
Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Congenital myopathies represent a clinically and genetically heterogeneous group of early-onset neuromuscular diseases with characteristic, but not always specific, histopathological features. They often present with stable or slowly progressive truncal and proximal weakness. The classification of congenital myopathies was initially based on pathological phenotype and clinical manifestations but has recently been based on their variable genetic basis (109). It is often not possible to make a diagnosis solely based on clinical findings. Additional extraocular, respiratory, distal involvement, scoliosis, and distal laxity may provide clues (25).
The “core myopathies” collectively represent the most common form of congenital myopathies, and the name pathologically corresponds to histochemical appearance of focally reduced oxidative enzyme activity and myofibrillar changes on ultrastructural studies. Subtypes of core myopathies are determined by the location, shape, size, and number of cores. Centrally located cores are called central core disease. Numerous small scattered cores are commonly found in multinucleated disease (MmD). Irregular areas of myofibrillar disarray with patchy oxidative staining and a lack of ATPase activity have been identified, and this has been called as dust nucleus disease (DuCD) (40).
Because of the clinical, pathological, and molecular overlaps, central core disease and multiminicore disease will be discussed together. Of note, cores can be seen in combination with nemaline rods and are termed “core-rod myopathies,” which is associated with a variety of different mutations (19).
Mutations in the skeletal muscle ryanodine receptor 1 (RYR1) gene are associated with dominantly inherited central core disease and subgroups of recessively inherited multiminicore disease, centronuclear myopathy (CNM), and congenital fiber type disproportion. Malignant hyperthermia susceptibility trait is a dominantly inherited allelic trait and is described as a pharmacogenetic predisposition to severe and potentially life-threatening reactions in response to halogenated anesthetic agents and depolarizing muscle relaxants.
RYR1-related malignant hyperthermia susceptibility is allelic to central core disease and has also been described as a common cause of induced and episodic phenotypes such as exertional rhabdomyolysis or periodic paralysis, which is present throughout life. Late-onset presentations in the adulthood period highlight the relevance of congenital myopathies for adult neuromuscular practice.
Mutations in another gene, CACNA1S, inherited in an autosomal dominant manner, can cause malignant hyperthermia in patients with core myopathy (08).
A number of distinct phenotypes are seen in multiminicore disease, which is most commonly caused by recessive mutations in the RYR1 and SEPN1 genes. It has been linked to dominant mutations in the gene for beta-myosin heavy chain protein (MYH7) and autosomal recessive mutations of titin (TTN). Recessive mutations of satellite cell gene (MEGF10) are defined in patients with early-onset myopathy, areflexia, respiratory distress, and dysphagia (EMARRD). A few other genes have also been identified within this group (Table 1).
|
• Core myopathies represent the most common form of congenital myopathies and are characterized pathologically by the absence of oxidative enzyme activity in the central area of myofibers representing mitochondrial depletion. | |
|
Central core disease and multiminicore disease are the most common congenital myopathy forms. | |
|
• Central core disease is dominantly inherited in most cases. Recessive central core disease is less common and often more severe, and it presents at a younger age. Most cases of central core disease are due to mutations in the gene for ryanodine (RYR1), a skeletal muscle calcium channel receptor. | |
|
• RYR1-related malignant hyperthermia susceptibility trait is an allelic condition to central core disease. RYR1 mutations may present in the adulthood period as induced and episodic phenotypes, such as exertional rhabdomyolysis and periodic paralysis. | |
|
• Major causes of multiminicore disease are recessively inherited mutations in the genes encoding selenoprotein N (SEPN1) and RYR1. Other rare causes are recessive mutations in TTN, MEGF10, and CACNA1S, and dominant mutations in MYH7 and CACNA1S. There are more (Table 1). | |
|
• Also, mutations in CACNA1S, inherited in an autosomal dominant manner, can cause malignant hyperthermia in patients with multiminicore disease. | |
|
• There may be a continuum between multiminicore disease and central core disease; patients and carriers with certain missense mutations in RYR1 are at risk of malignant hyperthermia. |
In 1956, Magee and Shy described “a new congenital nonprogressive myopathy” characterized by a distinctive microscopic appearance on skeletal muscle biopsy (63). It was the first recognition of the congenital myopathies, the diagnosis of which was based on the distinct structural or histochemical changes in biopsied skeletal muscle, as a distinct group of diseases.
This disorder was then named central core disease (44), and an absence of oxidative enzyme and phosphorylase reactivity in the cores were demonstrated on muscle biopsy in these patients (27).
In 1971, Engel and colleagues described a patient with multiple small cores within muscle fibers (29), which were later renamed as “multicores,” “minicores,” “focal loss of cross-striation,” “target-like lesions,” and “miniature cores;” multiminicore disease is now the preferred terminology (33).
There is a wide clinical spectrum of core myopathies, with severity ranging from mild to severe. Hypotonia, joint laxity, delayed motor milestones, hip girdle or axial weakness, and congenital hip dysplasia may be among early clinical presentations. Arthrogryposis represents the severe end of the spectrum. There may be variability even within the same family; some of the individuals are asymptomatic and others present with hyperkalemia, exertional myalgia, rhabdomyolysis or periodic muscle stiffness, and paralysis.
Typical central core disease patients present with mild and symmetrical weakness, hypotonia, and delayed motor milestones, and although late, achieve independent ambulation. The course is usually nonprogressive or slowly progressive. Patients presenting with severe neonatal weakness, arthrogryposis, and respiratory failure require early respiratory support and corrective scoliosis surgery. More significant respiratory muscle weakness is seen in infants with recessive core disease (71; 89; 02). Extraocular muscles are spared in the dominant forms. Because of musculoskeletal deformities including congenital hip dislocation, kyphoscoliosis, pes cavus, pes planus, and thoracic deformities, patients are frequently referred from orthopedic clinics. Heart disease is not considered as a part of the typical “core myopathy” spectrum. Some cardiac abnormalities described include mitral valve prolapse, arrhythmias, and asymptomatic right bundle branch block (98). Intelligence is generally normal in central core disease.
Genetic resolution of core myopathies has further led to “mutation-specific” clinical presentations and phenotype-genotype correlations. The availability of next-generation sequencing (NGS) techniques has improved the detection rate for mutations and expanded the clinical spectrum (26; 02; 10).
