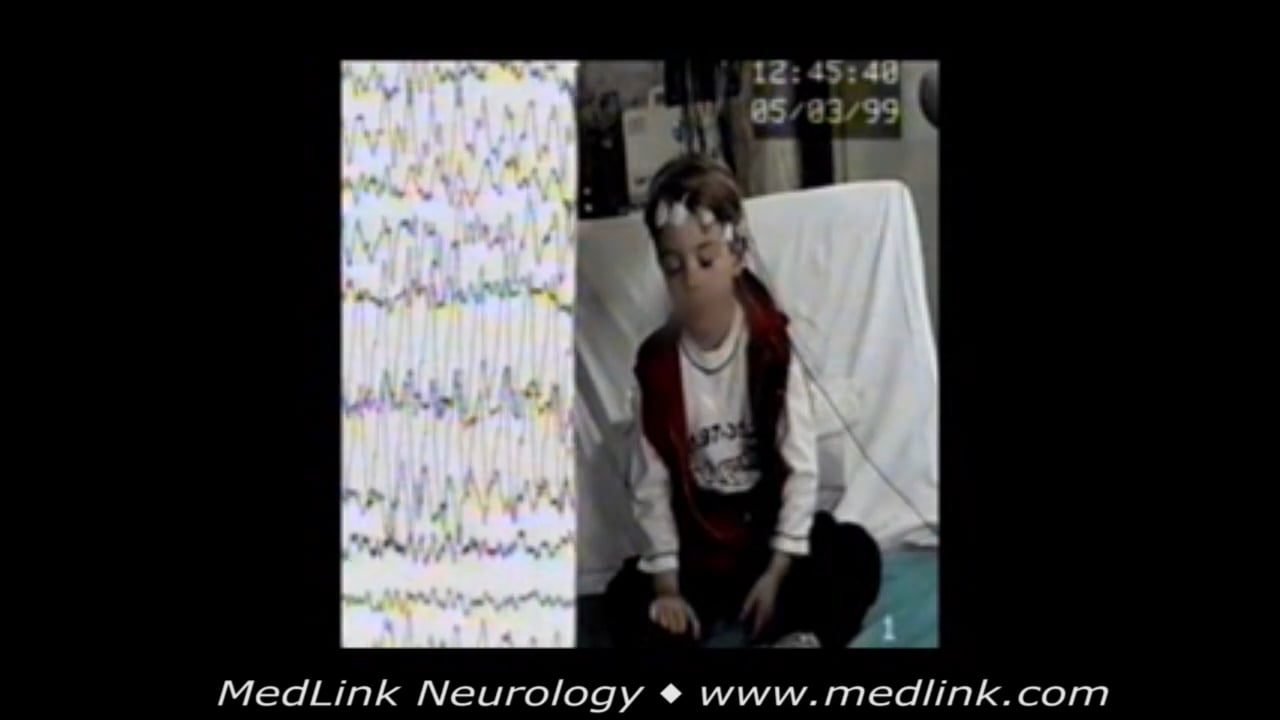
Typical absence seizures in a 4.8-year-old boy with normal development and a history of absence seizures occurring at a frequency of over 10 per day for the last 3 months. During hyperventilation, he suddenly stopped, his eyes ...
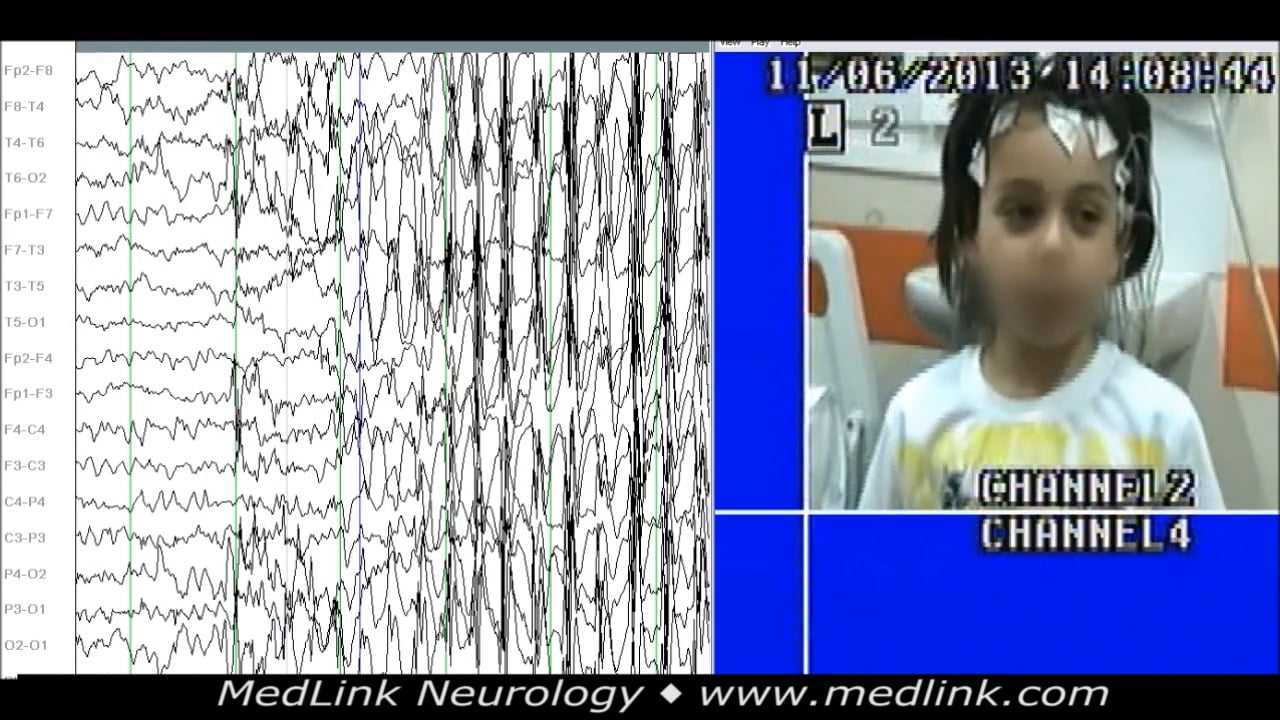
A 6.3-year-old girl with a history of a few staring episodes per day. During sleep video-EEG, frequent generalized spike-wave discharges of around 3 Hz, or occasionally fragmented, brief, double-spike or spike-wave generalized ...
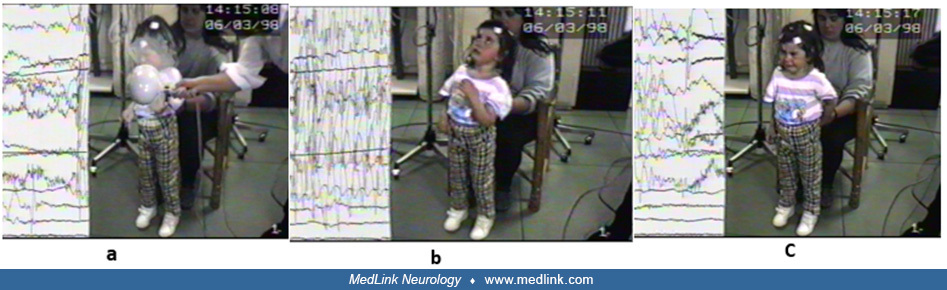
Typical absence seizures in a 5.5-year-old boy. During hyperventilation, he stopped overbreathing, and his eyes opened and stared within 1 second of discharge onset. After about 3 seconds, he started rhythmic-marked horizontal ...
A 3.8-year-old girl who, for the past 6 months, had episodes of staring with eyes up, head back, and some eyebrow jerking. Occasionally she would lose her balance. She was given phenobarbitone by her pediatrician. A few weeks b...
A diagnosis of childhood absence epilepsy should be seriously questioned in an untreated child who does not have an attack on hyperventilation (239). In a series from Strasbourg, hyperventilation provoked typical absence seizures in 100% of untreated patients with childhood absence epilepsy (109). Typical absence seizures that are consistently elicited by specific stimuli, such as light or patterns, do not belong to childhood absence epilepsy (49; 109; 142; 143; 167; 169; 111; 54).
Other types of seizures. Generally, other types of epileptic seizures are not part of childhood absence epilepsy. In particular, generalized tonic-clonic seizures or myoclonic jerks do not usually occur in childhood absence epilepsy preceding or concomitant with the stage of active absence seizures (49; 109; 142; 143; 54; 169). Generalized tonic-clonic seizures as a presenting seizure or during childhood are incompatible with the syndrome of childhood absence epilepsy. However, infrequent generalized tonic-clonic seizures in adolescence or adult life have been reported (49; 142; 143; 144; 202). In a Swedish population-based study, patients with absence epilepsy who only had absences had a 91% remission rate (108). Conversely, in another study comparing 11 patients with childhood absence epilepsy and generalized tonic-clonic seizures (preceding or concurrent with absence seizures) and 30 patients with typical childhood absence epilepsy, the two groups showed a similar prognosis (234). Absence status may occur in 5% to 16% of cases, with typical absence seizures starting before the age of 10 years (73), but this is probably incompatible with the syndrome of childhood absence epilepsy (04; 166; 167; 169).
Kessler and associates studied pretreatment seizure semiology in video of 1932 electrographic absence seizures from 416 patients with childhood absence epilepsy (120). Median seizure duration was 10.2 seconds; median time between electrographic seizure onset and clinical manifestation onset was 1.5 seconds. For individual seizures and by participant, the most common semiology features were pause/stare (seizure 95.5%, participant 99.3%), motor automatisms (60.6%, 86.1%), and eye involvement (54.9%, 76.5%). Variability of semiology features between seizures even within participants was high. Clustering analyses revealed four patterns (involving the presence/absence of eye involvement and motor automatisms superimposed on the nearly ubiquitous pause/stare). Most participants experienced more than one seizure cluster pattern. No individual semiologic feature was individually predictive of short-term outcome. One seizure subtype, pause/stare and eye involvement but no motor automatisms, was specifically associated with a worse treatment outcome (120). Absences are usually frequent throughout the day: if attacks do not recur daily, the diagnosis may be questioned. They begin in driblets, the parents noting short episodes of immobility or eye-rolling but passing it off as daydreaming or an emotional display. In time, however, the blackout periods increase in frequency or in duration and cannot be disregarded any longer. At this moment, they range from 10 to 200 per day (134).
An interesting observation is the occurrence of childhood absence epilepsy in an undetermined but probably small number of patients with benign childhood focal epilepsies (rolandic epilepsy, Panayiotopoulos syndrome, and idiopathic childhood occipital epilepsy of Gastaut) (16; 213; 37; 168; 228; 229). In a study of 277 children with self-limited epilepsy with centrotemporal spikes and 93 children with childhood absence epilepsy, nine children were identified to have overlapping EEG findings of both epileptic syndromes (116). This overlap is mostly in electrographic features with or without the clinical features seen in both syndromes.
Absence seizures occur in various epilepsy syndromes, including childhood and juvenile absence epilepsy and juvenile myoclonic epilepsy. When children present with absence seizures at ages when syndromes overlap, initial syndrome designation is not always possible, making early prognostication challenging. In a retrospective study, children with new-onset absence seizures between 8 and 11 years of age with at least 5 years of follow-up data were studied through the review of medical records and initial EEG tracings (69). Ninety-eight patients were included in the study. Forty-six percent developed generalized tonic-clonic seizures, and 20% developed myoclonic seizures. On initial EEG, bifrontal and background slowing and myoclonic seizures and anxiety are associated with developing generalized tonic-clonic seizures, which is of prognostic significance when early syndrome designation is difficult (69).
Age and sex at onset. The onset of childhood absence epilepsy is classically considered to be between 4 and 10 years old, with a peak at 5 to 7 years old (see above).
Childhood absence epilepsy is clearly more frequent in girls than in boys. Sixty percent to 70% of affected children are girls (134; 20; 27). In 124 children with typical absence epilepsy, the female-to-male ratio was 1.3 to one for children between 4 and 8 years old, two to one for children under the age of 4 years, and one to one for children over the age of 8 years (62). The duration was around 10 seconds (range 4 to 20 seconds, exceptionally longer), and the events occurred many times a day.
In general, childhood absence epilepsy has a good prognosis. Sixty-five percent of patients with absence epilepsy will outgrow their seizures. On the other hand, 18% to 35% will go on to develop pharmacoresistant epilepsy (10; 132). Children with the syndrome of childhood absence epilepsy do not, as a rule, show neurologic deficits, and in the majority of cases, seizures cease spontaneously with ongoing maturation. However, some children with childhood absence epilepsy demonstrate a less favorable response to therapy, exhibit cognitive deficits and long-term psychosocial difficulties, and have variable remission rates. A high incidence of learning and behavioral problems has been reported in absence seizures of frontal onset (130).
