
Infectious Disorders
Arboviral encephalitis
May. 15, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author reviews Creutzfeldt-Jakob disease, a member of the group of diseases known as prion diseases or subacute spongiform encephalopathies. Creutzfeldt-Jakob disease has a subacute clinical course and distinctive gray matter pathology. This disease is transmissible and is induced by an abnormal form of the prion protein that is extremely resistant to physical and chemical inactivation. The unusual nature of the transmissible agent and the emergence of variant Creutzfeldt-Jakob disease (as a result of ingesting contaminated beef) have significantly impacted public health in addition to science and medicine. The most recent epidemiological information suggests there will not be an epidemic of variant Creutzfeldt-Jakob disease. Diagnostic tests, such as protein misfolding cyclic amplification and real-time quaking-induced conversion (RT-QuIC), and ideas about treating Creutzfeldt-Jakob disease are discussed.
|
• The transmissible subacute spongiform encephalopathies, also known as prion diseases, have a similar noninflammatory spongiform pathology and are caused by a similar transmissible agent -- an abnormal (“scrapie-like”) protease-resistant conformation of the prion protein (PrP), which is designated PrPSc | |
|
• Creutzfeldt-Jakob disease is a subacute fatal disease with a clinical triad of dementia, myoclonus, and EEG abnormalities that is usually associated with other neurologic signs, along with neuropathological evidence of neuronal loss, spongiform changes, and astrocytosis. | |
|
• A test that makes use of protein misfolding cyclic amplification (real-time quaking-induced conversion, RT-QuIC) has high sensitivity and specificity and is extremely valuable in making the diagnosis. | |
|
• Iatrogenic cases of Creutzfeldt-Jakob disease have been described as related to corneal transplantation, implantation of intracerebral electrodes (inadequately sterilized with formaldehyde), growth hormone injection (from pooled pituitary glands), and dura mater implantation (from pooled dura mater). | |
|
• A variant form of Creutzfeldt-Jakob disease caused by bovine spongiform encephalopathy (mad cow disease) is distinctive because of the relatively young age of the patients, frequent behavioral abnormalities early in the disease, few EEG abnormalities, an MRI finding of increased signal in the pulvinar, and abundant “florid” plaques in the brain (similar to those seen with kuru). | |
|
• Prion diseases are transmissible after a prolonged incubation period by inoculating the infected CNS into nonhuman primates and some other species via multiple routes of inoculation; however, transmission is most efficient with an intracerebral inoculation into a species identical to the source of the infected CNS tissue. |
Jakob deserves credit for being one of the first to draw attention to a clinical and pathologic syndrome that we now refer to as Creutzfeldt-Jakob disease. Although Jakob believed that a previously published case by Creutzfeldt had the same clinical and pathological condition, this patient is now thought to have probably suffered from another disease process. In 1959, Klatzo and colleagues remarked on the similarity between the neuropathology of Creutzfeldt-Jakob disease and kuru, a noninflammatory ataxic spongiform encephalopathy found among the Fore tribesmen of the Eastern Highlands of Papua, New Guinea (39). In 1966, kuru was transmitted to nonhuman primates following intracerebral inoculation of CNS tissue (25); this prompted the inoculation of nonhuman primates with Creutzfeldt-Jakob disease-infected brain and its successful transmission (31).
Creutzfeldt-Jakob disease, kuru, Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, sporadic fatal insomnia, and a number of animal diseases (including scrapie, bovine spongiform encephalopathy, and chronic wasting disease of deer and elk) are grouped as prion diseases, or transmissible subacute spongiform encephalopathies, because of their similar clinical and pathologic features and their transmissibility (25).