Central core disease is associated mainly with dominant RYR1 mutations, and multiminicore disease is genetically a more heterogenous condition (55).
The central core disease is associated mainly with dominant RYR1 mutations, whereas the multimonitor disease is genetically more heterogeneous (54).
Dominantly inherited RYR1-related central core disease is characterized by mild to moderate muscle weakness presenting from infancy to childhood. Congenital hip dislocation, scoliosis, and generalized joint laxity are common. In contrast to the recessive forms with a more severe clinical phenotype, there is no extraocular muscle involvement. Bulbar, respiratory, and cardiac involvement is uncommon. Myalgia may be prominent. Central core disease tends to be stable over long periods with a possible progression in adulthood, and due to intrafamilial variability, there may be a delay in diagnosis.
Malignant hyperthermia is a disorder of calcium metabolism. Central core disease is associated with an increased risk of malignant hyperthermia. Most patients with malignant hyperthermia have normal muscle biopsy features, and less than 30% of the patients with central core disease have malignant hyperthermia susceptibility (58). Two well-known malignant hyperthermia-related syndromes are King-Denborough syndrome and Native American myopathy (NAM).
The association between malignant hyperthermia and central core disease patients was first described in 1973 and has since been confirmed in many other reports (20; 36; 98).
King-Denborough syndrome is characterized by facial and skeletal dysmorphism, malignant hyperthermia susceptibility, and myopathy (87; 17; 24). All genetically solved patients to date are due to RYR1 mutations.
Malignant hyperthermia susceptibility-related RYR1 mutations have been described as a common cause of exertional rhabdomyolysis, accounting for up to 30% of presentations in otherwise healthy individuals after exclusion of other causes (23; 50). A predisposing genetic background should be considered if episodes are familial, recurrent, out of context to the exercise performed, or preceded by other symptoms such as cramps, myalgia, and weakness. RYR1-related rhabdomyolysis may occur up to 72 hours after exercise and may mimic viral myositis, and in contrast to other metabolic myopathies, fasting does not appear to be a triggering factor (23; 69).
In a retrospective cohort study including 277 pediatric and adult patients referred for malignant hyperthermia and inherited myopathies, RYR1 mutations were detected in 77 unrelated patients with a detection rate of 28%, and exertional rhabdomyolysis phenotype was prominent in this Dutch series (99).
RYR1-related malignant hyperthermia susceptibility is allelic to central core disease, and some patients with central core disease may also have malignant hyperthermia susceptibility. Exertional rhabdomyolysis and periodic paralysis may present throughout life (23; 55). Late-onset presentations in the adulthood period highlight relevance of the congenital myopathies for adult neuromuscular practice.
In an ENMC workshop on RYR1-related myopathies, the phenotypical spectrum expanded further, particularly in relation to mutations associated with recessive inheritance, and induced or late myopathic manifestations of dominant mutations previously mainly associated with the malignant hyperthermia susceptibility trait (50).
Highlights from this workshop in terms of clinical phenotypes and genotype-phenotype correlations are summarized below:
|
A. Early-onset RYR1-related phenotypes | ||
|
1. Dominant RYR1 mutations typically present with congenital hypotonia, weakness, and hip dislocation at birth. Motor milestones are delayed; independent ambulation is eventually achieved. Weakness tends to involve hip girdle and quadriceps with sparing of facial and extraocular muscles. | ||
|
2. Dominant and recessive RYR1 mutations with a severe neonatal presentation leading to death are described. Fetal akinesia, arthrogryposis, severe weakness, poor feeding, and respiratory failure in the neonatal period can be the initial presentation. | ||
|
3. Recessive RYR1 mutations have a tendency of earlier and more severe presentation compared to most patients with dominant mutations; however, they are also associated with a wide range of clinical phenotypes and pathological changes. | ||
|
4. RYR1-related centronuclear myopathy (CNM) present with external ophthalmoparesis of variable degree, frequently associated with facial weakness. | ||
|
5. Histopathological pattern may resemble congenital fiber type disproportion and congenital muscular dystrophy. | ||
|
6. Recessive RYR1-associated phenotypes can be grouped as clinical groups with and without ophthalmoparesis. | ||
|
7. Bilateral congenital lumbar hernias in an infant with RYR1-related central core disease. | ||
|
8. Gain-of-function RYR1 mutations may lead to prolonged bleeding by altering vascular smooth muscle cell function. | ||
|
B. Late-onset RYR1-related phenotypes | ||
|
1. Malignant hyperthermia susceptibility | ||
|
C. SEPN1-related multiminicore myopathy | ||
|
1. Head lag and neck flexor weakness may evolve into rigid spine deformity. Patients with rigid spine deformity and scoliosis should be managed with early respiratory insufficiency and nocturnal hypoventilation. | ||
Bilateral congenital lumbar hernias in an infant with central core disease, severe clinical phenotype, multiple joint contractures, decreased muscle bulk, symptoms of malignant hyperthermia, and RYR1 gene mutation have been described (59). Congenital lumbar hernias are rare malformations caused by defects in the development of posterior abdominal wall. Authors hypothesize that RYR1 mutations may affect posterior wall development by causing inappropriate calcium fluctuations and influencing downstream signaling pathways (59).
Mild bleeding abnormalities are described in patients with malignant hyperthermia carrying gain-of-function RYR1 mutations (62). Knock-in mice with the malignant hyperthermia susceptibility RYR1 mutation Y522S had longer bleeding times than their wild-type littermates, and these mice exhibited higher frequency of subplasmalemmal Ca+2 sparks, leading to a more negative resting membrane potential. The bleeding defect of mice and one patient was reversed by treatment with RYR1 antagonist dantrolene, and Ca+2 sparks in primary vascular smooth muscle cells were blocked in the mice model.
Authors conclude that RYR1 mutations may lead to prolonged bleeding by altering vascular smooth muscle cell function, and they emphasize the a) reversible nature of the bleeding phenotype and b) potential therapeutic value of dantrolene in the treatment of bleeding disorders (62).