Treatment response. In a study of 124 cases, 102 presented with typical absence seizures only, eight presented with typical absence seizures followed by generalized tonic-clonic seizures, and 14 presented with generalized tonic-clonic seizures followed by typical absences (62). Of the 102 cases, 11 (11%) presented under the age of 4 years, 57 (56%) between the ages of 4 and 8 years, and 34 (33%) between the ages of 8 and 13.5 years. In 65 (52%) of the 124 cases, school performance was assessed as low average, and the duration of absence seizures prior to the start of an appropriate and effective treatment varied from 0.1 to 7 (mean 1.5 ± 1.6) years. In contrast, the duration of epilepsy in the remaining 59 (48%) cases with normal school performance was 0.1 to 3 (mean 0.5 ± 0.5) years. The low average performance of the 65 cases was attributed to a longer duration of epilepsy and the high frequency of daily absence seizures prior to starting an appropriate and effective treatment. Furthermore, the outcome was favorable in those cases between 4 and 8 years old with absence seizure onset without aberrant electroclinical features (Table 2). The relapse percentage of childhood absence epilepsy with aberrant electroclinical features was 33%. These were mainly cases that started before 3 years of age and had a mixture of typical absence seizures, myoclonic seizures, and generalized tonic-clonic seizures; cases of juvenile absence epilepsy starting in childhood; or even cases diagnosed as juvenile myoclonic epilepsy on relapsing after treatment was discontinued (62; 55; 54). The percentage of females in the relapse group was 77% compared to 43% in the nonrelapse group, and relapses occurred around menarche. Females appear to have a less favorable outcome on relapse, which is possibly related to the proconvulsive effect of female hormones compared to the antiseizure activity of testosterone (212). The predictors of long-term outcome were assessed in a study of 106 children with childhood absence epilepsy and a mean follow-up of 5 years (07). A history of headache or of generalized tonic-clonic seizures, along with the cumulative number of antiseizure medications utilized, predicted seizure recurrence on antiseizure medication discontinuation.
In a study, 63 patients were diagnosed with childhood absence epilepsy according to the Panayiotopoulos criteria (214). Young age was an advantage for treatment response (age at diagnosis: 7.48 ± 1.62 responsive versus 8.50 ± 1.64 nonresponsive). Occipital, intermittent, rhythmic delta activity and early treatment were determined to be good prognostic factors in childhood absence epilepsy, whereas high amplitude and high frequency of spike slow-wave discharge were poor prognostic factors.
A gradual withdrawal of medication is recommended after a seizure-free and EEG normalization period of at least 3 years. For cases with combined treatment, the last antiseizure drug introduced is withdrawn first. Some children with aberrant features subsequently develop generalized tonic-clonic seizures in adolescence or early adulthood, or juvenile myoclonic epilepsy.
Most of the available evidence is inconclusive regarding evolution and prognosis of childhood absence epilepsy (27; 111; 168; 169; 153). This is because classification criteria are markedly different in the relevant reports or of insufficiently short follow-up periods. Most authors include in childhood absence epilepsy any child with absences before the age of 10 years, which may not be childhood absence epilepsy (62; 55; 167; 168; 169; 56; 54). Retrospective studies in adults may lack accurate initial data. Patients must be followed beyond 18 or 20 years of age (145; 143; 141).
Childhood absence epilepsy, if properly defined, may have an excellent prognosis. In 1924, at a time when no anti-absence drug existed, Adie concluded that even if absence seizures in pyknolepsy persisted for a long time, they ultimately ceased, never to return (02). This is consistent with findings that absences of childhood absence epilepsy, even if they may persist several years, finally disappear with age in more than 90% of cases (145; 142; 143). In adults, absences of idiopathic generalized epilepsy (not necessarily childhood absence epilepsy) generally tend to be infrequent and milder, and they may occur with precipitating factors (92; 111), though in some of them absences may be extremely severe as in juvenile absence epilepsy (04; 167; 169).
However, cessation of absence seizures may not mean remission. This again depends on diagnostic inclusion and exclusion criteria. Considering absences with onset in childhood as childhood absence epilepsy, prognosis is uncertain and has great variations (62; 142; 27; 169; 107; 103; 203; 222; 54). Generalized tonic-clonic seizures may appear mainly between 8 and 15 years of age (73) or sometimes even later, between 20 to 30 years of age (92), and patients may develop juvenile myoclonic epilepsy (238; 70) or, more rarely, eyelid myoclonia with absences (91). Absence seizures in these patients may persist, improve, or disappear. With an early institution of effective therapy, generalized tonic-clonic seizures occurred in 30% of cases, whereas occurrence was 68% after incorrect therapy (20).
When stricter criteria are applied for childhood absence epilepsy, generalized tonic-clonic seizures are infrequent and easily controlled by medication (145; 142; 141; 107; 103; 222; 153). Loiseau and colleagues studied 53 patients older than 20 years at last follow-up. Inclusion criteria were age at onset (3 to 10 years) of daily and EEG-recorded typical absences as a presenting sign of normal children who were seen within the first year of onset or treatment and with no history of preceding seizures other than febrile convulsions (141; 142). EEG with multiple or irregular spike-waves or photosensitivity were excluded. Absences persisted in five (less than 10%) and in two of them as the only type of seizure. Generalized tonic-clonic seizures occurred in 14 patients (26%), but in 11 of them generalized tonic-clonic seizures were isolated or rare. Generalized tonic-clonic seizures were more common among patients with onset of absences from 9 to 10 years and without posterior delta rhythms. Control of absences with treatment varied; it was achieved in 12 patients within weeks, but in most cases they persisted for years. The later the onset of typical absence seizures, the higher is the risk of convulsive seizures. Generalized tonic-clonic seizures developed in 16% of patients with onset of typical absence seizures before 9 years of age; this raised to 44% for those with onset of typical absence seizures between 9 and 10 years. (142).
Forty-five of the 124 cases (female:male = 1.4:1) reported in 1992 were followed after treatment was withdrawn (62). At the time of diagnosis, none of the 45 cases had any obvious aberrant electroclinical features. Four female cases relapsed, and all had spaniolepsy absences. In certain cases, spaniolepsy may also coexist with other risk factors, such as briefer generalized spike-wave discharges and generalized tonic-clonic seizures. An additional trigger factor for females that has subsequently been identified is that some females relapse during menarche (212). This may be due to the reported proconvulsive effect of estrogens.
Grosso and colleagues studied 119 patients with childhood absence epilepsy (103); 57 patients were diagnosed on the basis of the broad ILAE criteria and 62 with the exclusion criteria in Table 2 (group 1). Patients in group 1 had significantly higher rates of seizure control (95% vs. 77%), higher rates of terminal remission (82% vs. 51%), fewer generalized tonic-clonic seizures (8% vs. 30%), and shorter mean periods of treatment (2.2 vs. 3.8 years); group 1 was also less likely to be on polytherapy (11% vs. 47%) or suffer seizure relapses at antiseizure drug discontinuation (0 vs. 22%). Valentin and colleagues classified patients with idiopathic generalized epilepsy and absences according to (1) the ILAE criteria of 1989 and (2) the exclusion criteria for childhood absence epilepsy in Table 2 (222). Among the 171 patients with absences, 46 (27%) had only typical absences; 73 (43%) had absences and generalized tonic-clonic seizures; 33 (19%) had absences, myoclonic jerks, and generalized tonic-clonic seizures; fiev (2%) had absences and myoclonic jerks only; 14 (8%) had absences with generalized tonic-clonic seizures or myoclonus with other seizure types. Of 129 patients with absences without significant myoclonus, 50 had juvenile absence epilepsy, 44 had childhood absence epilepsy according to the 1989 criteria, and 35 could not be classified. Thirty of these patients had childhood absence epilepsy per the inclusion and exclusion criteria in Table 2, and these had a significantly better outcome than those classified according to the 1989 classification.
In another approach, Agathonikou and colleagues studied 39 adults with idiopathic generalized epilepsy and typical absence seizures starting before 10 years of age (03). All were older than 18 years (31.5 ± 10.5; range 18 to 56) and all had EEG-recorded (15 with video-EEG) typical absences. Typical absences had onset at 6.2 ± 1.9 years (range 2 to 9) and still persisted in 28 (71.8%). Generalized tonic-clonic seizures occurred in 87.2% (onset 13 ± 7.2 years; range 2 to 36). Myoclonic jerks occurred in 38.5% (onset 2.6 ± 4.1 years; range 7 to 18). Sex (women, 82%) and photosensitivity (56.4%) were markedly predominating factors. Only one of them fulfilled strict criteria of childhood absence epilepsy and was well controlled on medication. Of the others, eight were classified as eyelid myoclonia with absences, five as juvenile absence epilepsy, four as perioral myoclonia with absences, three as juvenile myoclonic epilepsy, three as absences with single myoclonic jerk, three as predominantly photosensitive idiopathic generalized epilepsy with typical absence. Twelve patients had unclassified idiopathic generalized epilepsy (eight with photosensitivity). Typical absence seizures with an onset in childhood are heterogeneous and occur in all idiopathic generalized epilepsies in adults; they should not be included in childhood absence epilepsy.
In a report, 47 children with newly diagnosed childhood absence epilepsy were followed for 12 to 17 years (29). Overall prognosis was excellent with few children (7%) still having seizures after 12 to 17 years. The children were subdivided into three groups for the analysis: those becoming seizure-free (I) within 1 month after enrollment; (II) 1 to 6 months after enrollment; and (III) more than 6 months after enrollment or having seizures continuing during follow-up. No significant differences were observed between groups in sex, age at onset, occurrence of febrile seizures, and positive first-degree family history for epilepsy. All groups had high remission rates after 12 to 17 years. Significantly more relapses occurred in group III than in group I. Total duration of epilepsy and mean age at final remission were 3.9 and 9.5 years, respectively, being significantly longer and higher in group III than in groups I and II. In all groups, only a small number of children (total 13%) developed generalized tonic-clonic seizures. Remission rate in children with childhood absence epilepsy cannot be predicted on the basis of baseline and EEG characteristics. The early clinical course (ie, the first 6 months) has some predictive value with respect to the total duration of absence epilepsy.
Neuropsychiatric comorbidities. Attention, cognitive, linguistic, behavioral, social, and vocational disturbances in children with typical absence seizures have been noted for many years (62; 164; 175) and have been re-emphasized (152; 216; 33; 147; 43). These are of no surprise considering the high frequency of absence seizures every day. Many types of psychological testing used during generalized spike-wave discharges since 1939 have documented transient cognitive dysfunctioning that may start just before the onset and tend to get worse with longer duration of the discharge (196; 158; 101; 197; 179; 28). Mirsky and colleagues described it as follows:
|
The transitory bursts of spike-wave activity represent the tip of an iceberg. Below the surface, there may be a more or less continuously active pathophysiological process, which is reflected in impaired performance on tests of attention… (157). |
In a study, 69 patients diagnosed with childhood absence epilepsy were compared to 101 normal children (35). Twenty-five percent of the patients had subtle cognitive deficits; 43% had linguistic difficulties; 61% were assigned a psychiatric diagnosis, particularly attention deficit hyperactivity disorder and anxiety disorders; and 30% had clinically relevant Child Behavior Checklist broadband scores. The most frequent Child Behavior Checklist narrow band factor scores in the clinical or borderline range were attention and somatic complaints, followed by social and thought problems. Duration of illness, seizure frequency, and antiseizure drug treatment were related to the severity of the cognitive, linguistic, and psychiatric comorbidities. Only 23% of the subjects with childhood absence epilepsy received intervention for these problems, heralding the need for better and more comprehensive early intervention and treatment.
Conant and colleagues used a multivariate analysis of variance to compare the neuropsychological functioning of 16 children with childhood absence epilepsy with that of 14 children with type 1 diabetes mellitus and 15 healthy children (51). The childhood absence epilepsy group did not perform differently from the other groups on measures of intellectual functioning, memory, academic achievement, fine motor speed, or processing speed. In contrast, significant differences were found in problem solving, letter fluency, complex motor control, attention/behavioral inhibition, and psychosocial functioning. These results suggest that children with childhood absence epilepsy show difficulties in neuropsychological functions thought to be subserved by the basal ganglia-thalamocortical circuits.