|
Human prion diseases |
Description |
Etiology |
|
Kuru |
Cerebellar disease of New Guinea, now eradicated |
Infection from endocannibalistic ritual |
|
Creutzfeldt-Jakob disease | ||
|
• Sporadic |
Dementia, myoclonus, and other neurologic signs. Positive CSF RT-QuIC test. MRI DWI can show cortical ribboning. EEG can show periodic sharp wave complexes. |
Unknown |
|
• Familial |
Autosomal dominant with clinical features similar to sporadic CJD |
Mutation in PrP gene, PRNP |
|
• Iatrogenic |
Clinical features similar to sporadic CJD, except frequent cerebellar signs |
Infection from corneal transplants, intracerebral electrodes, dura mater grafts, growth hormone administration |
|
• Variant |
Early age of onset with psychiatric/behavioral symptoms. Declining incidence. |
Infection from bovine spongiform encephalopathy tissue |
|
Gerstmann-Sträussler-Scheinker disease |
Autosomal dominant, usually with cerebellar dysfunction and slower tempo than CJD. |
Mutation in PrP gene, PRNP |
|
Fatal insomnia (either sporadic or familial) |
Insomnia, autonomic abnormalities |
Sporadic – unknown |
The transmission of these diseases is of special interest because of the noninflammatory nature of the clinicopathologic syndrome, the long incubation period (ie, a “slow” infection), and the unusual nature of the transmissible agent, the prion (67; 69). The importance of these diseases was demonstrated by the award of the Nobel Prize to Gajdusek and Prusiner in 1976 and 1997, respectively. The prion diseases became more visible because of an epidemic of bovine spongiform encephalopathy (“mad cow” disease), found primarily in the United Kingdom, with the subsequent emergence of a variant of Creutzfeldt-Jakob disease. Variant Creutzfeldt-Jakob disease is thought to result from oral transmission of the bovine spongiform encephalopathy agent to humans. The recognition of chronic wasting disease as a prion disease of deer and elk in the United States has also focused attention on prion diseases.
Creutzfeldt-Jakob disease generally occurs from 50 to 70 years of age (although cases in the teens and 80s have been described); a mean age at death of 67 years has been reported (08; 43). The most common syndrome has a subacute course with a "clinical triad" of dementia, myoclonus, and a characteristic EEG (periodic or pseudoperiodic paroxysms of sharp waves or spikes against a slow background) (76). This triad is not always seen in its entirety, and portions of the triad may emerge over time. However, the absence of all three features is distinctly unusual. Various other neurologic abnormalities are frequently seen, including behavioral abnormalities, sensory complaints, higher cortical dysfunction, and signs of cerebellar, pyramidal, extrapyramidal, and lower motor neuron dysfunction (29). At times, patients have sleep-wake disturbances. Patients usually decline virtually every week, leading to a mute akinetic state and eventually to death within 6 months to 1 year. Less commonly, patients may have a slow progression with a late onset of dementia, myoclonus, or EEG abnormalities. Abnormalities occur on MRI and in the spinal fluid, including the very specific and ultrasensitive RT-QuIC test. Published criteria for diagnosing sporadic Creutzfeldt-Jakob disease are detailed in the diagnostic workup section. A typical case is described below.
About 10% of cases are familial with an autosomal dominant inheritance pattern. Familial cases are classified based on clinical and pathologic features and the particular mutation in the sequence of the prion protein gene as either familial Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker syndrome, or fatal familial insomnia (49; 22; 47; 54; 74; 18; 81). Patients with Gerstmann-Sträussler-Scheinker syndrome generally manifest a slower course than those with Creutzfeldt-Jakob disease, with prominent cerebellar findings but less dementia and fewer EEG abnormalities; the pathology shows prominent amyloid plaques in the cerebellum. Patients with fatal familial insomnia manifest insomnia, autonomic abnormalities, and fewer EEG abnormalities than do those with Creutzfeldt-Jakob disease. A noninherited disease with a similar clinical and pathological phenotype to fatal familial insomnia has also been described and referred to as sporadic fatal insomnia (50). A report of a large kindred with Gerstmann-Sträussler-Scheinker syndrome highlights the phenotypic heterogeneity in this disease (89). Patients with familial cases that manifest a Creutzfeldt-Jakob disease clinicopathologic picture without features of Gerstmann-Sträussler-Scheinker syndrome or fatal familial insomnia are classified as having familial Creutzfeldt-Jakob disease.