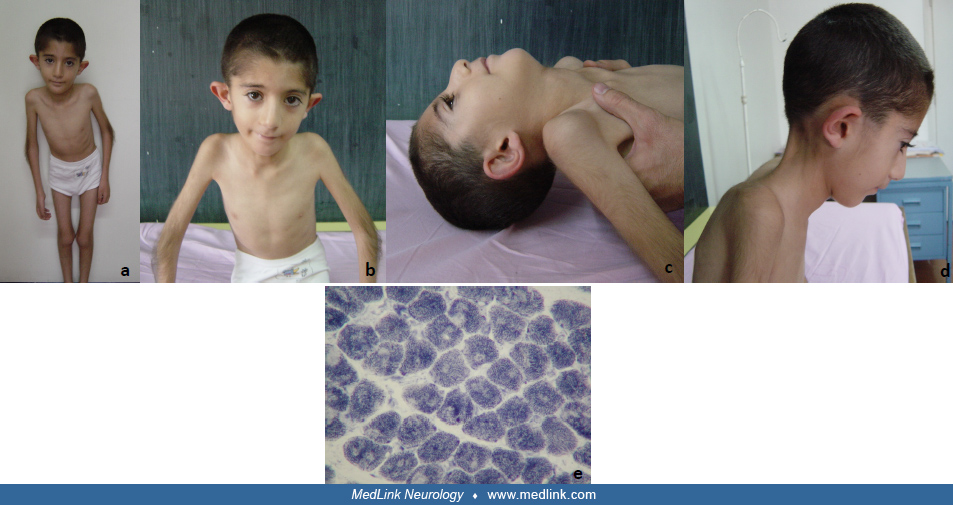
Multiminicore disease is a clinically heterogeneous disorder. Four major clinical subgroups are recognized (31; 32; 76; 55; 54). Core phenotype due to SEPN1-related myopathies can be defined as predominant axial weakness, early spinal rigidity, scoliosis, and respiratory involvement. There is a disproportion between axial and skeletal muscle weakness. This is the most common (approximately 75% of all cases) “classic” form in the neonatal period or first year of life. A history of reduced fetal movements can be elicited in as many as 30% of cases (53). Affected infants are hypotonic and weak and have delayed motor development. Some children have associated congenital abnormalities such as cleft palate, dislocated hips, or arthrogryposis (31). Physical examination reveals generalized hypotonia, joint hyperlaxity, and asthenic phenotype with decreased muscle bulk. Short stature and failure to thrive are common in children with significant weakness. Intelligence is normal. Weakness in the classic form is predominantly axial. The neck flexors are usually the most affected muscles, with poor or absent head control in infancy being characteristic of this disorder. There is mild to moderate weakness of the proximal limb muscles. Facial weakness is common but of variable severity, whereas the extraocular muscles are spared. The deep tendon reflexes are absent or diminished. There is often a Gowers sign. Extremity weakness may be static or slowly progressive or may even appear to improve slightly with increasing age (81). Progressive weakness of the axial musculature often results in kyphoscoliosis and spinal rigidity of variable severity (31; 101; 16; 76). Paraspinal rigidity and kyphoscoliosis frequently lead to early-restrictive respiratory dysfunction with progressive respiratory insufficiency, which may be rapid in onset. Weakness and spinal deformity are usually static after adolescence. Minor contractures of the elbows, knees, and hips may develop after infancy but are generally not problematic. Because limb weakness is often relatively mild, most patients are ambulant well into adulthood, even in the presence of significant respiratory compromise. Up to two thirds of patients with the classic form of minicore disease develop respiratory insufficiency in late adolescence or early adulthood (31; 32). Cardiac involvement is usually in the form of cor pulmonale secondary to respiratory insufficiency, rather than primary myocardial involvement. About 50% of cases of “classic” multiminicore disease are caused by recessive mutations in the SEPN1 gene (33).
Adult-onset “classic” multiminicore disease is seen in a minority of cases, often in association with the rigid spine syndrome (31), which may be associated with respiratory impairment and development of cor pulmonale (106). Cardiomyopathy may be more common in late-onset cases (97; 65; 79). Occasional cases of adult-onset multiminicore disease presenting with myalgia have been reported (100; 26).
The moderate form of multiminicore disease with hand involvement is relatively rare (fewer than 10% of all cases) and is characterized by relatively mild distal weakness of the upper limbs with hand amyotrophy and marked joint hyperlaxity. The lower extremities are mildly affected with proximal pelvic girdle weakness. Scoliosis and respiratory involvement are minimal or absent (31).
Fewer than 10% of cases of multiminicore disease are of the ophthalmoplegic form, in which there is external ophthalmoplegia in addition to proximal limb weakness (42; 53; 71). This form of minicore disease may be associated with recessive mutations in RYR1 (48).
Less than 10% of all cases present in the neonatal period with arthrogryposis. Affected infants have severe generalized joint contractures as a consequence of markedly decreased movement in utero. Other characteristic findings, also related to decreased fetal motion, include a high-arched palate, low-set ears, short neck, and clinodactyly (31). The genetic basis of this form of multiminicore disease is unknown (48).
There is a huge phenotypic variability in multiminicore disease. Spanish kindreds with a dominantly inherited distal myopathy with weakness of the great toe and ankle dorsiflexors and often associated neck flexor, finger extensor, and mild facial weakness were found to have minicore myopathy caused by the common mutation in the MYH7 (beta-myosin heavy chain protein) gene, which is more commonly known to cause Laing distal myopathy (74). A report described two kindreds with minicore disease caused by dominant mutations in MYH7, presenting in childhood with proximally predominant weakness with progression in adulthood to distal weakness and dilated cardiomyopathy (16).
Recessive mutations of a satellite cell gene (MEGF10) are also implicated in multicore disease (11). Clinical phenotype is characterized by early-onset myopathy, areflexia, respiratory distress, and dysphagia (EAMRDD) (61).