Vega and colleagues examined the specific types of attention-related problems in 38 children with childhood absence epilepsy experience and the role of disease factors in the development of attention-related problems (226). These were compared with 46 matched healthy controls. The Behavior Assessment System for Children (BASC) was completed by parents, and the Attention Problems and Hyperactivity subscales were used to characterize the problems of children with childhood absence epilepsy. Item analysis within the subscales revealed that children with childhood absence epilepsy demonstrate higher rates of hyperactive (overactivity and fidgetiness) and inattentive (forgetfulness and distractibility) problems and require more supervision. Analyses within the childhood absence epilepsy group revealed that those who were actively having seizures were more impatient and those with a longer duration of illness were less proficient in completing homework. The authors concluded that children with childhood absence epilepsy are at risk for certain inattentive and hyperactive problems, which can differ depending on duration of illness and active seizure status. In another report, the same authors examined the specific anxiety and depression symptoms children with childhood absence epilepsy experience and explored the role of disease factors in the severity of their presentation (225). Forty-five subjects with childhood absence epilepsy and 41 healthy matched controls, aged 6 to 16 years, participated in the study. The Behavior Assessment System for Children was completed by parents, and the Anxiety and Depression subscales were used to characterize problems. Item analysis within the subscales revealed that children with childhood absence epilepsy demonstrated higher rates of symptoms of anxiety (nervousness and thought rumination) and depression (sadness and crying), as well as more general psychosocial problems, including isolation and low self-esteem. Disease duration, intractability, and medication effects were not associated with higher rates of affective problems in this limited patient sample. The authors suggested that “screening of patients with childhood absence epilepsy for comorbid psychiatric disorders early by focusing on specific symptom profiles unique to this population may enhance overall treatment and developmental outcomes.”
D'Agati and colleagues investigated whether specific executive functions and attention deficit patterns were present in a well-defined group of 15 children with childhood absence epilepsy who were taking valproate (66). These were compared with 15 healthy controls aged 8 to 15 years and matched for sex, age, and IQ. Comparisons were made in the following neuropsychological domains: planning and problem solving, verbal fluency, verbal short-term memory, verbal working memory, visuospatial memory, and sustained and divided attention. No differences were found between the two groups on measures of intellectual functioning, verbal short-term memory, and visuospatial memory. By contrast, significant differences were found in total time of planning task, phonological and category fluency, and sustained and divided attention.
Cerminara and associates investigated several components of attention in children with childhood absence epilepsy using a unique computerized test battery for attention performance (39). Participants included 24 patients with childhood absence epilepsy (all in medication and mainly with sodium valproate) and 24 controls matched for age and sex. The test battery included the following tasks: selective attention, impulsivity, focused attention, divided attention, alertness, and vigilance. Compared with healthy controls, patients with childhood absence epilepsy made more commission errors in the Go/No-Go task and more omission errors in the divided attention task. Patients with childhood absence epilepsy also showed decreased reaction times in measures of selective attention and a great variability of reaction times in alertness and Go/No-Go tasks. The authors concluded that their findings suggested that patients with childhood absence epilepsy were impaired in tonic and phasic alertness, divided attention, selective attention, and impulsivity. However, drugs, particularly valproate, may have contributed to these findings and particularly attentional disturbances, which were seen in the valproate but not in the ethosuximide cohort (98; 99).
Vanderwiel and associates found a high risk of comorbid ADHD in patients with absence epilepsy; children with childhood absence epilepsy are approximately 5.7 times more likely to have a diagnosis of ADHD compared to children without epilepsy (223). They determined that there was no significant relationship between the presence of ADHD and the duration of active epilepsy, age of seizure onset, age of epilepsy diagnosis, or lag in diagnosis. They also determined that both groups had favorable responses to ADHD medication, and use of ADHD medication was not associated with seizure exacerbations. There is also an increased risk of deficits in other neurocognitive functions, including linguistic skills, visual-spatial skills, and sleep disorders. It is suspected that there may be a common underlying neurobiological etiology, perhaps involving abnormal neuronal circuitry or genetic factors.
Masur and associates assessed pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy (152). Subjects with newly diagnosed childhood absence epilepsy entering a double-blind, randomized controlled clinical trial had neuropsychological testing, including assessments of general intellectual functioning, attention, memory, executive function, and achievement. Attention was reassessed at the week 16 to 20 visit. At study entry, 36% of the cohort exhibited attention deficits despite otherwise intact neurocognitive functioning. Structural equation modeling of baseline neuropsychological data revealed a direct sequential effect among attention, memory, executive function, and academic achievement. At the week 16 to 20 visit, attention deficits persisted, even if seizure freedom was attained. More subjects receiving valproic acid (49%) had attention deficits than subjects receiving ethosuximide (32%) or lamotrigine (24%) (p = 0.0006). Parental assessment did not reliably detect attention deficits before or after treatment (p < 0.0001). The authors concluded that children with childhood absence epilepsy have a high rate of pretreatment attentional deficits that persist despite seizure freedom. Rates are disproportionately higher for valproic acid treatment compared with ethosuximide or lamotrigine. Parents do not recognize these attentional deficits. These deficits present a threat to academic achievement. Vigilant cognitive and behavioral assessment of these children is warranted (152).
Gencpinar and colleagues evaluated the executive function of 19 children with typical absence epilepsy and 19 healthy subjects (93). There was a significant difference between the groups on the Serial Digit Learning Test and on a subtest of the Wisconsin Card Sorting Test. The authors concluded that long-term risk for learning impairments, failure in executive abilities, and short-term memory and attention disorders can occur in children with absence epilepsy.
Absence epilepsy and attention deficits show some patterns of pathophysiological association. This relationship may account for dysfunctions in everyday activities in the pediatric population. Particular metrics, such as the risk related to biking in children with absence epilepsy, should be used in future studies to address the problem in a novel way and to impact clinical indications (15). The risk for accidental injuries during absence seizures compared with controls is well reported (38).
Α systematic review and meta-analysis synthesizing human and animal models investigated mood disorders and absence seizures (104). Patients with absence seizures had greater odds of developing depression and anxiety when compared to controls (odds ratio = 4.93; 95% CI = 2.91–8.35; p < 0.01). These studies further suggest a strong correlation between absence seizures and depression and anxiety in the form of a bidirectional relationship sharing underlying mechanisms, such as genetic predisposition, neurophysiology, and anatomical pathways. Additionally, antiseizure drugs and antidepressant drugs affect both absence seizures and mood disorders, suggesting a common psychopathology.
Sleep patterns may also be disturbed in children with untreated or uncontrolled childhood absence epilepsy (74). Children with controlled absences show significant improvement in total sleep time, REM-sleep latency, REM-sleep, arousals-number/hour, and arousals-duration/hour (74).
Social adaptation is poor in one third of patients with childhood absence epilepsy, even when in remission (145; 73; 237). The effect of antiseizure drugs, the impact of stigmatizing because of “epilepsy,” parental and mates’ attitudes, bias in selection of the most serious cases, inclusion of idiopathic generalized epilepsies other than childhood absence epilepsy, and other factors must be considered when assessing the true dimension of these problems prior to concluding that childhood absence epilepsy is “misconceived as a benign syndrome.”
According to Camfield and colleagues, approximately 15% of patients with childhood absence epilepsy (probably not the classical childhood absence epilepsy) who initially remit during their childhood years later develop juvenile myoclonic epilepsy as teenagers (32). These patients will have many issues for continuing medical care and transition because their seizure disorder generally persists into adulthood.
In a Danish cohort study, school performance and psychiatric comorbidity in 114 children with childhood absence epilepsy was compared with both population controls and non-neurologically chronically ill children (26). Children with childhood absence epilepsy had increased psychiatric comorbidity, and a considerable proportion of these children receive special needs education in primary and secondary school. However, this is insufficient to normalize their considerably lower grade point average in the ninth grade by 1.7 grade points.
In a comparative cross-sectional study of 30 children with typical childhood absence epilepsy—15 on antiepileptic drugs (group I), 15 newly diagnose-drug naive (group II), and 15 healthy children, control group (group III)—both group I and group II patients with childhood absence epilepsy (onset above 4 years of age, no generalized tonic-clonic and myoclonic seizures) demonstrated significant executive dysfunction, putting them at risk of learning impairment, in comparison with the controls (192). In another study, reduction in density and duration of sleep spindle that is associated with the mechanisms underlying childhood absence epilepsy was found substantially lower in the cognitively impaired group than in the cognitively unimpaired group, which is a useful predictive indicator of cognitive impairment in children with absence epilepsy (247). Furthermore, in juvenile absence epilepsy, the reported widespread cognitive dysfunction was attributed to a detrimental effect on cognitive performance that have the common prolonged “subclinical” discharges on EEG of patients with juvenile absence epilepsy (71).
In a study, pretreatment EEG features of patients with newly diagnosed childhood absence epilepsy were assessed in relation to the baseline neuropsychological function and treatment outcome (75). Occurrence of a seizure greater than or equal to 20 seconds, but not overall seizure frequency, was associated with differential baseline measures of attention. Patients whose shortest pretreatment EEG seizure was longer in duration were more likely to achieve seizure freedom, regardless of treatment.
In childhood absence epilepsy, as in any type of epilepsy, a comprehensive neuropsychological assessment for concomitant cognitive and other comorbidities should be routinely applied in addition to early diagnosis and treatment, and appropriate early intervention and treatment should be introduced for better prognosis and quality of life. Further, when the word “epilepsy” is disclosed at critical stages of development, as a rule, it leads to poor self-esteem, lack of self-confidence, social withdrawal, and poorer subsequent quality of life (46), even in cases of self-limited epilepsies.
Genetic etiologies. It is thought that typical absence seizures are genetic in origin with polygenic inheritance factors, although most patients do not have a family history and may have a de novo mutation or exhibit complex inheritance (132). Childhood absence epilepsy is a genetically determined idiopathic generalized epilepsy; the complex polygenic genetic component has not yet been clearly identified. Positive family history for epilepsy in first-degree relatives has been found to vary, from 15% to 44% (64; 148). A 20% positive family history of epilepsy and a 7% positive family history of febrile seizures, with a 74% concordance rate, has been found in monozygotic twins (62; 54).
Six-year-old monozygotic twins presented with frequent (>10) typical absence seizures. Sleep-awake video-EEGs showed normal background activity and typical generalized spike-wave discharges with concomitant absences provoked...
In two independent series, 17% of patients with childhood absence epilepsy had first-degree relatives with epilepsy manifesting with absence seizures, generalized tonic-clonic seizures, or both (142; 30). In studies on twins, 84% of monozygotic twins had 3 Hz spike-waves; typical absence seizures developed in 75% of pairs and 16 times less often in dizygotic twins (134). However, as concordance in monozygotic twins is not 100%, nongenetic factors are likely (21; 22). Bianchi and colleagues found that in 24 families with a childhood absence epilepsy proband, there was a high concordance (33.3%) for the same clinical form in first-degree relatives, whereas febrile convulsions (46.7%) and generalized tonic-clonic seizures (30%) were more common in distant relatives (25). Thus, a genetic abnormality is responsible for a predisposition and for the EEG trait, but childhood absence epilepsy, which is its clinical expression, is likely to be multifactorial. Acquired factors may be considered (233).
Although childhood absence epilepsy is genetically determined, the precise mode of inheritance and the genes involved remain largely unidentified. Various chromosomal loci have been identified in families with absences of childhood onset (not necessarily equated with childhood absence epilepsy). Linkage to chromosome 1 was found in families with absences starting in childhood and later developing myoclonic jerks and generalized tonic-clonic seizures as in juvenile myoclonic epilepsy (70; 154). Linkage analysis of a 5-generation family in which affected patients had childhood absences and generalized tonic-clonic seizures provided evidence for a locus on chromosome 8q24 (88; 70). The candidate region for this locus, designated ECA 1, has been refined, but a gene remains to be identified. According to the criteria proposed in this chapter, neither of these groups is childhood absence epilepsy. Mutations linked to the GABA(A) receptor (GABRG2 gene) on chromosome 5q3,1–33 have been found in childhood absence epilepsy with febrile seizures (235). In another subset of childhood absence epilepsy, mutations were found in GABA(A) receptor gene GABRB3 in Mexican families (211). The expression of this gene in the developing brain may explain the age-related expression of childhood absence epilepsy.
A genome-wide high-density SNP-based linkage analysis of 41 nuclear pedigrees with at least two affected members with childhood absence epilepsy identified a susceptibility locus on chromosome 3p23-p14 (45). The linked region harbors the functional candidate genes TRAK1 and CACNA2D2. Fine-mapping using a tagSNP approach demonstrated disease association with variants in TRAK1.
There are reports focusing on genes encoding GABA receptors or brain expressed voltage-dependent calcium channels that may underlie childhood absence epilepsy. Feucht and colleagues (85) found a significant association between a polymorphism in GABRB 3 in chromosome 15q11 and patients of 50 families with childhood absence epilepsy. Marini and colleagues (149) found GABA-A receptor gamma2 subunit gene (GABRG2) mutations on chromosome 5 in a large family with childhood absence epilepsy and febrile seizures (including febrile seizures plus and other seizure phenotypes). This gene mutation segregated with febrile seizures and childhood absence epilepsy and also occurred in individuals with the other phenotypes.