Some cases of Creutzfeldt-Jakob disease are iatrogenic with known sources of contamination occurring from corneal transplants, intracerebral recording electrode implants, growth hormone injections, and dura mater implants (58; 09; 01; 59). Data strongly suggest that cases of a variant form of Creutzfeldt-Jakob disease result from transmission of bovine spongiform encephalopathy to humans. Over 225 cases of variant Creutzfeldt-Jakob disease have been identified (93). Fortunately, the cases of variant Creutzfeldt-Jakob have significantly declined; few cases of variant Creutzfeldt-Jakob disease are reported in the United Kingdom:www.cjd.ed.ac.uk. Variant Creutzfeldt-Jakob patients had a number of distinctive features compared to classical Creutzfeldt-Jakob disease. The average age of onset was less than 30 years, and disease duration was longer. Anxiety and depression were common early symptoms; other early signs were ataxia and sensory disturbances. Characteristic EEG abnormalities were absent. A number of the cases had an abnormal MRI with increased signal in the posterior thalamus on T2-weighted images (the “pulvinar sign”). Postmortem examination of the brain revealed a much higher than usual occurrence of widespread amyloid plaques. These plaques, known as "florid" plaques, are prion protein-immunoreactive with an eosinophilic core and a pale periphery surrounded by spongiform change; the RT-QuIC test for Creutzfeldt-Jakob disease was not available at this time.
Creutzfeldt-Jakob disease patients usually become mute and bedridden with little movement except myoclonus. Death usually ensues from respiratory complications within 6 to 12 months of disease onset. Survival is rarely prolonged over a year or 2, except in atypical and some familial prion disease cases.
This 75-year-old man was in excellent health until 3 months before admission when, shortly after returning from a trip to Asia, he noticed difficulty in walking and in balance. The patient’s difficulties were initially subtle, confined to his lower extremities, and worsened when attempting to turn. Over the ensuing weeks, however, his balance became increasingly worse, and the patient began to notice incoordination of both legs, such that he needed to use a cane and then a walker. This was followed by difficulties with coordination in his upper extremities, complaints of muscle twitching in both arms, and writing difficulty. Initial evaluation by the patient’s physician 2 months after onset of symptoms revealed only incoordination: MRI studies of both brain and spine were unremarkable, and EMG was normal, as were all laboratory studies, including vitamin B12 level and serology for dengue virus, West Nile virus, and enteroviruses. The patient was suspected of having a paraneoplastic disorder and was admitted to the hospital for lumbar puncture and further evaluation.
On admission, the patient was alert, oriented, and afebrile. General physical examination was unremarkable. Neurologic examination revealed normal cranial nerves, normal strength and tone, and normal sensation. Deep tendon reflexes were brisk but without Babinski sign or other pathological reflexes. Cerebellar examination, however, showed slowing of rapid alternating movements and a bilateral perpendicular tremor most prominent in the lower extremities but also present in the arms. Balance was severely impaired: the patient walked on a widened base with significant unsteadiness and inability to perform tandem gait. Routine blood studies were normal, as were angiotensin-converting enzymes in both serum and CSF. Repeat viral serologies were unremarkable. CSF analysis showed one white blood cell/mm3, protein of 43 mg/dL, glucose of 56 mg/dL, absent oligoclonal bands, and negative CSF VDRL. CSF cultures for bacteria, Mycobacterium tuberculosis, and fungi were negative. Repeat MRI studies of brain and thoracic spine were unremarkable. Studies of serum and CSF for paraneoplastic and anti-GAD65 antibodies were negative. The patient’s coordination and balance did not appear to change, but he was noted to have become forgetful late in his hospital course. On outpatient follow-up 3 weeks after discharge the patient was found to have become completely unable to stand without assistance and to have become significantly cognitively impaired. MRI at that time showed increased cortical signal on diffusion-weighted magnetic resonance imaging, suggesting Creutzfeldt-Jakob disease. CSF was positive for both 14-3-3 and tau proteins, consistent with the diagnosis.