At the beginning of 2020, TRIP4 mutations leading to loss of the coactivator protein ASC1, which direct binds to transcription factors, have been shown to cause multiminicore disease with a contractural phenotype (107).
|
Gene |
CCD |
MmD |
DuCD |
Core-rod |
|
RYR1 |
+++ |
+ |
+ |
+ |
|
SELENON |
++ | |||
|
MYH2 |
+ | |||
|
MYH7 |
+ | |||
|
TTN |
+ | |||
|
CCDC78 |
+ | |||
|
UNC45B |
+ | |||
|
ACTN2 |
+ | |||
|
MEGF10 |
+ | |||
|
NEB |
+ | |||
|
ACTA1 |
+ | |||
|
KBTBD13 |
+ | |||
|
CFL2 |
+ | |||
|
TRIP4 |
+ | |||
|
TNNT1 |
+ | |||
|
| ||||
The course is static or slowly progressive in most central core disease patients. Involvement of the respiratory muscles may be of insidious onset and may remain subclinical until unmasked by intercurrent illness or anesthesia. Lung function should be monitored with serial pulmonary function tests, and where indicated, polysomnography. Cardiac complications are uncommon, but baseline electrocardiography and echocardiography are appropriate in most cases of suspected myopathy. Children should be monitored for the development of scoliosis and other skeletal deformities (111).
Many patients with central core disease are at risk of developing malignant hyperthermia during a general anesthetic. As the first exposure to trigger substances elicits an event in only 50% of malignant hyperthermia susceptibility patients, a previous history of tolerance of halogenated anesthetic agents or depolarizing muscle relaxants does not guarantee that these agents can be used safely in future anesthetics. Appropriate anesthetic precautions should be taken in all instances (111).
Nonskeletal muscle presentations of RYR1-related myopathies are described because RyR1 receptors are expressed in a range of other tissues (50). These can be summarized as bleeding abnormalities (62): severe CNS involvement in an adolescent suffering a fatal malignant hyperthermia episode, olanzapine as a triggering agent, with a cerebellar involvement showing similarities to deceased heat stroke victims (35), and evidence of cardiac involvement described in the form of sudden death at a relatively early age by a patient with a late-onset myopathy who died suddenly at the end of running a marathon in the Dutch cohort of patients (50).
Smooth muscle cell involvement in the arteries and arterioles could be an explanation, and these complications and possible CNS and cardiac phenotypes should be investigated in detail.
Minicore myopathy follows a variable course. In many patients, the condition remains benign with static or slowly progressive weakness of the extremities and retention of independent mobility. In some, spinal rigidity becomes a clinically predominant feature in late childhood or adolescence. Severe progressive scoliosis is apparent in a minority. Surgical fixation of the spine is required in most such cases. Minor contractures of the elbows, knees, and hips may develop after infancy and are generally amenable to physiotherapy.
Progressive scoliosis or respiratory insufficiency is seen in up to two thirds of patients with the “classic” form of multiminicore disease, although the ability to walk independently is often preserved even in adults with respiratory failure. There is a marked discrepancy between profound respiratory impairment and preserved ambulation (55). Mortality from multiminicore disease is usually related to complications of respiratory disease. Mortality related to cardiac involvement is uncommon but has occasionally been reported (97; 65; 16). Significant cardiomyopathy in minicore myopathy should prompt consideration of short-chain acyl-CoA dehydrogenase deficiency (102).
Clinical discordance in monozygotic twins has been reported in RYR1 myopathy, denoting other factors beyond nuclear (83).
Case 1. A 10-year-old boy was referred from the pediatric gastroenterology department with a previous diagnosis of celiac disease. He had an asthenic phenotype and growth failure, which was noticed at 2 years of age. Prenatal and natal histories were uneventful. Parents noted that his head control was at 2 years of age, although he was able to walk without support at the age of 14 months. Onset of scoliosis was around the age of 7 years. Parents were first cousins. Family history was negative for a neuromuscular disease.
An asthenic phenotype, decreased muscle bulk, achieving head control later than the ability to walk unsupported, and consanguinity were clues for a neuromuscular disease primarily involving the axial muscles.
Physical examination revealed an asthenic phenotype, decreased muscle bulk, cervical spinal rigidity, mild scoliosis at the thoracic level, neck flexor muscle weakness (MRC 2 of 5), proximal skeletal weakness in upper and lower extremities (MRC 4 of 5), and Gower sign. There was no extraocular involvement and facial weakness.
Asthenic phenotype, rigid spine deformity, and scoliosis necessitate including a neuromuscular disease in the differential diagnosis. Head lag and discrepancy between head control and skeletal muscle weakness were further clues for SEPN1-related myopathies, congenital muscular dystrophy with rigidity of spine (RSMD1), and laminopathies.
Serum creatine kinase level was normal. Muscle biopsy was consistent with minicore myopathy. Molecular genetic analysis revealed a homozygous c.817G-A (p.G273R) mutation in SEPN1. Respiratory function tests (forced vital capacity (FCV): 31% and polysomnography) were consistent with restrictive respiratory dysfunction and nocturnal hypoventilation syndrome. The patient was followed with pediatric chest diseases specialists and remained stable on nocturnal noninvasive ventilatory support (BiPAP).
Central core disease is caused by mutations in the RYR1 gene on chromosome 19q13.1. The RYR1 gene encodes a protein product, the calcium release channel ryanodine receptor (RyR1), which is highly expressed in skeletal muscle sarcoplasmic reticulum, B lymphocytes, and lymphoblastoid cells (41). The skeletal muscle ryanodine RyR1 is a 560 kDa tetrameric structure that, when triggered, allows release of calcium from the lumen of the sarcoplasmic reticulum into the sarcoplasm, triggering muscle contraction. Proteins associated with the RyR1 include the dihydropteridine receptor, FK506 binding protein, calmodulin, triadin, and calsequestrin. Skeletal muscle excitation-coupled calcium entry relies on the interaction between the sarcolemmal 1,4-dihydropyridine receptor and the RyR1 on the sarcoplasmic reticulum membrane. Excitation-coupled calcium entry is strongly enhanced in cells from patients with central core disease compared with individuals with malignant hyperthermia and controls (103).
The RYR1 gene is large (containing 106 exons), rendering genotype-phenotype correlation difficult.
Mutations in the ryanodine gene can be identified in more than 90% of patients with central core disease when all parts of the RYR1 are carefully sequenced (114). Mutations causing central core disease and malignant hyperthermia cluster to three regions of the RYR1 gene. Approximately 50% of patients with clinical evidence of a myopathy and central cores on muscle biopsy have a mutation in region 3 in exons 93 to 104 of RYR1 (73).