A T-type calcium channel CACNA1H gene encoding Ca(v)3.2 has been associated with childhood absence epilepsy in Chinese individuals (42; 138) but not in Europeans (44). Another report found evidence that genes encoding brain-expressed voltage-gated calcium channels, including CACNG3 on chromosome 16p12–p13.1, are a susceptibility locus in a subset of childhood absence epilepsy in European patients (82). The dichotomy between thalamocortical versus neocortical may contribute to the discovery of susceptibility genes for photoparoxysmal response associated with absence epilepsy in certain cases (61).
Contrary to these findings, a previous study of 33 nuclear families, each with two or more individuals with childhood absence epilepsy, found no evidence that genes encoding GABA-A and GABA-B receptors, voltage-dependent calcium channels, and the ECA1 region on chromosome 8q account independently for the childhood absence trait in a majority of the families (186). Further, in five consanguineous Tunisian families with at least two siblings with childhood absence epilepsy, linkage analyses or direct sequencing excluded CACNG2, CACNA1A, CACNB4, and CACNA2D2, orthologs of genes responsible for autosomal recessive absence seizures in mice (01).
Overall, it is thought that the majority of identified generalized epilepsies are related to mutations in ion channel functions or genes that play a role in cell excitability, and ion channel function is known to be an important factor in the development of immature brains. Therefore, mutations affecting ion channels may alter brain maturation and result in epileptogenesis. For example, some mutations implicated in absence epilepsy include calcium channel mutations (eg, CACNA1A), which are consistent with the town pathophysiology of T-type calcium channels controlling the firing rate of neurons, thus, affecting pathogenic oscillations in neural networks. Other implicated genes include GABA receptor channels and glutamate receptor channels (132).
Cortical structures and networks. Imaging modalities, including functional magnetic resonance imaging (fMRI) and magnetoencephalography (MEG), have been used to study the physiological generation of absence seizures.
fMRI analysis of children with typical absence seizures showed changes in blood oxygenation level dependent (BOLD) signals in multiple brain regions, including the cortex, thalamus, and subcortical structures, and these changes can start even before the onset of the absence seizures, often seconds before the clinical behavioral changes (132). There is also evidence to support cerebellar involvement, including imaging analysis that shows cerebellar volume loss and changes in BOLD signal during synchronized absence seizure activity (132).
Neuromagnetic activity has also been studied in patients with childhood absence epilepsy using magnetoencephalography (MEG) analysis. This tool identified frequency-dependent changes in neuronal activity related to absence seizure manifestations and showed changes in cortical structures, including frontal, temporo-parietal, and parietal lobes, during the ictal period. These studies support the cortical focus theory that suggests that cortical regions dominate the onset of absence seizures (135). Li and colleagues also showed that during absence seizures, there is anteriorization of the alpha rhythm, similar to the frontal alpha rhythm induced by sleep and anesthesia. They also showed that gamma2 band activity was silent during absence seizures, whereas all other frequency bands were involved in seizure onset and termination.
Functional imaging with positron emission tomography demonstrates normal cerebral glucose metabolism and benzodiazepine receptor density in absence epilepsies with diffuse hypermetabolism during 3 Hz spike-and-wave discharges (189; 78). There is no evidence of any interictal overall abnormality of opioid receptors, though typical absences have been found to displace 11C-diprenorphine from the association areas of the neocortex. In contrast, binding of 11C-flumazenil to central benzodiazepine receptors has been shown not to be affected by serial absences (78). Kapucu and colleagues (117) evaluated 23 patients of childhood absence epilepsy with 99mTc-hexamethylpropylenamine oxime brain single photon emission computed tomography. Interictally, 10 of them had relative hypoperfusion in the frontal lobes that could involve neighboring parietal and temporal regions. The activation study during absence seizures revealed that 13 of 23 patients had relative hyperperfusion in the same brain regions that were relatively hypoperfused in the baseline study. These hyperperfused regions occupied larger areas than baseline hypoperfused regions. All patients had global increased perfusion in the ictal study.
In a report, 15 drug-naive children with newly diagnosed childhood absence epilepsy were studied using continuous EEG-fMRI (136). Blood oxygen level-dependent (BOLD) signal changes associated with interictal (nine sessions with six patients) and ictal (eight sessions with six patients) generalized spike-wave discharges were analyzed at the individual and group levels. Generalized spike-wave discharges were associated with widespread and symmetrical deactivation in the cortex and caudate nucleus, with a frontal maximum, and predominant activation in the thalamus bilaterally during ictal generalized spike-wave discharges and in the cortex during interictal generalized spike-wave discharges. The BOLD response was characterized by a higher statistical significance and a more widespread extent at the time of the ictal generalized spike-wave discharges, as compared to the time of interictal generalized spike-wave discharges.
In a study, source localization and functional connectivity was used to characterize neuronal networks underlying absence seizures and subclinical discharges (128). The strong connections of the thalamus with other brain regions that are important for consciousness, and with components of the default mode network, suggest severe impairment of consciousness in ictal generalized spike-wave discharges versus weaker connectivity between thalamus and these brain regions for subclinical discharges. This may suggest prevention of impairment of consciousness and may benefit future therapeutic targets and improve the management of patients with childhood absence epilepsy (128).
Berman and colleagues performed EEG-fMRI with simultaneous behavioral testing in 37 children with typical childhood absence epilepsy (24). Attentional vigilance was evaluated by a continuous performance task (CPT), and simpler motor performance was evaluated by a repetitive tapping task (RTT). Generalized spike and wave discharges were obtained during fMRI scanning from nine patients among the 37 studied. fMRI signal increases during the EEG discharges were observed in the thalamus, frontal cortex, and primary visual, auditory, somatosensory, and motor cortex, and fMRI decreases were seen in the lateral and medial parietal cortex, cingulate gyrus, and basal ganglia. Omission error rate (missed targets) with generalized discharges during fMRI was 81% on CPT and 39% on RTT. For those seizure epochs during which CPT performance was impaired, fMRI changes were seen in cortical and subcortical structures typically involved in spike-wave discharges whereas minimal changes were observed for the few epochs during which performance was spared. The authors concluded that their findings suggest typical absence seizures involve a network of cortical-subcortical areas necessary for normal attention and primary information processing. Identification of this network may improve understanding of cognitive impairments in childhood absence epilepsy and may help guide development of new therapies for this disorder. In a similar study of the same group of investigators, Bai and colleagues reported the dynamic time course of typical childhood absence seizures (14). They acquired simultaneous EEG-fMRI in 88 typical childhood absence seizures from nine pediatric patients. They investigated behavior concurrently using a continuous performance task or simpler repetitive tapping task. EEG time-frequency analysis revealed abrupt onset and end of 3 to 4 Hz spike-wave discharges with a mean duration of 6.6 seconds. Behavioral analysis also showed rapid onset and end of deficits associated with electrographic seizure start and end. In contrast, they observed small early fMRI increases in the orbital/medial frontal and medial/lateral parietal cortex more than 5 seconds before seizure onset, followed by profound fMRI decreases continuing more than 20 seconds after seizure end. This time course differed markedly from the hemodynamic response function (HRF) model used in conventional fMRI analysis, consisting of large increases beginning after electrical event onset, followed by small fMRI decreases. Other regions, such as the lateral frontal cortex, showed more balanced fMRI increases followed by approximately equal decreases. The thalamus showed delayed increases after seizure onset followed by small decreases, most closely resembling the HRF model.
Also, the same team of authors investigated whether the abnormal bisynchrony of ictal absence seizures in childhood absence epilepsy may extend to the interictal period using a blood oxygen level-dependent resting functional connectivity approach (13). EEG-fMRI data were recorded from 16 patients with childhood absence epilepsy and 16 age- and gender-matched controls. Through appropriate analyses they found significantly increased resting functional connectivity between hemispheres in the lateral orbitofrontal cortex of patients with childhood absence epilepsy compared to normal controls. The significance of these findings has been discussed in a companion editorial (245).
Using advanced brain imaging techniques, Chan and colleagues found that, compared with controls, patients with childhood absence epilepsy showed areas of grey matter decrease in both thalami and in the subcallosal gyrus (41). White matter decrease was found in the extranuclear subcortical area and in the white matter of the basal forebrain. Grey and white matter increase was restricted to small clusters of cortical and subcortical areas. The authors hypothesized that bilateral thalamic atrophy may be either a result of damage from seizures (as in hippocampal sclerosis) or a reflection of a primary underlying pathology as the cause of absence seizures.
In another relevant MRI study, a group of 26 children with childhood absence epilepsy had significantly smaller gray matter volumes of the left orbital frontal gyrus, as well as both left and right temporal lobes, compared to age- and gender-matched children without epilepsy (35). In the childhood absence epilepsy group, these volumes were related to age, gender, ethnicity, and pregnancy complications but not to seizure, IQ, and psychopathology variables. In the group of children without epilepsy, however, the volumes were related to IQ. The authors concluded that childhood absence epilepsy impacts brain development in regions implicated in behavior, cognition, and language. The same group of authors used MRI scans at 1.5 Tesla to compare frontotemporal brain volumes in 26 children with childhood absence epilepsy to 37 age- and gender-matched children without epilepsy (34). They also examined the association of these volumes with seizure, demographic, perinatal, IQ, and psychopathology variables. The childhood absence epilepsy group had significantly smaller gray matter volumes of the left orbital frontal gyrus as well as both left and right temporal lobes compared to the age- and gender-matched children without epilepsy. In the childhood absence epilepsy group, these volumes were related to age, gender, ethnicity, and pregnancy complications but not to seizure, IQ, and psychopathology variables. In the group of children without epilepsy, however, the volumes were related to IQ. The authors concluded that their “findings suggest that childhood absence epilepsy impacts brain development in regions implicated in behavior, cognition, and language. In addition to supporting the cortical focus theory of childhood absence epilepsy, these findings also imply that childhood absence epilepsy is not a benign disorder.” In another report of the same group, Tosun and colleagues used surface-based morphometry to examine whether age-related changes in gray matter tissue thickness and depth of sulcal regions at high spatial resolution across the cortex differed in children with childhood absence epilepsy compared to healthy control subjects (221). In addition, the possibility of variable brain-cognition relationships in the childhood absence epilepsy group compared to the control group was investigated. The main findings were: (1) From the developmental perspective, children with childhood absence epilepsy did not demonstrate the normal regional age-related changes involving a decrease in cortical thickness and increase in sulcal depth. (2) None of the seizure variables, including age of onset, seizure frequency, and antiseizure drugs had a significant effect on the association between age and cortical morphometry measures in the childhood absence epilepsy population. (3) Even though the childhood absence epilepsy group had mean VIQ and PIQ scores in the average range, these findings suggest that they use different brain regions to perform these cognitive functions compared to healthy controls.
Additionally, in a voxel-based morphometry study with MRI at 3.0T, Wang and associates examined 20 drug-naive patients with childhood absence epilepsy and 20 age- and gender-matched healthy subjects (236). Compared with controls, the patients showed less grey matter volume in the bilateral thalami, and this was negatively correlated with disease duration and age of onset in the patient group. Furthermore, structural impairment in the default mode network regions was documented in children with childhood absence epilepsy using diffusion tensor imaging (182).
Killory and colleagues investigated the brain networks involved in impaired interictal attention of childhood absence epilepsy (121). The authors used the continuous performance task of attentional vigilance and the repetitive tapping task, a control motor task, to examine interictal attention in 26 children with childhood absence epilepsy and 22 matched healthy controls. Each subject underwent simultaneous 3T functional magnetic resonance imaging-electroencephalography (fMRI-EEG) and continuous performance task/repetitive tapping task testing. Areas of activation on fMRI during the continuous performance task were correlated with behavioral performance and used as seed regions for resting functional connectivity analysis. All behavioral measures reflecting inattention were significantly higher in patients. Correlation analysis revealed that impairment on all measures of inattention on the continuous performance task was associated with decreased medial frontal cortex activation during continuous performance task. In addition, analysis of resting functional connectivity revealed an overall decrease within an “attention network” in patients relative to controls. Patients demonstrated significantly impaired connectivity between the right anterior insula/frontal operculum and medial frontal cortex relative to controls. These results suggest that there is impaired function in an attention network comprising anterior insula/frontal operculum and medial frontal cortex in patients with childhood absence epilepsy.