Over the ensuing weeks, the patient became increasingly more ataxic, declined rapidly in terms of memory and cognition, and eventually became anarthric. By 4 months after admission, he had become bedridden and rigid, with prominent startle myoclonus. The patient died 5 months after the onset of his initial symptoms. Autopsy revealed extensive spongiform change and astrogliosis, confirming the diagnosis of Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease, like other spongiform encephalopathies, is transmissible via multiple routes of inoculation, although the intracerebral route is the most efficient. The transmissible agent is an isoform of the prion protein that is proteinase-resistant, which is thought to result from a change in conformation from the normal cellular form of prion protein (67; 69). The abnormal prion protein isoform (PrPSc) is generally considered to be all or part of a proteinaceous infectious agent, the prion, which apparently lacks nucleic acid. Interactions of the abnormal prion protein with the normal cellular form of prion protein (PrPC) are critical for disease pathogenesis (78). The abnormal prion protein aggregates to produce amyloid.
The amino acid sequence of PrP varies in the normal population, with either a methionine or valine at codon position 129 of PrP. This polymorphism affects the infectivity and phenotype of prion disease. Interestingly, variant Creutzfeldt-Jakob disease seems to be generally confined to patients that are homozygous for methionine at codon 129. The function of PrPC is still somewhat unclear, but it has a role in myelination (40).
Different PrPSc “strains” can be identified or typed on the basis of Western blots; affected tissues show varying glycosylation patterns and electrophoretic mobility of PrP after proteinase K digestion, presumably because of their different conformation. The Creutzfeldt-Jakob disease agent can be classified into six subtypes (MM1 or 2, VV 1 or 2, and MV or 2) based on the PrP gene haplotype (methionine [M], or valine [V], homozygosity or heterozygosity [MV] at codon 129) and the electrophoretic mobility and degree of glycosylation of proteinase K-digested fragments (61; 72). Clinical phenotypes of prion diseases are thought to correlate with these molecular subtypes. For example, most patients with MM1 or MV1 subtype have a rapid disease with progressive dementia along with myoclonus and visual disturbance, whereas patients with MV2 or VV2 have an atypical disease with a longer duration and late dementia (62). Variant Creutzfeldt-Jakob disease manifests a distinctive electrophoretic subtype also seen in bovine spongiform encephalopathy.
In some forms of human prion disease resembling Creutzfeldt-Jakob disease, PrP accumulates and is not sensitive to protease treatment (26). These cases of variably protease-resistant prionopathy can have very atypical disease courses that lack the usual EEG and MRI findings.
The pathogenesis of Creutzfeldt-Jakob disease is not completely understood. It is known that this disease is transmissible to nonhuman primates and other animals following filtration of the inoculum, indicating that the agent is small and "replicating" (31). The intracerebral route of inoculation is more efficient than the peripheral route, with a mean incubation period of 24 months in squirrel monkeys. The transmissible agent is unusually resistant to chemical and physical agents (eg, heat inactivation, UV irradiation, X-irradiation), which led to suggestions that it does not contain nucleic acid.
A distinctive protein that corresponds to an abnormal scrapie-like isoform of prion protein, PrPSc (66), is present in Creutzfeldt-Jakob disease CNS tissue. As noted above, this protein differs from its normal cellular prion protein counterpart, PrPC, because it is protease-resistant, presumably owing to a change in conformation of the protein (although it has an identical amino acid sequence). It is believed that PrPSc deposits in the CNS of Creutzfeldt-Jakob patients, causing dysfunction. Importantly, PrPSc acts as a template, converting PrPC to PrPSc. In the case of experimentally transmitted Creutzfeldt-Jakob disease, the inoculated PrPSc is believed to enter the CNS and thereby start the conversion of PrPC to more PrPSc and the induction of disease. In the case of familial Creutzfeldt-Jakob disease (as well as Gerstmann-Sträussler-Scheinker syndrome and fatal familial insomnia), a mutated form of the prion protein gene appears to lead to prion protein deposition. In a landmark experiment, the presence of a mutated prion protein gene (from a patient with Gerstmann-Sträussler-Scheinker syndrome) as a transgene in mice was found to induce a spongiform neuropathology (36). This latter result suggested that the mutant PrPSc is sufficient to produce disease (ie, a mutant form of prion protein replicates and induces disease).