The RyR1 controls calcium release during excitation-contraction coupling. In patients with central core disease, RYR1 mutations result in muscle weakness by either causing abnormal “leakiness” of calcium channels, with augmented release of calcium from the sarcoplasmic endoplasmic reticulum (04), or by altering excitation-contraction coupling. Channel “leakiness” appears to be more profound with mutations causing muscle weakness than in those causing only malignant hyperthermia susceptibility (12). Some central core disease-causing RYR1 mutations, however, do not significantly modify calcium release and likely cause muscle weakness by altering excitation-contraction coupling in muscle contraction (84; 05). This hypothesis has been supported by studies showing abnormal distribution of calcium-handling proteins, including those of the sarcoplasmic reticulum (calsequestrin, SERCA1/2, and triadin) and the T-tubule (dihydropyridine receptor-alpha1subunit) in RYR1-associated core myopathies (46). RYR1 mutations associated with both malignant hyperthermia susceptibility and central core disease may cause both calcium depletion from the sarcoplasmic reticulum and increased basal calcium levels, whereas those associated with malignant hyperthermia alone may result in a more modest increase in basal calcium levels without an overall increase in net sarcoplasmic reticulum calcium content (22). Mutations causing dominant central core disease alone tend to result in leaky channels, leading to depletion of calcium from sarcoplasmic reticulum stores. In contrast, mutations causing malignant hyperthermia susceptibility, without central core disease, render the RyR1 hypersensitive to activation by electrical and pharmacological stimuli. A minority of RYR1 mutations is unveiled by silencing of the wild type allele, causing a decrease in expression of mutant RYR1 channels on sarcoplasmic reticulum membrane. Recessive RYR1 mutations may cause a marked decrease in sarcoplasmic reticulum calcium release during excitation-contraction coupling (115; 39). The majority of gene mutations in recessive central core disease are either missense or nonsense mutations (02).
Interestingly, RyR1 are also expressed in the brain, with high levels in the cerebellum, hippocampus, and hypothalamus, where they play a role in voltage-induced calcium release in nerve terminals (18).
Central core disease has traditionally been regarded as an autosomal dominant disease with variable penetrance. De novo mutations are relatively common (73). The new mutation rate is estimated to be about 10%.
Autosomal recessive inheritance of central core disease has been recognized in a number of families (33; 51; 71; 89; 57). Recessive central core disease links to RYR1 but demonstrates more clinical and pathologic heterogeneity than that is seen with dominant inheritance. Protein expression studies variably suggest a correlation between specific mutations, protein levels, and phenotype. Although patients with dominant or recessive mutations associated with typical central core disease phenotypes appear to have normal RyR1 expression, individuals with more generalized weakness, multiminicores, and external ophthalmoplegia have a pronounced depletion of RyR1 expression (118; 02). Recessive core disease may be more common than has previously been recognized. A specific genetic mechanism of recessive core disease may be monoallelic RYR1 transcription, or limitation of RNA transcription from one of the mutated RYR1 alleles, which appears to occur as a tissue-specific phenomenon in some individuals with central core disease. In such cases, the nonexpressed allele is often maternally derived (117).
An ENMC International workshop is dedicated to RYR1-related myopathies; clinical spectrum, genotype-phenotype correlations, basic science, animal models, therapy development, clinical trials, and challenges have been discussed extensively (50).
The pathological hallmark of central core disease is the presence of well-demarcated cores (round or oval shaped regions within a muscle fiber that lack oxidative enzyme activity on histochemical stains) within type 1 muscle fibers. There is usually type 1 fiber predominance, which may be most marked in those with RYR1 mutations at the C-terminal (114). Cores usually extend along most or all of the length of the fiber. There may be one or more cores within a fiber. Although the biopsy may show other myopathic features such as fiber size variability, increased internal nuclei, and fiber splitting, the presence of cores as the predominant pathological feature in the biopsy establishes the diagnosis of central core disease. Necrosis and significant fiber regeneration are uncommon, but extensive fibrosis and fatty infiltration are seen occasionally (96).
Cores may be centrally or eccentrically placed and, in a minority of cases, may resemble minicores, raising the possibility of the separate but related disorder minicore myopathy, which is generally autosomal recessive in inheritance. In such cases, microscopic evaluation of cross sections may not distinguish between central core disease and multiminicore disease, whereas longitudinal sections reveal the cores of central core disease to run the whole length of the fiber. Those of multiminicore disease are usually shorter and are seen in both type 1 and type 2 fibers. Diagnosis is also difficult in the small number of patients whose muscle biopsy demonstrates uniformity of type 1 fibers without cores. This entity, now known as congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation (CNMDU1), is caused by mutations in RYR1 in 40% of cases (92). Cores and nemaline rods are seen in tandem in some kindreds with dominant RYR1 mutations (72; 93; 47).
Desmin (an intermediate filament protein found in muscle fibers) reactivity, detected by an indirect immunofluorescence assay, is abnormal in cores (104). Accumulations of desmin are seen in many other myopathies. Myotilin, a Z-disc protein that binds alpha-actinin, gamma-filamin, and F-actin, is also present in central cores (94). Immunocytochemistry is helpful in demonstrating cores but shows no other specific abnormalities in central core disease (96).
Electron microscopic examination of cores shows an absence of mitochondria, the anatomical correlate to the loss of oxidative enzyme activity in histochemical reactions. The appearance of the myofibrillar contractile apparatus is variable in the cores; it ranges from essentially normal (structured) to significantly disrupted (unstructured) (75).
There is no clear correlation between biopsy findings and clinical phenotype in central core disease (02).
In an ENMC workshop, a summary of the main pathological features of muscle biopsies from patients with RYR1 mutations are presented, with an emphasis on heterogeneity of pathology and limitations in our understanding of their meaning (50). In some young cases, increase in adipose and connective tissue can be extensive resembling congenital muscular dystrophies. Of note, sampling may be a factor because of differential involvement of muscles, there may be evolution of biopsy findings, and patients presenting with exertional myalgia and malignant hyperthermia often show mild, nonspecific myopathic pathological features (50).
Muscle MRI may help to define distinct pattern of involvement and can be used as a potential biomarker for disease severity in neuromuscular conditions (48; 112).