In another study, Yang and colleagues used the EEG-functional MRI to measure the resting state functional connectivity during interictal generalized spike-wave discharges in drug-naïve childhood absence epilepsy, and they evaluated three different brain networks: the default mode network, cognitive control network, and affective network (244). They found decreased functional connectivity within each of the three networks in the patients with childhood absence epilepsy during interictal generalized spike-wave discharges. There were also some regions, primarily the parahippocampus, which showed increased connectivity. These findings demonstrate that interictal generalized spike-wave discharges can cause dysfunction in specific networks important for psychosocial function.
Masterton and associates examined absence epilepsy subnetworks revealed by event-related independent components analysis of fMRI (151). EEG-fMRI recordings from eight patients with childhood absence epilepsy were studied using event-related independent components analysis (eICA). Six eICA components were identified, representing distinct generalized spike-wave–related subnetworks. Activations were detected in a number of brain regions, including the striatum. These findings suggest that the earliest activity associated with generalized spike-wave may be in posterior cortical regions and provide new evidence that the thalamostriate network may play a more important role in the generation of generalized spike-wave than suggested by previous studies.
Xue and colleagues used diffusion tensor imaging tractography to map the white matter structural network, composed of 90 cortical and subcortical regions, in 18 patients with childhood absence epilepsy and 18 age- and gender-matched healthy controls (243). Graph theoretical methods were applied to investigate the alterations in the topological and nodal properties of the networks in these patients. It was found that both the patients with childhood absence epilepsy and the controls showed small-world properties in their white matter networks. However, the network connection strength, absolute clustering coefficient, and global/local efficiency were significantly decreased. Characteristic path length was significantly increased in the patients with childhood absence epilepsy compared with the controls. Significantly decreased white matter connections, nodal properties, and impaired subnetworks were found in the subcortical structures, orbitofrontal area, and limbic cortex in the childhood absence epilepsy patients. Moreover, network connection strength, local efficiency, and nodal features in some regions were significantly negatively correlated with the duration of epilepsy.
Curwood and colleagues investigate whether structural connectivity changes are present in the brains of patients with childhood absence epilepsy in young adulthood (65). Cortical thickness measurements were obtained for 30 subjects with childhood absence epilepsy (mean age: 21 ± 2 years) and 56 healthy controls (mean age: 24 ±4) and regressed for age, sex, and total intracranial volume. Structural connectivity was evaluated by measuring the correlation between average cortical thicknesses in 915 regions over the brain. Maps of connectivity strength were then obtained for both groups. It was found that when compared to controls, the childhood absence epilepsy group showed overall increased "connectivity" with focal increased connection strength in anterior regions, including the anterior cingulate and the insula and superior temporal gyrus bilaterally, the right orbito-frontal and supramarginal regions, and the left entorhinal cortex. Decreased connection strength in the childhood absence epilepsy group was found in the left occipital lobe, with a similar trend in right occipital lobe. The authors concluded that a) brains in young adults whose childhood absence epilepsy was resolved had abnormal structural connectivity b) their findings suggest that frontal regions correlate most with cortical thickness throughout the brain in childhood absence epilepsy patients, whereas occipital regions correlate most in well-matched normal controls, and c) this is evidence of a developmental difference in childhood absence epilepsy that emphasizes these frontal lobe regions, perhaps driven by frontal lobe epileptiform activity (65).
Studies in the interictal state of drug-naïve children with childhood absence epilepsy found abnormal cortico-cortical network with highly significant increase of outgoing connectivity involving frontal and central cortical areas (187) and frequency-specific alteration in the effective connectivity network (242). Tenney and associates performed a prospective observational study in order to determine how pretreatment ictal network pathways, defined using a combined EEG-functional magnetic resonance imaging (EEG-fMRI) and magnetoencephalography effective connectivity analysis, were related to treatment response (217). Sixteen children with newly diagnosed and drug-naive childhood absence epilepsy had 31 typical absence seizures during EEG-fMRI and 74 during magnetoencephalography. Patterns of pretreatment connectivity demonstrated strongest connections in the thalamus and posterior brain regions (parietal, posterior cingulate, angular gyrus, precuneus, and occipital) at delta frequencies and the frontal cortices at gamma frequencies (P < .05). Ethosuximide treatment nonresponders had pretreatment connectivity, which was decreased in the precuneus region and increased in the frontal cortex compared to ethosuximide responders (P < .05). The authors concluded that pretreatment ictal connectivity differences are associated with response to antiseizure treatment, which may be a possible mechanism for the variable treatment response seen in patients with childhood absence epilepsy (217). The same group of investigators used an established thalamocortical computer model to determine how T-type calcium channels work in concert with cortical excitability to contribute to pathogenesis and treatment response in childhood absence epilepsy (124). The model is comprised of cortical pyramidal, cortical inhibitory, thalamocortical relay, and thalamic reticular single-compartment neurons, implemented with Hodgkin-Huxley model ion channels and connected by AMPA, GABAA, and GABAB synapses. Network behavior was simulated for different combinations of T-type calcium channel conductance, inactivation time, steady state activation/inactivation shift, and cortical GABAA conductance. It was found that decreasing cortical GABAA conductance and increasing T-type calcium channel conductance converted spindle to spike and wave oscillations; smaller changes were required if both were changed in concert. In contrast, left shift of steady state voltage activation/inactivation did not lead to spike and wave oscillations, whereas right shift reduced network propensity for oscillations of any type. These results provide a window into mechanisms underlying polygenic inheritance in childhood absence epilepsy, as well as a mechanism for treatment effects and failures mediated by these channels (124).
Autopsy and MRI studies found microdysgenesis and other cerebral structural changes in some patients with childhood absence epilepsy that may be inconceivable for this benign, age-dependent, and age-limited epileptic syndrome. Meencke reviewed autopsy findings in childhood absence epilepsy and confirmed his previous reports on microdysgenesis, with the frontal lobe more severely affected in 12 patients (156; 155). Using quantitative MRI, Woermann and colleagues found that patients with idiopathic generalized epilepsy had significantly larger cortical grey matter volumes than control subjects (240). Abnormalities of the regional distribution of cerebral grey and subcortical matter were frequent in patients with idiopathic generalized epilepsy but not so frequent in patients with childhood absence epilepsy.
It should be noted that all cases of Meencke had frequent absences from childhood to adulthood and generalized tonic-clonic seizures, which would not conform to a strict diagnosis of the syndrome of childhood absence epilepsy (155). Similar may be the situation with the newer studies on childhood absence epilepsy (35; 34; 221) and the single patient with abnormal MRI of Woermann and colleagues (240).
Neurophysiology. The pathophysiologic mechanism of absence seizure generation is an ongoing topic of investigation. Studies have shown evidence of cortical focal-onset triggers that cause an abrupt activation followed by sustained activity of cortico-thalamo-cortical loops, which may be progressive throughout the course of the seizure (10). Leitch describes evidence that spike-wave discharges may originate in the somatosensory cortex before very rapidly spreading across bilateral cortico-thalamo-cortical networks (132). Furthermore, Aud’hui and colleagues describe how external sensory stimulations, including loud and painful stimuli, can stop absence seizures in rat models, and acoustic stimuli are most effective if applied in the first 3 seconds of seizure onset (10). This is thought to be possibly due to the progressive involvement of the cortico-thalamo-cortical loop reaching a critical threshold, thus, resulting in increased connections and bilateral, oscillatory hyper-synchronization, after which sensory stimuli are not effective (132). Overall, thalamic recruitment is thought to be a key step necessary for the generalization of absence seizures (132).
The pathophysiological mechanisms of absence seizures have been studied in various animal models with generalized spike and wave discharges associated with behavioral arrest. In a publication, it was demonstrated that absence seizures in the WAG/Rij rat, an established rodent model of absence epilepsy, were highly sensitive to arterial carbon dioxide, suggesting that seizure-generating circuits are sensitive to pH (193). Moreover, hyperventilation consistently activated neurons within intralaminar nuclei of the thalamus, a structure implicated in seizure generation. It was shown that intralaminar thalamus also contains pH-sensitive neurons. Collectively, these observations suggest that hyperventilation activates pH-sensitive neurons of the intralaminar nuclei to provoke absence seizures.
It appears that generalized spike and wave discharges are generated and sustained by highly synchronized abnormal oscillatory rhythms in thalamocortical networks that mainly involve neocortical pyramidal cells, the reticular thalamic nucleus, and the relay nuclei of the thalamus. Neither the cortex nor the thalamus alone can sustain these discharges, indicating that both structures are involved in the discharges’ generation.
Children with childhood absence epilepsy show some autonomic differences from control subjects as assessed by interictal cardiorespiratory measurements (224). Patients present a reduced/increased parasympathetic/sympathetic modulation during seizure-free activity, in comparison to the control group. Patients display a less adaptive heart rate and a modified cardiorespiratory interaction (224).
In a study, fMRI informed magnetoencephalography multilayer network analyses showed beta/gamma cross-frequency and within-frequency coupling in the frontoparietal and frontofrontal regions during childhood absence seizures that has not previously been reported for any primary generalized seizure (218). These results provide a possible mechanistic explanation for previous fMRI and neurophysiology studies showing that focal areas of the precuneus and frontal lobe are critically important for the initiation and maintenance of generalized rhythmic spike-wave discharges characterizing absence seizures. As nonpharmacologic treatments, such as neuromodulation, become available for generalized epilepsies, a detailed mechanistic understanding of how ‘‘diffuse’’ seizures are generated and maintained will be necessary to provide optimal outcomes (218).
In another report, 22 children with childhood absence epilepsy were recruited to study the neural source activity during ictal-onset and interictal periods at frequency bands of 1 to 30 Hz and 30 to 80 Hz with magnetoencephalography scanning (210). Most of the participants had more active source activity locations in the ictal-onset period than in the interictal period, both at 1 to 30 Hz and 30 to 80 Hz. The frontal lobe, the temporoparietal junction, and the parietal lobe became the main active areas of source activity during the ictal period, whereas the precuneus, cuneus, and thalamus were relatively inactive. Some brain areas become more excited and have increased source activity during seizures, providing a basis for exploring the network mechanism of childhood absence epilepsy development.
Because focal EEG abnormalities are frequent in childhood absence epilepsy (146), Koutroumanidis and associates investigated the nature of the focal spike-wave discharges (FSWDs) and focally led generalized spike-wave discharges (GSWDs) in typical childhood absence epilepsy (127). They analyzed 24 abnormal video-EEGs from 13 consecutive children with childhood absence epilepsy and good response to appropriate antiseizure drugs. The conclusion was that in childhood absence epilepsy, focal EEG paroxysms reflect a system of multifocal, nonlocalizing, electrically unstable cortical areas that, under the facilitator influence of exogenous or endogenous factors like sleep instability, can foster a corticothalamic response of sufficient strength to generate 3 Hz generalized spike-wave discharges that are conditionally sustainable and potentially ictal. Focal spike-wave discharges can be viewed as incomplete forms of the generalized spike-wave discharges. Together they define the EEG identity of idiopathic ‘‘generalized’’ epileptogenesis.
Kokkinos and colleagues also studied the spatiotemporal profiles of focal and generalized spikes in childhood absence epilepsy (126). Spatiotemporal analysis demonstrated a variety of mainly frontal and occipital locations for pre-generalization focal spike-wave discharges with propagation along the longitudinal axis in either direction and across homologous sites. Interictal focal spike-and-wave discharges demonstrated similar spatiotemporal characteristics. In contrast, the topography and propagation patterns of the first generalized spike of the spike-wave discharge showed less variability, mainly involving the frontotemporal/temporal areas, and correlated poorly (< 10%) with that of the pre-generalization focal spike-wave discharge.
In another report, the same group of investigators studied the spatial distribution during generalized ictal spikes in 12 children with childhood absence epilepsy who had more than two typical absences during their routine video-EEG (125). Seizures were identified, and ictal spikes were marked over the maximum electronegative peak, clustered, waveform-averaged, and spatiotemporally analyzed in 2D electrode space. It was found that consistency of spatiotemporal patterns of ictal spikes was high between the absences of the same child but low between children. Three main discharge patterns were identified: (1) anterio-posterior propagation, (2) posterio-anterior propagation, and (3) confined to the frontal/prefrontal regions. In four patients, the propagation patterns transformed during the seizure into either a lateralized diminished or a nonlateralized reverse direction form. Most spikes originated frontotemporally; all maximized over the frontal/prefrontal electrodes and mostly decayed prefrontally. In four patients, lateralized propagation patterns were identified. It was concluded that ictal spike propagation patterns suggest that epileptogenic childhood absence epilepsy networks are personalized, interconnected distal areas in the brain – not the entire cortex – with a tendency to generate bilateral symmetrical discharges, sometimes unsuccessfully. The transformation of propagation patterns during the seizure indicates the existence of dynamic interplay within epileptogenic networks.