The importance of interactions between PrPSc and PrPC in disease was further supported by prion protein knock-out mice (10). These knock-out mice do not develop prion disease following inoculation of (mouse-adapted) PrPSc; reintroduction of PrPC as a transgene into the knock-out mouse leads to a return in susceptibility of these mice. Transgenic mice with human PrPC expressed have an enhanced susceptibility to human prion disease, such as Creutzfeldt-Jakob disease, further supporting the importance of interactions of PrPC and PrPSc for disease development. Several observations demonstrate the potential of a broad spectrum of abnormalities that can be caused by PrP. One study found that overexpression of PrPC in muscle leads to a primary myopathy in transgenic mice (37). In addition, cardiac disease can be induced by overexpression of PrPSc in the heart (84).
The pathogenesis of sporadic Creutzfeldt-Jakob disease remains unclear. It has been hypothesized that a spontaneous somatic change in prion protein conformation in the CNS initiates the disease. Of note, transgenic mice that overproduce wild-type rodent PrPC, or part of PrPC, develop neurodegeneration (90; 80; 23). This suggests that prion disease can be induced by excessive amounts of PrPC or particular peptides of PrPC, presumably because the chance of misfolding of the overexpressed PrPC into an abnormal PrPSc conformation is more likely than if there is a normal amount of PrPC expressed. Some of the presumed sporadic cases of Creutzfeldt-Jakob disease may actually represent an unrecognized transmission from infectious material, either as result of iatrogenic exposure or an environmental focus.
Interestingly, a number of reports have been published that demonstrate the presence of prion-like elements in yeast (92). Experiments show that these elements lead to aggregation and amyloid formation of a protein. The transmission of these abnormal properties is related to the presence of the altered protein rather than nucleic acid (42). These studies suggest that prions (ie, abnormally folded proteins), as a cause of abnormal phenotypes, are more widespread than realized. Misfolded proteins have also been hypothesized to underlie a number of neurodegenerative diseases (68; 87; 12). Relevant to this topic is a report of transmission of multiple system atrophy to transgenic mice that express a mutant alpha-synuclein transgene (70), as well as presumed transmission of amyloid-beta pathology and cerebral amyloid angiopathy to recipients of pituitary-derived growth hormone who died of Creutzfeldt-Jakob disease (38). Although these diseases may not be transmissible in the same way that the subacute spongiform encephalopathies are, the pathogenic protein specific for these diseases is misfolded and thought to spread throughout the CNS by means of a prion-like mechanism, in which the misfolded protein leads to misfolding of the wild type protein. In fact, a CSF RT-QuIC assay has been used to detect misfolded proteins associated with non-prion neurodegenerative diseases (11).
Creutzfeldt-Jakob disease occurs worldwide, with an annual incidence of about 1.2 cases per million population (43). About 5% to 10% of Creutzfeldt-Jakob disease cases are familial and are caused by particular mutations of the prion protein gene (53). The number of inherited prion disease mutation carriers in the United Kingdom has been estimated (17). Clusters of Creutzfeldt-Jakob disease cases have been reported, at times in a particular geographic region. In some of these cases, the geographic focus has been found to actually represent a cluster of familial cases, as in a focus among Libyan Jews (35). In other cases, the cluster can represent horizontal transmission from a common source (eg, contaminated beef, as is the case of transmission of variant Creutzfeldt-Jakob disease).
Iatrogenic Creutzfeldt-Jakob disease has been reported as a result of a number of different procedures involving transmission of contaminated material from prions of an affected individual: intracerebral electrodes that were incompletely sterilized and implanted, cornea or dura mater that was transplanted, and pools of human growth hormone that were inoculated (09; 01; 59). For unclear reasons, cases related to contaminated preparations of growth hormone, in contrast to sporadic cases, have a far more frequent occurrence of cerebellar and basal ganglia signs with relatively little mental deterioration, myoclonus, or typical EEG abnormalities (08; 09). In the UK, patients with iatrogenic Creutzfeldt-Jakob disease due to pituitary-derived growth hormone had incubation periods of up to 40 years (77). Interestingly, patients were initially predominantly valine homozygous at codon 129, but later, patients had a mixed picture over time, with methionine homozygous and methionine-valine heterozygous. These data may suggest that the infecting prion contaminating the pituitary pool was from an individual with valine-valine homozygosity and an individual with methionine-valine heterozygosity. Disease among members of the medical profession has been reported only rarely.