Selenoprotein N, a glycoprotein, localizes to the endoplasmic reticulum and is found at low levels in virtually all body tissues. Other selenoproteins are involved in reduction-oxidation process or processing of thyroid hormone. SEPN1 has a calcium binding motif similar to that seen in calmodulin, suggesting a possible role in calcium homeostasis (70), although SEPN1 mutations have not been associated with abnormal localization of calcium-related proteins in or around minicores (46). Selenoprotein N mutations have also been implicated in myogenesis, with work suggesting a possible role in muscle sarcomeric organization and myofiber attachment (21; 60). SEPN1 mutations cause 40% to 50% of cases of classic minicore myopathy (33a).
Mutations in two genes are responsible for approximately 50% of cases of minicore disease. In a study from Italy, mutations of SEPN1 represented 6% of congenital muscular dystrophy patients (43). The proportion related to MYH7 mutations is not known but likely to be low given that dominantly inherited minicore myopathy is uncommon (74; 16; 14).
Recessive mutations in the gene for SEPN1 account for 30% of all multiminicore disease and represent 40% to 50% of all “classic” form (33). Until recently, all identified mutations have been located in the 5’ UTR and coding sequence of SEPN1, consisting of microdeletions or insertions leading to frameshift, splice site mutations causing aberrant RNA splicing, or single nucleotide changes causing missense mutations. A novel mutation in the 3’UTR region of the gene has been reported. This mutation in the selenocysteine insertion sequence of selenoprotein messenger RNA alters binding of a crucial binding protein (SBP2) and decreases SEPN production (01). Most identified mutations result in premature termination of translation; the 5’ remainder representing missense amino acid substitutions. Homozygous mutations of the same gene were originally described in congenital muscular dystrophy with rigid spine (rigid spine muscular dystrophy) (70), an entity that is now felt to represent severe “classic” minicore myopathy presenting in early childhood (33a).
SEPN1 mutations have also been shown to cause desmin-related myopathy with Mallory body-like inclusions, a congenital myopathy characterized by intrasarcoplasmic desmin aggregates (30).
Minicore myopathy is described in subjects with mutations in MYH7, a gene for beta-myosin heavy chain protein usually associated with Laing distal myopathy (74; 16; 14). Weakness in such cases may be of childhood or adult onset, may be proximally or distally predominant, and may be associated with an adult-onset cardiomyopathy.
Recessive RYR1 mutations associated with minicore disease appear to be distributed throughout the RYR1 gene, resulting in abnormalities of excitation-contraction coupling and calcium homeostasis in skeletal muscle (28; 117). RYR1 mutations have been shown to be associated with depletion of ryanodine channels in minicores, with local accumulation of other proteins of the sarcoplasmic reticulum (calsequestrin, SERCA1/2, and triadin) and the T-tubule (dihydropyridine receptor-alpha1subunit) (46). RYR1 mutations have been linked to the moderate form of minicore disease with hand involvement (33b; 51), in a single patient, to the classic form of minicore myopathy (71), and to a tendency to malignant hyperthermia syndrome with pathological (but not clinical) changes consistent with minicore disease (45).
The MYH7 gene encodes slow/beta cardiac myosin heavy chain, a myosin isoform found in cardiac and type I skeletal muscle fibers. Mutations in MYH7 are an established cause of hypertrophic cardiomyopathy and less commonly cause dilated cardiomyopathy and left ventricular noncompaction. MYH7 gene mutations have been linked to a number of skeletal myopathies, including Laing distal myopathy, myosin storage myopathy, congenital fiber-type disproportion, and now minicore myopathy (16; 14).
Recessive mutations of a satellite cell gene (MEGF10) are also implicated in multicore disease (11). Patients present with early-onset myopathy, areflexia, respiratory distress, and dysphagia (EAMRDD) (61).
With a new era in the genetics of neuromuscular diseases and an availability of NGS technologies, being cost-effective and available, a new terminology “diagnosis by sequencing” has been introduced (85). The latest European Malignant Hyperthermia Group diagnostic guidelines have introduced the option of primary DNA screening for the index case. During an ENMC workshop, in a United Kingdom cohort of 126 unrelated patients, RYR1 variants were found in 72 samples, but only 20 of these were the variants known to be pathogenic or likely to be pathogenic, and 16 variants were known polymorphisms or unlikely to be pathogenic, whereas 50% of the RYR1 variants found were of unknown significance (50).
Importance of in depth analysis correlating genotypes with phenotypes, histotypes, and pathophysiological consequences-physiotype is addressed to understand complex relationships (50; 54).
Central core disease is probably the most common form of the congenital myopathies (02; 64). Recessive SEPN1-related minicore myopathy is the second most common core myopathy (55). The exact prevalence of the condition is unknown. In prevalence study of congenital myopathies in a representative pediatric United States population, overall point prevalence of congenital myopathies was one of 26,000, with mutations in RYR1 being the most common cause of congenital myopathies at one of 90,000 (03). SEPN1 mutations represent 6% of congenital muscular dystrophy patients in a published Italian cohort (43). Cases have been reported in a variety of ethnic and racial groups.
Genetic counseling and prenatal diagnosis are possible in families with a known mutation in the index patient. Information to the families can be given according to the inheritance pattern.
The differential diagnosis varies depending on the symptoms and the patient’s age group at the time of presentation. In children presenting at birth or within the first 2 years, the differential diagnosis is confined mainly to other congenital myopathies, anterior horn cell disease, congenital neuropathies, neuromuscular junction defects including congenital myasthenic syndromes, congenital muscular dystrophy, infantile myotonic dystrophy, and metabolic myopathies (07). For patients presenting at a later age, muscular or myotonic dystrophy, autoimmune myasthenic conditions, and more benign variants of motor neuron disease take a higher position on the list of differential diagnoses. Reader is referred to elegant articles on congenital myopathies to review clinical phenotypes and clues, including a detailed differential diagnosis (76; 54).
The traditional approach to diagnosing a congenital myopathy is combining detailed clinical and family history with physical examination findings to recognize clues to clarify a phenotype. The International Standard of Care Committee for Congenital Myopathies provides a diagnostic approach and highlights the fact that other than muscle biopsy, investigations are rarely specific for congenital myopathies but are widely used to exclude other possible diagnoses (76).