More information can be found in the pathophysiology section of the article on typical absence seizures.
Childhood absence epilepsy is a common pediatric epilepsy syndrome. The annual incidence rate of childhood absence epilepsy in children less than 15 years of age has been estimated between 6.3/100,000 (163; 140) and 8.0/100,000 (25). Recruitment bias explains that the frequency of childhood absence epilepsy in childhood epilepsies has been differently assessed, ranging from 2.3% to 37.7% of cases. In two prospective community-based studies, prevalence of childhood absence epilepsy was 10% (30) and 12.3% (19) for children younger than 16 years of age with epilepsy. Among all cases of epilepsy in school-aged children, 10% to 17% are due to childhood absence epilepsy (153).
Absence epilepsy in early childhood (under 3 years old). The main presenting symptoms of early-onset absence seizures are absences or myoclonic jerks; in some cases, generalized tonic-clonic seizures are a presenting symptom. The age of onset varies from 0.6 to 2.9 years (mean 2.3 ± 0.7 years). Early-onset absence seizures are twice as common in females and are characterized by clinical and genetic heterogeneity (68; 55; 40). The nonmyoclonic group of absence seizures includes cases with spanioleptic and pyknoleptic absence seizures reminiscent of juvenile absence epilepsy and childhood absence epilepsy-like epilepsy, respectively. The myoclonic group of absence seizures is usually pyknoleptic, and 68% of cases are associated with automatisms if EEG discharges last more than 5 seconds (55). Briefer discharges of 1 to 3 seconds are associated with inconspicuous or conspicuous absences and require meticulous video-EEG analysis to identify them. The absences in the myoclonic group are associated with marked clonic or myoclonic, tonic, and atonic components; facial myoclonias; or even segmental or massive jerks. These are probably not a specific expression of distinct syndromes seen later in childhood, such as facial myoclonia with absences, eyelid myoclonia and absences, myoclonic atonic seizures, and juvenile myoclonic epilepsy (54), or more severe forms of idiopathic and symptomatic generalized epilepsies (36; 95; 231; 05; 06; 96). Doose and colleagues proposed a syndrome of absence epilepsy of early childhood or myoclonic astatic (atonic) seizures (77; 76). Absence seizures of early onset (a few months to 4 years) are particularly demanding in their diagnosis and management (112; 169).
Absence seizures associated with glucose transporter-1 (GLUT1) deficiency syndrome. Of clinical importance is the diagnosis of absence seizures associated with GLUT1 deficiency syndrome. Seizures begin between the ages of 1 and 4 months in 90% of cases. The frequency and severity of seizures varies among affected individuals. Typical or atypical absences, mainly of early onset, are the most prominent seizure types. Typical absences may imitate various syndromes of idiopathic generalized epilepsy with absences such as epilepsy with myoclonic absences, childhood absence epilepsy, or juvenile absence epilepsy (188; 209; 131; 95; 207; 204). The ketogenic diet is highly effective in controlling the seizures (123). An early diagnosis and early start of a ketogenic diet may prevent deterioration. Phenobarbital is contraindicated.
Molecular genetic testing for SLC2A1, which is the only gene known to be associated with GLUT1 deficiency, is now clinically available. Families with members manifesting various types of idiopathic generalized epilepsy with absences (early-onset absence seizures, childhood or juvenile absence epilepsy, juvenile myoclonic epilepsy) should be tested for SLC2A1 gene mutations (207). However, in a study of 84 children with rigorous diagnosis of early-onset absence seizures, no mutations in the SLC2A1 gene were detected (06). Similarly, SLC2A1 gene analysis in 12 children with onset of absences in the first year of life failed to reveal glucose transporter 1 deficiency (96).
Childhood absence epilepsy with aberrant features. The syndrome of childhood absence epilepsy should be distinguished from childhood absence epilepsy with aberrant electroclinical features (Table 2).
Juvenile absence epilepsy. In juvenile absence epilepsy, typical absence seizures are, by definition, always present and usually appear at a later age with less frequent (spaniolepsy) and less severe absence seizures as compared to typical absence seizures in the syndrome of childhood absence epilepsy. The onset of absence seizures is less abrupt, and the loss of consciousness is less profound, than in childhood absence epilepsy. Generalized tonic-clonic seizures and myoclonic jerks are part of the phenotype in 80% and in 15% to 30%, respectively (54). Absence status epilepticus occurs in about 20% of cases. EEG features may be similar, but polyspikes (more than three to four) and fragmented discharges of longer duration favor juvenile absence epilepsy. There may be an overlap with juvenile absence epilepsy starting before 10 years of age (111; 54). Severe impairment of consciousness and high daily frequency are the main characteristics of childhood absence epilepsy, but these may also be features of juvenile absence epilepsy. Further, frequent daily absence seizures may also occur in juvenile absence epilepsy (162), and generalized tonic-clonic seizures are nearly unavoidable in juvenile absence epilepsy. Onset of juvenile absence epilepsy varies between 7 and 17 years of age (peak age 12 to 13 years). However, age at onset alone is not an absolute criterion for differentiation between childhood absence epilepsy and juvenile absence epilepsy. The distinction between childhood absence epilepsy and juvenile absence epilepsy is necessary for prognostic reasons; the latter persists for many years (54).
Typical absence seizures and generalized tonic-clonic seizures may be part of the phenotype.
Early-onset myoclonic absence epilepsy (under 3 years old). See Absence epilepsy in early childhood (under 3 years old).
Myoclonic epilepsy in infancy and childhood. The ILAE has recognized myoclonic epilepsy in infancy as a syndrome (80). However, the age of onset has gradually been extended from infancy to childhood to include cases that present between the ages of 4 months and 5 years. The following phenotypes can be identified: myoclonic seizures only; myoclonic seizures and generalized tonic-clonic seizures; myoclonic seizures with absences; and myoclonic seizures with absences and generalized tonic-clonic seizures. Differences in clinical expression are relative to the developmental stage of the child. If present, the absence seizures are conspicuous or inconspicuous and may precede or follow the jerk. The eyes may open and stare with the jerk, a brief absence may be associated with rhythmic jerking of the head, or head dropping may be associated with open eyes and a stare (54b).
Myoclonic atonic (astatic) epilepsy (formerly known as Doose syndrome). Myoclonic-atonic epilepsy is classified as a developmental encephalopathy in the ILAE Classification and Definition of Epilepsy Syndromes with Onset in Childhood (206). Myoclonic-atonic seizures were first reported as akinetic seizures by Doose in 1964 and represent 1% to 2% of all childhood epilepsies (105). Seizures start between the ages of 6 months and 5 years in otherwise normal children. Genetics play an important role. The main seizure types in myoclonic-atonic seizures are atonic and myoclonic, which involve the proximal muscles or are massive, causing a fall. Generalized tonic-clonic seizures, absences, and tonic seizures often coexist in at least two thirds of cases. Interictal EEG shows generalized spike-wave discharges of 2 to 3 Hz, which are irregular on normal background activity. Ictal myoclonias are associated with irregular 2.5 to 3 Hz spike-wave discharges, followed by a high-amplitude slow wave that correlates with the atonic component. Sodium channel blockers are contraindicated.
Juvenile myoclonic epilepsy (Janz syndrome). By definition, myoclonic jerks occur in all cases in juvenile myoclonic epilepsy. Typical absence seizures are observed in at least 48%, generalized tonic-clonic seizures in 60%, and positive response to intermittent photic stimulation in 75% of cases. Myoclonic and generalized tonic-clonic seizures, as well as photosensitivity, are exclusion criteria for the syndrome of childhood absence epilepsy. In a study, absence seizures occurred in one third of juvenile myoclonic epilepsy patients, but these were usually highly mild, often inconspicuous, and had different EEG patterns (173). The main seizure type in juvenile myoclonic epilepsy is myoclonic jerks on awakening. The problem occurs when juvenile myoclonic epilepsy starts with absences prior to the onset of myoclonic jerks; however, there are a number of clinico-EEG differences, such as milder impairment of consciousness, frequent polyspikes, and fragmentations of EEG discharges, in juvenile myoclonic epilepsy (168). Some authors consider these cases as absence epilepsy evolving or progressing to juvenile myoclonic epilepsy (238; 70). Another opinion is that juvenile myoclonic epilepsy starting with absences in childhood. In the majority of these patients with juvenile myoclonic epilepsy starting with typical absences in childhood, video-EEG studies would clearly differentiate them from the typical absence seizures of childhood absence epilepsy (173; 174). However, there may be cases in which their differentiation is difficult and juvenile myoclonic epilepsy is not diagnosed until many years later. In the pediatric population, this is seen in cases that present with myoclonic seizures and typical absence seizures before the age of 3 to 4 years. The characteristic electroclinical features of juvenile myoclonic epilepsy are demonstrated when relapses occur, after discontinuation of treatment.
Generalized tonic-clonic seizures may be part of the phenotype. These types of absence seizures are betrayed by their predominant ictal myoclonic manifestations and concomitant absence seizures, which do not feature in childhood absence epilepsy (168; 169; 54).
Epilepsy with myoclonic absences (formerly known as Tassinari syndrome). Epilepsy with myoclonic absences is recognized by the ILAE as an epileptic syndrome (49; 79; 80).
Facial myoclonic epilepsy with absences. In some children, typical absence seizures may be associated with perioral or eyebrow myoclonias, or both. Among the different clinical phenotypes (absences with perioral myoclonias, absences with eyebrow myoclonias, and absences with both eyebrow and perioral myoclonias), there are no differences regarding presentation, response to therapy, and prognosis. The onset is between 1 and 13 years of age.
Eyelid myoclonia and absences (formerly known as Jeavons syndrome). Eyelid myoclonia and absences was recognized as a syndrome by the ILAE under the name of “epilepsy with eyelid myoclonia” (206). It is a type of photosensitive epilepsy. The mean age of onset is 7 ± 2.7 years. There is a marked jerking of the eyelids, often with upward deviation of the eyes, which is associated with 3 to 5 Hz spike-and-polyspike-wave discharges that are often irregular, immediately after eye closure and invariably evoked on intermittent photic stimulation (57). Eyelid myoclonia and absences is an eye closure, not a closed-eye, phenomenon. In the past, the ILAE recognized eyelid myoclonia as a different seizure type (79; 86).
Typical absence seizures and myoclonic jerks may be part of the phenotype.
Generalized tonic-clonic seizures. Generalized tonic-clonic seizures characteristically occur during awakening, drowsiness, relaxation, and excessive alcohol intake. The coexistence of myoclonic and typical absence seizures suggests an overlap among juvenile absence epilepsy and juvenile myoclonic epilepsy, and this has been hypothesized genetically.
Pure photosensitive epilepsy. The onset of photosensitivity is around puberty. The clinical expression of photosensitivity is seizures due to a flickering light source or pattern. The photoparoxysmal response is most commonly elicited at the moment of eye closure, between the frequencies of 10 to 30 f/s. The methodology is very important (118). The most common types of seizures are generalized tonic-clonic seizures, myoclonic seizures, and absences. The most common provocative factors, by far, are television, video and computer games, and natural and environmental lighting.
Absence-like episodes induced during hyperventilation with concomitant high-amplitude delta activity (HIHADA). Absence seizures should be distinguished from absence-like episodes induced during hyperventilation in association with high-amplitude delta activity (59; 56). These episodes have some similarities with typical absences. The age of onset and female preponderance are similar. In both, clinical and EEG events are provoked by hyperventilation and respond to valproic acid. In some cases, school performance may improve when treatment is given. However, in absence-like episodes, the onset is less abrupt, the children seem more alert, and real automatisms or autonomic symptoms are not observed. Unlike typical absence seizures, the absence-like episodes never occur during the basic recording, and the burst of slow waves is not associated with spikes, even if absence-like episodes and typical absence seizures coexist in the same patient. The two phenomena always appear separately, even in the same recording. Absence-like episodes during HIHADA should be considered as an idiopathic low-threshold phenomenon induced by hyperventilation; they have a benign course and do not occur spontaneously (56).
Focal epilepsies. The differential diagnosis of childhood absence epilepsy from focal impaired awareness is detailed in the section on typical absence seizures. Automatisms may be common in both. However, typical absence seizures with a frontal lobe origin may also have concomitant, more or less regular, bilateral spike-wave discharges (84). Focal motor components, asymmetrical ictal discharges, or interictal frontal foci on EEG may help in their differentiation. MRI may demonstrate frontal abnormalities (84) or subependymal grey matter heterotopia (183). Focal EEG abnormalities are frequent in childhood absence epilepsy (146).