The occurrence of an outbreak of bovine spongiform encephalopathy ("mad-cow disease") in Britain with the subsequent emergence of variant Creutzfeldt-Jakob disease raised public health concerns. Compelling evidence indicates that variant Creutzfeldt-Jakob disease resulted from oral ingestion of beef contaminated with the bovine spongiform encephalopathy agent (93). Fortunately, there has been a dramatic decrease in the number of variant Creutzfeldt-Jakob cases, with few cases reported in the United Kingdom, presumably due to the decline in cases of bovine spongiform encephalopathy.
Of note, a 2013 report from the United Kingdom found that 16 of 32,441 (approximately one in 2000) appendices that were surgically removed from 2000 to 2012 stained positive for PrP accumulation, indicating that prion infection may be more frequent than predicted from the very low incidence of variant Creutzfeldt-Jakob at that time (32). Although a patient with variant Creutzfeldt-Jakob disease who is homozygous for methionine at codon 129 of the prion protein gene is extremely rare (compared to the United Kingdom population, which includes a significant number of individuals who are valine homozygotes and heterozygotes at codon 129), the codon 129 genotype of the individuals with PrP positive-stained appendices was: eight with methionine homozygosity, four with methionine-valine, and four homozygous for valine. The results from this study of appendices suggest that in the case of bovine spongiform encephalopathy, the barrier for infection of human lymphoreticular infection is lower than for clinical disease, as also suggested from animal experiments (05). The data also suggest that although clinical disease seems to selectively target patients with a homozygous methionine genotype, this is not the case with lymphoreticular infection. On the other hand, individuals with valine and methionine-valine genotypes might eventually develop clinical disease. If these individuals do develop Creutzfeldt-Jakob disease, it is unclear whether the clinical disease will have a different phenotype from variant Creutzfeldt-Jakob disease in individuals who are homozygous for methionine.
A subsequent study of appendices collected before 1980 (before the epidemic of bovine spongiform encephalopathy) and after 1996 (a year when measures to remove bovine spongiform encephalopathy from the food chain were fully in place) was carried out to clarify the effect of prion dietary exposure (71). Surprisingly, two of 14,692 (approximately one in 7000) appendices collected before 1980 were positive, and five of 14,824 (approximately one in 3000) appendices were positive from patients born after 1996. Despite the difference in age and dietary exposure among the three groups, there was no statistical difference in the frequency of positive appendices collected before 1980, from 2000 to 2012, or from individuals born after 1996. These results are difficult to interpret with certainty. One suggestion is that human exposure to bovine spongiform encephalopathy occurred before 1980 and after 1996. Another interpretation is that there is a background of variant Creutzfeldt-Jakob disease infection that does not progress to disease, ie, there is a disconnection between a positive appendix result and variant Creutzfeldt-Jakob disease (71). In other words, prion infectivity can be separated from toxicity (04).
One publication reviewed evidence of prion infectivity in 29,516 appendices removed from individuals between 1962 and 1979 from persons born between 1891 and 1965 and from individuals born after 1996 who had been operated on from 2000 through 2014 (33). Seven appendices were positive for abnormal PrP, of which two were from the pre-bovine spongiform encephalopathy-exposure era and five from the post-bovine spongiform encephalopathy-exposure period. None of the seven positive samples were from appendices removed before 1977 or in patients born after 2000, and none came from individuals diagnosed with variant Creutzfeldt-Jakob disease. There was no statistical difference in the prevalence of abnormal PrP across birth and exposure cohorts. Two interpretations are possible: either there is a low background prevalence of abnormal PrP in human lymphoid tissues that may not progress to variant Creutzfeldt-Jakob disease; alternatively, all positive specimens are attributable to bovine spongiform encephalopathy exposure, a finding that would necessitate human exposure having begun in the late 1970s and continuing through the late 1990s.
The occurrence of chronic wasting disease, a prion disease of deer and farmed elk in the world, including the United States and Canada, provides another environmental risk (03; 83). Although there is no evidence that this disease can be transmitted to humans, experimental transmission into squirrel monkeys has been carried out following intracerebral inoculation (48). Chronic wasting disease has spread significantly and is documented in many states in the United States as well as internationally (65).