Evaluation of muscle biopsy is important; however, one should also keep in mind the overlap and morphological abnormalities seen in these conditions, marked variability in their clinical progression, and severity (86).
Serum creatine kinase is usually normal or mildly elevated.
Electromyography (EMG) and nerve conduction studies (NCS) are useful to exclude denervation disorders. EMG is typically normal or shows myopathic features, with short-duration, small-amplitude, polyphasic motor unit potentials.
Muscle imaging (ultrasonography and MRI) may be useful in diagnosis demonstrating a characteristic pattern of selective muscle involvement, which may be used in conjunction with clinical features to guide genetic testing (48; 76).
Relative sparing of the rectus femoris, with increased echogenicity (ie, atrophy) of the vastus intermedius, has been suggested to be characteristic of minicore myopathy (53). Muscle MRI in RYR1-related myopathies has shown involvement of the vasti with selective sparing of the rectus femoris in the thigh, and involvement of soleus and the gastrocnemii below the knee (48b). These findings are similar but more severe in minicore disease in association with ophthalmoplegia (48a), whereas minicore disease associated with SEPN1 mutations is associated with more significant involvement of the gastrocnemii than the soleus, and with selective involvement of the sartorius in the thigh (67).
Muscle biopsy and analysis of muscle histology, histochemistry, immunohistochemistry, and ultrastructure by light and electron microscopy (EM) have been the mainstay of reaching the diagnosis of a specific form of congenital myopathies (76).
There are now a few laboratories worldwide offering diagnostic mutational screening, and the paradigm is shifting from invasive procedures to the new era of next-generation strategies (85). The cost-effectiveness and increasing availability of these techniques in different centers give the opportunity to perform molecular analysis directly.
We believe that investigations at the moment should be complementary to understanding the underlying complex pathomechanisms.
Diagnostic work-up finally includes molecular genetic testing. In a patient with a phenotype consistent with core myopathy, the first RYR1 mutations, mini core myopathy's first SEPN1 mutations, and second RYR1 mutations should be screened (76). In case of associated cardiomyopathy, MYH7 and TTN analysis is recommended (76).
Whole-exome and whole-genome sequencing approaches, wherever available, may help diagnose cost-effectively (85).
Consensus statements on standard of care for congenital myopathies should be considered (111; 25).
The management of congenital myopathies has shown some progress in recent years. Potential therapeutic approaches for congenital myopathies can be divided into the following aspects: treatment of potential genetic defects, calcium/myosin regulation targeting muscle contraction, and therapies that target genes involved in a pathway (116).
Core elements of treatment include physical therapy, orthopedic interventions, management of respiratory complications, and feeding problems.
There are currently no approved treatments for the possible genetic defects in congenital myopathy. The management of the congenital myopathies is currently supportive and based on a multidisciplinary team approach involving a neuromuscular neurologist, respiratory physicians, orthopedic surgeons, physiotherapists, occupational therapists, and speech-language therapists (108) The primary focus is on managing associated symptoms, particularly respiratory dysfunction that can be latent and life-threatening, often ranging from noninvasive ventilation during nocturnal sleep to almost permanent mechanical ventilation via tracheostomy (116).
Involvement of the respiratory muscles may have an insidious onset and may remain subclinical until unmasked by intercurrent illness or anesthesia. Respiratory function should be monitored with serial pulmonary function tests, and where indicated, sleep studies.
Many patients have difficulty chewing and swallowing and require a gastrostomy for feeding. Tube feeding is necessary for infants and young children with poor sucking ability.
Patients with congenital myopathy and skeletal deformities such as clubfoot, pectus excavatum, scoliosis, and short ribs may require surgical correction or conservative treatment based on their condition.
Children should be monitored for the development of scoliosis and other skeletal deformities. Scoliosis may develop quickly and may be of a severity disproportionate to that of limb weakness. Congenital dislocation and dysplasia of the hip require orthopedic treatment. Gamble and colleagues reported a higher number of treatment failures and hip contractures after surgery in children with central core disease than in other myopathies (38).
Cardiac complications are uncommon in central core disease, but baseline electrocardiography and echocardiography are appropriate in most cases (98; 50).
Because malignant hyperthermia susceptibility is common in central core disease, malignant hyperthermia susceptibility should be considered in cases of RYR1, CACNA1S, and STAC3 mutations and in all cases of unknown genetic origin (108). All patients subjected to general anesthesia require preoperative anesthetic consultation and planning to ensure that a nontriggering anesthetic technique is used.
Treatment of malignant hyperthermia, which is a medical emergency, requires dantrolene and additional supportive care measures. Malignant hyperthermia susceptibility is suspected in an individual with congenital myopathy when (a) there is a positive family history of malignant hyperthermia susceptibility, (b) there have been previous difficulties with anesthesia, and (c) the patient has a documented RYR1 mutation (111). Of note, prophylactic dantrolene is not recommended prior to anesthesia, even in cases where malignant hyperthermia susceptibility has been established (111).
An ENMC workshop discusses current and future therapeutic approaches (50; 52). Possible therapeutic strategies include modifying RyR1 function, correcting associated oxidative abnormalities, using a pharmacological compound to enhance muscle contractility or neuromuscular transmission, and correcting a specific gene defect.
A range of more specific therapies are in development or have already reached the clinical trial stage, including antioxidants such as N-acetylcysteine (NAC). These treat the oxidative abnormalities documented in SELENON- and RYR1-associated myopathies, although their effects in clinical trials have been limited. There have also been reports of neuromuscular transmission defects associated with the response of some congenital myopathies to acetylcholinesterase inhibitors or other pharmacological agents used to treat myasthenic diseases (54; 108).
Individuals with multiminicore disease are usually able to care for themselves in all activities of daily life. Schooling is unaffected. A regime of physical therapy wherein muscle strength is maintained and range of motion preserved is generally appropriate. Minor contractures of the elbows, knees, and hips may develop after infancy and may respond to physical therapy or splinting. Surgical release is rarely necessary.