Symptomatic absence seizures. In genetically predisposed individuals, brain damage may precipitate typical absence seizure occurrence that is most often associated with neurologic signs or developmental impairment. In most cases, the association is probably coincidental. However, cerebral pathology may modify the expression of genetic seizure susceptibility (84). In these cases, the correct diagnosis is not childhood absence epilepsy but symptomatic absence epilepsy with a less favorable outcome.
A report on the “electroclinical features of absence seizures in childhood absence epilepsy” reflects well the misunderstanding of what childhood absence epilepsy is (190). The authors analyzed ictal video-EEG semiology in children aged 2 to 10 years having only typical absence seizures. They found 47 children that they diagnosed as having childhood absence epilepsy “as defined by the 1989 ILAE criteria”; only five of the children could be classified as childhood absence epilepsy according to our proposal. However, the 47 patients of this report constitute a heterogeneous group of children with onset of typical absence seizures between 2 and 10 years of age rather than childhood absence epilepsy (Table 3). Furthermore, no follow-up or prognosis is provided, despite the original presentation of the patients being between 1992 and 1997.
Analysis of the study criteria reveals that these are at variance even with the 1989 ILAE definition. Specifically, the authors lowered the age range to include children as young as 2 and 3 years old (from 2 to 10 years) and reduced the seizure frequency to include “daily” absences, which does not mean “several per day” (pyknolepsy). Furthermore, they defined typical absence seizures as “a clinical change associated with generalized spike-and-slow wave activity or multiple spike-and-slow wave activity with a frequency of > 2.5Hz at onset” even if this is not associated with an absence. Impairment of consciousness (absence) is a prerequisite of any definition of typical absence seizures. This may be associated with other clinical symptoms (myoclonic, atonic, clonic, autonomic), but these do not make typical absence seizures without clinically testable impairment of consciousness.
|
Electroclinical features included in the report's classification of CAE |
Number of patients |
Epileptic syndromes in which these features are more likely to manifest |
Exclusion criterion of CAE? |
|
A. Duration of seizure < 4 seconds |
21 |
JME, EMA |
Yes |
|
B. Age of child < 4 years |
7 |
EMA, PerMA, EpMA |
Probably yes (exceptional) |
|
C. No severe impairment of consciousness during seizure |
16 |
All other IGEs with absences but CAE and JAE |
Yes |
|
D. Myoclonic features in seizures except mild in eyes, eyebrows, and eyelids |
12 |
EMA, PerMA, EpMA |
Yes if severe and consistent during the absence* |
|
E. Photic induction of seizures |
8 |
EMA, IGE with photosensitivity |
Yes if this is a consistent provocation of clinical seizures |
|
F. > 3 spikes per wave |
13 |
JME, EMA |
Probably yes if consistent in the whole duration of the discharge* |
|
G. Disorganized discharges |
25 |
JME, EMA, PerMA |
Yes if consistent in the whole duration of the discharge* |
|
| |||
The diagnosis of childhood absence epilepsy is determined by the history, the typical absence seizures provoked by hyperventilation, and the correlation of clinical and generalized spike-wave discharges on EEG. The diagnosis of childhood absence epilepsy may be difficult, even for specialists, and may be misdiagnosed as attention disturbance or daydreaming and, therefore, inappropriately treated. Other conditions mistaken for absences in children include masturbation, Munchausen syndrome by proxy (reports of apnea), hyperventilation-induced absence-like episodes (59; 56), and nonepileptic absences in Rett syndrome (52). However, during video-EEG recordings, the abrupt onset and termination of typical absence seizures and the provocation by hyperventilation concomitant with generalized spike-and-wave discharges on EEG makes the diagnosis easy. Hyperventilation induces absence seizures in almost all untreated cases or cases treated with an inappropriate drug (109; 239). This procedure should preferably be videotaped for documentation of the clinical features (169). It is always more informative to have a sleep-awake video-EEG after sleep deprivation. In some cases, the sleep EEG is important for revealing aberrant EEG discharges that are incompatible with the diagnosis of the syndrome of childhood absence epilepsy (Table 2) (54).
In practical terms, a child suspected of typical absences should be asked to overbreathe for 3 minutes while counting his or her breaths. In the younger age group, hyperventilation is encouraged by either blowing a paper, a paper streamer, or a straw inside a cup with fluid, causing bubbles (54).
The EEG, preferably video-EEG, is the single most important diagnostic procedure in diagnosing childhood absence epilepsy. Ideally, all children with absence seizures should have video-EEG recordings in an untreated state. The EEG accompaniment of typical absence seizures is a bilaterally synchronous and symmetrical discharge of rhythmic spike-and-slow wave complexes (49).
Ictal EEG. The ictal EEG shows generalized, spike- or double-spike (no more than 3 spikes are allowed) and slow-wave complexes of 2.5 to 4 Hz (no less than 2.5 Hz and no more than 4 Hz) at the initial phase of the discharge, with a gradual and smooth decline in frequency from the initial to the terminal phase. The discharges are usually higher amplitude in the frontal regions, and some rhythmic slow waves may precede frontally or follow in the posterior regions (54). The response during an absence and the associated generalized spike-wave discharge is compared with the response during normal status and EEG activity for accuracy. For the briefer discharges, less profound or inconspicuous impairment of consciousness can easily be identified by asking the child to count in a rhythm faster than the duration of discharges or by training the child before the EEG to immediately repeat words given to him acoustically (54). Counting during hyperventilation is less successful in younger children because the pattern of breathing is interrupted, and effective overbreathing is rarely achieved (54).
Clinically, the eyes open early in the discharge, with a slight upward or side deviation of the eyeballs. A mild rhythmic eyelid fluttering at the same frequency as the spike-wave discharge is usually observed. The longer the discharge, the more likely it is to be accompanied by other clinical phenomena, usually de novo or perseverative automatisms (54). Exclusion criteria for the syndrome of childhood absence epilepsy include spaniolepsy, the coexistence of generalized tonic-clonic seizures, marked clonic or myoclonic components (except mild eyelid myoclonia), brief (under 4 seconds) spike-wave discharges with mild or no impairment of consciousness, and photosensitivity (Table 2) (174; 54).
Fragmentation of the ictal discharges (ie, transient discontinuation of the rhythmic spike-wave and multiple spike-and-slow wave discharges) is also considered an exclusion criterion for childhood absence epilepsy (174; 109; 54). However, according to a study, polyspike and wave discharges do not predict generalized tonic-clonic seizures in childhood absence epilepsy (232).
Interictal EEG. Background EEG is normal (49). However, some patients exhibit a rather particular posterior delta rhythm. This usually occurs in long runs of 3 Hz sinusoidal high activity, either symmetrical or more often asymmetrical, in the occipital and occipitoparietal areas. It is blocked by eye-opening and enhanced by hyperventilation. Asymmetrical posterior slow waves, usually with a right predominance, are physiological in children.
Interictal paroxysmal activity consisting of single or brief discharges of bilateral spike-waves may occur with important morphologic changes, particularly during non-REM sleep. Transient asymmetries of ictal or interictal spike-wave discharges are frequent, particularly in treated patients. Transient focal epileptiform abnormalities such as centrotemporal sharp waves (108; 168) or persistent focal abnormalities (146; 150) occur in childhood absence epilepsy.
Response to intermittent photic stimulation is not mentioned by the Commission on Classification and Terminology of the ILAE (49). Clinical photosensitivity has been proposed as an exclusion criterion (108; 62; 109; 110).
For the few cases in which an accurate complete diagnosis of absence seizures is difficult, particularly in early childhood, the use of low-cost, portable EEG device monitoring may be useful in facilitating diagnosis. Such EEG home monitoring is feasible in pediatric patients if an EEG wearable is easy to put on and is comfortable. Remote seizure monitoring is one of the elements of personalized treatment for childhood absence epilepsy and juvenile absence epilepsy, similar to personalized drug titration and determining the duration of pharmacotherapy (54; 169; 97), though this is well accepted by other authors (241). Mild EEG photosensitivity may occur (109).
A 2.5-year-old girl with normal development and a 1-month history of vague events. The frequency was initially one episode per day and, during the 7 days preceding EEG, had increased to five to seven absences per day. EEG showe...
Antiseizure medications. Due to frequent absence seizures in childhood absence epilepsy, treatment with the appropriate antiseizure drug is necessary. At present, the main anti-absence agents are ethosuximide, sodium valproate, and lamotrigine (166; 180; 54; 98; 169; 114; 153; 119). In a study of 124 cases with childhood typical absence epilepsy, 85% became seizure free, and EEG became normal on monotherapy (90% on valproate and 10% on ethosuximide) (62). The rest (15%) responded to a combination of sodium valproate and ethosuximide or levetiracetam, lamotrigine, and benzodiazepines. Lamotrigine monotherapy is less effective, with nearly half of the patients becoming seizure-free (89; 53; 113). In order to compare the efficacy, tolerability and neurophysiological effects of ethosuximide, valproate, and lamotrigine in children with newly diagnosed childhood absence epilepsy, Glauser and colleagues performed a double-blind, randomized, controlled clinical trial (98). Drug doses were incrementally increased until the child was free of seizures, the maximal allowable or highest tolerable dose was reached, or a criterion indicating treatment failure was met. The primary outcome was freedom from treatment failure after 16 weeks of therapy; the secondary outcome was attentional dysfunction. Differential drug effects were determined by means of pairwise comparisons. The 453 children who were randomly assigned to treatment with ethosuximide (156), lamotrigine (149), or valproate (148) were similar with respect to their demographic characteristics. After 16 weeks of therapy, the freedom-from-failure rates for ethosuximide and valproate were similar (53% and 58%, respectively; odds ratio with valproate vs. ethosuximide, 1.26; 95% confidence interval [CI], 0.80 to 1.98; P = 0.35) and were higher than the rate for lamotrigine (29%; odds ratio with ethosuximide vs. lamotrigine, 2.66; 95% CI, 1.65 to 4.28; odds ratio with valproate vs. lamotrigine, 3.34; 95% CI, 2.06 to 5.42; P < 0.001 for both comparisons). There were no significant differences among the three drugs with regard to discontinuation because of adverse events. Attentional dysfunction was more common with valproate than with ethosuximide (in 49% of the children vs. 33%; odds ratio, 1.95; 95% CI, 1.12 to 3.41; P = 0.03) (98).
The results of this study at 12 months have been published (99). By 12 months after starting therapy, only 37% of all enrolled subjects were free from treatment failure on their first medication. At the 12-month visit, the freedom-from-failure rates for ethosuximide and valproate were similar and were higher than the rate for lamotrigine. The frequency of treatment failures due to lack of seizure control (p < 0.001) and intolerable adverse events (p < 0.037) was significantly different among the treatment groups. Almost two-thirds of the 125 subjects with treatment failure due to lack of seizure control were in the lamotrigine cohort. The largest subgroup (42%) of the 115 subjects discontinuing due to adverse events was in the valproate group. The previously reported higher rate of attentional dysfunction seen at 16 to 20 weeks in the valproate group compared with the ethosuximide or lamotrigine groups persisted at 12 months (p < 0.01). Shinnar and colleagues reported the long-term outcomes of generalized tonic-clonic seizures in this childhood absence epilepsy drug trial (202). As of June 2014, the cohort had been followed for a median of 7.0 years since enrollment, and 12% (53) had experienced at least one generalized tonic-clonic seizure. The median time to develop generalized tonic-clonic seizures from initial therapy was 4.7 years. The median age at first generalized tonic-clonic seizure was 13.1 years. Fifteen patients (28%) were not on medication at the time of their first generalized tonic-clonic seizure. On univariate analysis, older age at enrollment was associated with a higher risk of generalized tonic-clonic seizures (p = -0.0009), as was the duration of the shortest burst on the baseline EEG (p = 0.037). Failure to respond to initial treatment (p < 0.001) but not treatment assignment was associated with a higher rate of generalized tonic-clonic seizures. Among patients initially assigned to ethosuximide, 94% (15/16) with generalized tonic-clonic seizures experienced initial therapy failure (p < 0.0001). A similar but more modest effect was noted in those initially treated with valproate (p = 0.017) and not seen in those initially treated with lamotrigine. Children initially treated with ethosuximide who are responders have a particularly low risk of developing subsequent generalized tonic-clonic seizures.
In a retrospective study of 128 patients with well-defined childhood absence epilepsy treated with ethosuximide, valproate, and lamotrigine as initial monotherapies: (1) the seizure-free rate of ethosuximide (84%) was significantly higher than that of valproate (62%) and lamotrigine (53%) at 3 months; (2) the seizure-free rate of ethosuximide (90%) was significantly higher than that of lamotrigine (63%) at 6 months; and (3) after 9 months, there was no significant difference in seizure-free rate among the three groups. Adverse-event rates were not significantly different (ethosuximide 25%, valproate 29%, and lamotrigine 14%) (114).