Of concern is the finding that chronic wasting disease is efficiently spread and very contagious among cervids in the field. Varied cervids have polymorphisms of PRNP depending on the species (63). Furthermore, the disease can be spread in several animal species. Studies demonstrated that this prion disease can be spread by blood and saliva from infected deer before they develop clinical signs of illness. In addition, transmission occurs from exposure to feed buckets, water, and bedding (presumably as a result of fecal, urine, and saliva contamination) from the housing areas of infected deer—without any direct animal-to-animal contact (51); of note, even earthworms are capable of taking up and then excreting infectious prions (65). The amount of PrPsc excreted in the feces into the environment can be very substantial and can even be comparable to the amount in the brain at terminal disease (64). Of concern is the finding that prions can persist and retain infectivity in the environment for at least 16 years (28). These observations underline the important role of the environment in prion transmission.
The pathogenesis of sporadic Creutzfeldt-Jakob disease remains unclear. It has been hypothesized that a spontaneous somatic change in the conformation of prion protein in the CNS leads to its aggregation and the subsequent induction of disease. It remains a possibility, however, that some cases of presumed sporadic Creutzfeldt-Jakob disease are unrecognized iatrogenic cases as a result of surgical transmission from prion-contaminated instruments, as suggested by some epidemiological studies (44). Little evidence exists of horizontal spread from person to person, with only rare reports of conjugal Creutzfeldt-Jakob disease. In addition, Creutzfeldt-Jakob disease among members of the medical profession has been reported only rarely. A number of epidemiologic studies have failed to find a relationship between the incidence of scrapie and the presence of Creutzfeldt-Jakob disease (08); however, there is less knowledge concerning the potential of human transmission of atypical scrapie strains or chronic wasting disease.
Studies have shown that in some prion diseases, a variety of body fluids are infectious, including spinal fluid, blood, saliva, urine, and feces (06). Studies have also reported infectious material in the blood of patients with variant and sporadic Creutzfeldt-Jakob patients (21). An ongoing United States surveillance study found no evidence of transmission of Creutzfeldt-Jakob disease in recipients of blood transfusions from donors who developed Creutzfeldt-Jakob disease (19); however, the latter result has to be considered tentative because one would anticipate an extremely prolonged incubation period if transmission from this source occurred, considering the low infectivity of blood and the peripheral route of entry. Secondary transmission of variant Creutzfeldt-Jakob disease, which is more lymphotrophic than classical Creutzfeldt-Jakob disease, is thought to have occurred through transfusion of blood from patients with variant Creutzfeldt-Jakob disease (86). A variety of tissues of prion disease patients are infectious. Although the skin of Creutzfeldt-Jakob patients was demonstrated to have PrPSc and to be infectious, there is no evidence that skin transmission of PrPSc leads to disease in humans (60). Another study demonstrated PrPSc in the cornea, ocular fluid, retina, choroid, sclera, and optic nerve of some patients with sporadic Creutzfeldt-Jakob disease; in some cases, PrPSc was demonstrated by immunohistochemical staining of retinal tissue (59). There is likely variable infectivity of peripheral tissues from both variant and sporadic Creutzfeld-Jakob patients, depending on when the tissue is obtained during the disease course and how long the assay is monitored (20).
The identification of particular mutations of the prion protein gene among cases of familial Creutzfeldt-Jakob disease signifies that in-utero identification of this disease and a subsequent elective therapeutic abortion are possible. Prevention of disease among individuals who are known to carry the mutation but are not yet affected is unfortunately not possible.
The transmissible agent of Creutzfeldt-Jakob disease is highly resistant to chemical and physical agents, prompting the publication of guidelines concerning inactivation of the agent's infectivity and safety precautions at the time of autopsy, which can be found on the Centers for Disease Control and Prevention website. Patients should generally be kept on "stool and needle" precautions in the hospital, and body fluids should be labeled as potentially dangerous. Invasive procedures should be carried out using precautions similar to those used with AIDS patients.