One study examined the effect of salbutamol, a beta-2 agonist, on effect on muscle strength in central core and minicore disease. Five children with minicore myopathy (average age 13.6 years) received 2 mg salbutamol orally four times a day for 6 months. One stopped the medication because of tremors and palpitations. The other four completed 6 months of therapy, at the end of which, they reported improved stamina and had a small but significant improvement in strength measured by myometry and MRC scores, and in their vital capacity, relative to baseline (68).
The two major complications of multiminicore disease are the development of progressive kyphoscoliosis and progressive deterioration of respiratory function. Monitoring of spinal growth is important, especially during growth spurts. Given the possibility of "malignant" kyphoscoliosis, it is advisable to obtain a surgical opinion as soon as kyphoscoliosis becomes evident, and early spinal operation should be considered in those with progressive respiratory involvement (82). The possible predisposition to malignant hyperthermia should be kept in mind in all patients with multiminicore disease (37).
Respiratory function should be monitored in all affected children, but especially those with significant scoliosis and spinal rigidity (90). The degree of respiratory insufficiency seen in minicore disease is often disproportionate both to the extent of peripheral weakness and to the severity of scoliosis, and in those with significantly diminished reserve, there is potential for life-threatening decompensation of respiratory function with intercurrent illness. Hypoventilation is usually first apparent at night. Prior to the onset of overt diurnal respiratory difficulties, hypoventilation may present with excessive daytime somnolence, early morning headaches, anorexia, and deteriorating school performance. Clues on physical examination include excessive use of accessory muscles and fixed deformities of the thoracic cage. Symptoms and signs of chronic hypoventilation may be difficult to ascertain or even lacking in affected children; however, all patients should undergo regular (annual or more frequent) formal assessment of respiratory function. This should include lung function tests specifically assessing the forced vital capacity, forced expired volume in 1 second, maximal inspiratory and expiratory pressures, and the cough peak flow (66). Significant falls in the vital capacity when measured supine, as opposed to sitting or standing, infer diaphragmatic dysfunction and identify patients with diminished respiratory reserve.
Polysomnographic evaluation should also be undertaken in order to evaluate for subclinical nocturnal hypoventilation. Patients with severe scoliosis often require ventilatory support, the first line of which is usually nocturnal assisted ventilation followed if necessary by daytime noninvasive positive pressure ventilation. Nocturnal ventilatory support may both prolong life and improve quality of life in children with chronic neuromuscular respiratory insufficiency (110).
Evaluation of cardiac function, in the form of a clinical assessment, ECG, and echocardiogram, should be undertaken at baseline and in patients with progressive skeletal weakness or respiratory insufficiency. Cardiac involvement in multiminicore disease is most commonly in the form of cor pulmonale as a sequela of progressive respiratory failure, but occasional cases have been associated with primary cardiomyopathy (65; 09; 90).
Currently, the MFM, CHOP INTENT, and Bayley are the recommended motor functions tests in physically evaluating an individual (113).
Genetic counseling should be provided to all affected families.
There are ongoing efforts to try oral dantrolene in RYR1-related myopathies. We should learn more in the future.
There are ongoing efforts to try oral dantrolene in RYR1-related myopathies. Even though there is no concise agreement, experts tend to use off label dantrolene in RYR1-related myopathies whether malignant hyperthermia risk is evident or not. We should learn more in the future.
A large proteomics study found a significant decrease in PGC1α, which could serve as potential therapy target and biomarker in CCD (105).
Several iPS lines have been generated from adult central core disease individuals with dominant variants in the RYR1 gene to create a baseline for future treatment efforts (15).
The congenital myopathies carry a slightly increased risk of breech and other abnormal presentations (63; 29). Specific complications of pregnancy in patients with central core disease have not been reported.
Precautions like those for other patients with muscle weakness apply to patients with minicore myopathy. Loss of respiratory reserve in advanced pregnancy may be of concern in those with significant pulmonary disease. Successful pregnancies have been reported in families with dominant inheritance. In these families, affected offspring have been symptomatic from birth (97). Successful delivery using epidural anesthesia has been reported in malignant hyperthermia-susceptible patients with multiminicore disease (91).
Malignant hyperthermia susceptibility is a common accompaniment of central core disease; all patients with central core disease who are to be subjected to a general anesthetic and whose susceptibility to malignant hyperthermia is unknown require preoperative anesthetic consultation and planning to ensure that a nontriggering anesthetic technique is used. A review discusses appropriate evaluation and anesthetic management in patients with core myopathies (13).
Of 297 unrelated malignant hyperthermia patients studied in the United Kingdom, 85 were shown to have RYR1 mutations; 10 patients also had central core disease (88). A similar study of 124 unrelated North American malignant hyperthermia patients identified RYR1 mutations in 29 cases (23%) (95).
Although not routinely performed, definitive diagnostic test for malignant hyperthermia involves an in vitro contracture test, which is based on increased sensitivity to stimuli usually inducing calcium release by the sarcoplasmic reticulum. The degree of muscle contraction after exposure to halothane and caffeine is a specific and sensitive marker of malignant hyperthermia susceptibility. The in vitro contracture test requires a fresh muscle biopsy and is not well standardized in children.
In a 25-year retrospective study from the Hospital for Sick Children, Canada; 13 patients were identified with malignant hyperthermia-like reactions (06). Nine patients were classified as having true malignant hyperthermia after review of anesthesia record, and genetic testing results were available for seven patients, five of whom had mutations in RYR1. Of the four patients who had severe reactions to anesthesia but did not meet the criteria for true malignant hyperthermia, two had Duchenne muscular dystrophy (06).
Limitations are those that apply to all myopathies wherein respiratory function may be reduced. At least a single death has occurred following an anesthetic (56). In common with central core disease, malignant hyperthermia has been reported in patients with multiminicore disease, so anesthetics should be given with caution (37; 80; 91). Formal testing for a predisposition to malignant hyperthermia should be considered prior to elective anesthesia.
Advanced kyphoscoliosis may pose a particular problem in anesthesia because of distortion of the thoracic cage.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Haluk Topaloglu MD
Dr. Topaloglu of Hacettepe Children's Hospital in Ankara, Turkey, has no relevant financial relationships to disclose.
See Profile
Bita Poorshiri MD
Dr. Poorshiri of Tabriz University of Medical Sciences has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neuroimmunology
Apr. 26, 2026