Whether to start with ethosuximide or valproate as first-choice drugs in absence epilepsies has long been a debated issue. They are equally effective as monotherapy in controlling the absences in around 80% of children with childhood absence epilepsy. Many clinicians today prefer valproate because this drug, as opposed to ethosuximide, also controls generalized tonic-clonic seizures and improves compliance when given once daily, usually at night (60). Ethosuximide should be preferred in childhood absence epilepsy with no aberrant electroclinical features, whereas sodium valproate should be preferred in cases with aberrant, or even no aberrant, features (Table 2). When both drugs fail as monotherapy, a trial combining both drugs at lower doses should be attempted before trying to combine each of them with levetiracetam or lamotrigine. As second monotherapy, ethosuximide and valproate demonstrate higher freedom from failure proportions and greater efficacy than lamotrigine. The hepatotoxicity of valproate practically does not exist in this age group and in idiopathic generalized epilepsies. However, valproate is associated with more attentional dysfunction (47) and worse behavioral outcomes than ethosuximide (201). In a retrospective study of 124 cases of absence epilepsy in infancy and childhood in which the majority of cases used valproate, children with longer durations of symptoms prior to appropriate and effective treatment had greater cognitive dysfunction and educational problems (62). In another study, the combination treatment of valproate and small doses of lamotrigine was reported as most effective, possibly because of a pharmacodynamic interaction between the two drugs (170). Small doses of lamotrigine added to valproate have a dramatic beneficial effect; small doses are also mandated because of increased adverse effects associated with this combination (166; 169). The high efficacy of low-dose valproate-lamotrigine combination therapy in childhood absence epilepsy has also been confirmed by Kim and colleagues (122). After 48 months of therapy of children with well-defined childhood absence epilepsy, the rate of freedom from treatment failure was significantly higher and with fewer side effects for the valproate-lamotrigine combination therapy group than for the three monotherapy groups (ethosuximide, valproate, lamotrigine). Further, two cases of childhood absence epilepsy who showed seizure disappearance after ethosuximide was added to valproate and ethosuximide drug eruption occurred were reported, suggesting the possibility that the epilepsy disappears due to immune responses to antiseizure medication (160).
Clonazepam, clobazam, and acetazolamide are only second-line drugs; they share a similar risk of tolerance and adverse effects.
Levetiracetam, because of its efficacy in primarily generalized tonic-clonic seizures (23), myoclonic jerks (161), photosensitivity, and safer ADR profile, appears to be the most promising substitute for valproate in at least juvenile myoclonic epilepsy (161; 200; 227; 230; 169). Levetiracetam should be the first choice for adolescent females, particularly those with juvenile myoclonic epilepsy. However, its role in syndromes with predominant absence seizures has been unclear though in one report, three of four patients with childhood absence epilepsy and all five patients with juvenile absence epilepsy became seizure-free with levetiracetam alone or in combination with other antiseizure drugs (72). In a series by Verrotti and colleagues, the efficacy of levetiracetam was prospectively studied in newly diagnosed patients with childhood and juvenile absence epilepsy (230). Half of them were seizure-free, and most of the remaining patients significantly improved at 1 year on levetiracetam monotherapy without significant adverse effects. Of interest is that this beneficial outcome on typical absence seizure control was mainly seen in childhood rather than juvenile absence epilepsy. These are promising findings, but the study was small and unblinded, and typical absence seizures were assessed on mainly historical counts by parents (168). Thus, the results need confirmation by double-blind, randomized controlled trials. In a report, aggravation of absences with levetiracetam has been reported in six patients resistant to other antiseizure drugs (11). These six patients were retrospectively found in an unspecified number of a probably large population of children with absence epilepsy from three pediatric neurology departments in France. As with any drug, abrupt or fast titration of levetiracetam is not advisable, particularly in childhood.
Topiramate has a weak effect on absence seizures (63), and it is not recommended in childhood absence epilepsy because of lack of efficacy (178).
In 20 patients with childhood absence epilepsy and incomplete seizure control in therapy with the first antiseizure medication (seven ethosuximide, five valproate, five levetiracetam, and three lamotrigine), perampanel was added as the first add-on antiseizure drug (165). None of the patients had generalized tonic-clonic seizures. Fifteen cases became seizure free, and video EEG was negative for epileptic abnormalities (five valproate, five levetiracetam, three ethosuximide, and two lamotrigine). The retention rate after 12 months of follow-up was 75%. The tolerability profile was favorable. The patients who were seizure-free with adjunctive perampanel were switched to perampanel monotherapy (mean follow-up on perampanel monotherapy = 5.56 ± 1.59 months). Nine of 15 remained seizure free in monotherapy with perampanel, whereas the remaining six relapsed, and double antiseizure therapy was restored (165).
Contraindicated antiseizure drugs, vigabatrin and tiagabine are strongly pro-absence drugs that generate absences and absence status epilepticus (166; 169; 54). Of other antiseizure drugs, carbamazepine, oxcarbazepine, phenytoin, phenobarbital, gabapentin, and pregabalin are also contraindicated because they are either ineffective or exaggerate absence seizures (54; 169). An antiseizure drug is contraindicated not only when it exaggerates seizures but also when it is ineffective in controlling the seizures that it is supposed to treat. It may cause unnecessary adverse reactions and deprives the patient of the therapeutic effect that could be provided by another antiseizure drug.
Berg and associates examined the possible association between long-term seizure outcome in childhood absence epilepsy and the initial treatment choice (18). Children with childhood absence epilepsy were prospectively recruited at initial diagnosis and followed in a community-based cohort study. Children presenting with convulsive seizures or significant imaging abnormalities, or who were followed less than 5 years, were excluded. Early outcomes included success of initial medication, early remission, and pharmacoresistance. The primary long-term outcome was complete remission: both seizure free and medication free for 5 or more years. Survival methods were used for analyses.
The first medication was ethosuximide in 41 (69%) and valproate in 18 (31%) children. Initial success rates were 59% (ethosuximide) and 56% (valproate). Early remission and pharmacoresistance were similar in each group. Apart from atypical EEG features (61% [valproate], 17% [ethosuximide]), no clinical features varied substantially between the treatment groups. Complete remission occurred in 31 children (76%) treated with ethosuximide and seven (39%) who received valproate (p = 0.007). Children with versus without atypical EEG features were less likely to enter complete remission (50% vs. 71%, p = 0.03). In a Cox regression, ethosuximide was associated with a higher rate of complete remission than valproate (hazards ratio [HR] 2.5, 95% confidence interval [CI] 1.1 to 6.0; p = 0.03). Atypical EEG features did not independently predict outcome (p = 0.15). Five-year and 10-year remission, regardless of continued treatment, occurred more often in children initially treated with ethosuximide versus valproate. The authors concluded that their findings are congruent with results of studies in genetic absence models in rats and provide preliminary evidence motivating a hypothesis regarding potential disease-modifying effects of ethosuximide in childhood absence epilepsy.
In a study of 164 children with childhood absence epilepsy, the first antiseizure medication administered was ethosuximide in 63.4%, valproic acid in 23.2%, and lamotrigine in 6.7%; 22% of the children had treatment-resistant seizures, 32.9% had learning disabilities, 28% had attention deficit hyperactivity disorder, and 12.8% had anxiety (129). Statistical differences between the response groups included developing a second seizure type, specifically generalized tonic-clonic seizure; the second and third antiseizure medication; and absence of EEG normalization.
Masur and associates assessed pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy (152). Their study provided class I evidence that valproate is associated with more significant attentional dysfunction than ethosuximide or lamotrigine in children with newly diagnosed childhood absence epilepsy. It has been reported that there are few studies concerning pharmacological-resistant childhood absence epilepsy and the possible neuropsychological implications for affected children (185).
Some suggest that in the pure form of childhood absence epilepsy, drug therapy can be gradually withdrawn (within 3 to 6 months) after 2 to 3 years free of seizures (111; 169).
There is a trend towards improved seizure freedom off medications in published childhood absence epilepsy studies after 1985 (159). Seizure freedom off medications was higher in studies after 1985 versus those before 1975 (82% vs. 35%; p < 0.001). Ethosuximide and valproate were more commonly used after 1985, and patients previously treated with ethosuximide or valproate had higher seizure freedom off medications than those treated only with other medications (64% vs. 32%; χ2> 10; p < 0.001) (159).
Genetic implications. A study attempted to determine whether common polymorphisms in CACNA1G, CACNA1H, CACNA1I, and ABCB1 are associated with differential short-term seizure outcome in childhood absence epilepsy (100). In a randomized double-blind trial of ethosuximide, lamotrigine, and valproate, 446 children with childhood absence epilepsy children had short-term seizure outcome determined. Associations between polymorphisms (minor allele frequency > /= 15%) in four genes and seizure outcomes were assessed. In vitro electrophysiology on transfected CACNA1H channels determined impact of one variant on T-type calcium channel responsiveness to ethosuximide. Eighty percent (357 of 446) of subjects had informative short-term seizure status (242 seizure-free; 115 not seizure-free). In ethosuximide subjects, two polymorphisms (CACNA1H rs61734410/P640L, CACNA1I rs3747178) appeared more commonly among not-seizure-free participants (p = 0.011, odds ratio [OR] = 2.63, 95% confidence limits [CL] = 1.25-5.56; p = 0.026, OR = 2.38, 95% CL = 1.11-5.00). In lamotrigine subjects, one ABCB1 missense polymorphism (rs2032582/S893A; p = 0.015, OR = 2.22, 95% CL = 1.16-4.17) was more common in not-seizure-free participants, and two CACNA1H polymorphisms (rs2753326, rs2753325) were more common in seizure-free participants (p = 0.038, OR = 0.52, 95% CL = 0.28-0.96). In valproate subjects, no common polymorphisms were associated with seizure status. In vitro electrophysiological studies showed no effect of the P640L polymorphism on channel physiology in the absence of ethosuximide. Ethosuximide's effect on rate of decay of CaV 3.2 was significantly less for P640L channel compared to wild-type channel. The authors concluded that 4 T-type calcium channel variants and one ABCB1 transporter variant were associated with differential drug response in childhood absence epilepsy. The in vivo P640L variant's ethosuximide effect was confirmed by in vitro electrophysiological studies. This suggests that genetic variation plays a role in differential childhood absence epilepsy drug response (100).
Predictors of treatment response. Looking forward, there is a growing interest in the development of biomarkers of treatment response and side effects (119).
In a publication, various EEG features were analyzed to predict responsiveness to valproic acid in patients with childhood absence epilepsy before antiseizure medication use (137). Alpha band power was significantly higher and delta wave power was significantly lower in the responder group than in the nonresponder group. The ability to predict patient responses to first-line antiseizure medication would help clinicians to select suitable medication for patients with childhood absence epilepsy at an early stage, thereby decreasing the pill burden of patients as well as the risk of side effects.
A study of 24 drug-naive patients with childhood absence epilepsy investigated the differences between antiseizure drug responders and nonresponders using magnetoencephalography, which evaluated whether neuromagnetic signals of the brain neurons were correlated with response to therapy (246). All patients were treated with appropriate antiseizure drugs. Outcome was assessed at least 1 year after their magnetoencephalography recordings, and they were consequently divided into responders and nonresponders. The results displayed abnormal dynamic networks involving the frontal cortex, especially the medial frontal cortex at the alpha band, that might be relevant to patients with childhood absence epilepsy who are nonresponsive to antiseizure drugs. However, one single mechanism cannot explain resistance to antiseizure drugs because resistance could be due to multiple factors that coexist in the same patient.
In summary, for children with childhood absence epilepsy who only present with typical absence seizures, ethosuximide is not only a more effective, but also a safer, drug. In children with childhood absence epilepsy and aberrant electroclinical features, valproate should be preferred. This is also suggested in a publication (205).
Outcome is usually excellent for the syndrome of childhood absence epilepsy if properly diagnosed and treated, provided diagnosis is based on strict inclusion criteria (Table 2). Spanioleptic typical absence seizures rarely have an onset in childhood, thereby indicating juvenile absence epilepsy. If such cases are included in studies of childhood absence epilepsy, among others with aberrant electroclinical features, this may explain the rare, generalized tonic-clonic seizures reported in adolescence and the variable outcomes.
Future studies comparing the response of childhood absence epilepsy with and without aberrant electroclinical features to appropriate anti-absence medications are needed. This will reveal specific electroclinical features related to treatment response and related neuropsychiatric problems.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Donya Eizadkhah MD
Dr. Eizadkhah of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Apr. 08, 2026