Attempts to decrease the incidence of iatrogenic disease have been implemented. Intracerebral electrodes are no longer sterilized by paraformaldehyde vapor, and guidelines regarding the transplantation of tissues and transfusion of blood are in continuing discussion. Genetically engineered material, rather than purified human extracts, is used in growth hormone treatments. Generally, there is an effort to avoid transplantation of tissues or delivery of biological products that were collected from a pool of donors. If the pool of donors is unavoidable as a source, it is important to try to prevent the inclusion of prions and to screen for prions in the final product.
Changes in the way that cattle are fed and slaughtered have been implemented to eradicate bovine spongiform encephalopathy and subsequent transmission as variant Creutzfeldt-Jakob disease to humans. Of note, secondary transmission of variant Creutzfeldt-Jakob disease may occur through transfusion of blood from a patient with variant Creutzfeldt-Jakob disease; white cells are also known to carry the transmissible agent.
Another direction for preventing prion disease involves targeting the amino acid sequence variation at codon position 129 of PrP, which normally has either a methionine or valine residue. This amino acid affects the susceptibility and phenotype of prion diseases, including both scrapie and variant Creutzfeldt-Jakob disease. Also, studies of Kuru have demonstrated that the amino acid coded for by this codon affects the length of the incubation period (53). These findings may have an impact on preventing prion disease in animals and humans.
Knocking down PrP appears to be a promising route for protecting and treating Creutzfeld-Jakob disease. This plan has led to the identification of antisense oligonucleotides that knockdown PrP as well as other ways of knockdown (13). Unfortunately, cases of Creutzfeldt-Jakob disease have very severe impairment by the time they are diagnosed. Therefore, PrP knockdown may need to be utilized for patients who carry PrPSc but are not yet affected with disease. The methods of PrP knockdown are discussed in the management section of this article.
The usual manifestations of Creutzfeldt-Jakob disease are fairly distinctive, and other diseases rarely produce a similar picture; however, Creutzfeldt-Jakob disease can, at times, have atypical clinical features. In addition, early in the disease course, Creutzfeldt-Jakob disease may be confused with other disorders. The availability of the CSF test, RT-QuIC, and the identification of MRI abnormalities in Creutzfeldt-Jakob disease are important tools with excellent sensitivity and specificity to clarify the diagnosis.
Many diseases in the differential diagnosis of Creutzfeldt-Jakob disease fall into a group of rapidly progressive dementias (30). Paraneoplastic syndromes (including, for example, limbic encephalitis or cerebellar degeneration) and autoimmune encephalitis should be included in the differential diagnosis. Infectious or granulomatous processes (eg, neurosyphilis, CNS fungal disease, sarcoid, HIV-1-related diseases, Lyme disease), tumors, and vasculitis (possibly confined to the CNS) may present a clinical picture similar to that seen with Creutzfeldt-Jakob disease; however, imaging studies and blood and CSF investigations usually identify neoplastic or inflammatory processes. More confusing may be disease processes related to toxins, especially inorganic mercury, a primary central nervous system vasculitis with little or no laboratory evidence of systemic inflammation, Hashimoto encephalopathy, and "mixed" neurodegenerative processes (eg, Lewy body dementia, frontotemporal dementia, Parkinson disease with dementia, amyotrophic lateral sclerosis with dementia); however, imaging and CSF studies should help clarify the diagnosis (85). A study from the U.S. National Prion Disease Pathology Surveillance Center noted that 352 (32%) of 1106 brain autopsies from cases that were suspicious for prion disease had another diagnosis, most frequently Alzheimer disease (154 cases) or vascular dementia (36 cases) (14). Of note, these cases were diagnosed before RT-QuIC. Other diagnoses of “incurable” neurologic disease included: unspecified degenerative brain disease (10), frontotemporal lobar dementia (9), mesial temporal lobe sclerosis (5), diffuse Lewy body disease (4), tauopathy (4), hereditary diffuse leukoencephalopathy with spheroids (3), progressive supranuclear palsy (3), corticobasal ganglionic degeneration (1), adult polyglucosan body disease (1), Huntington disease (1), Marchiafava-Bignami disease (1), and superficial siderosis (1). Of special concern was the finding that 71 individuals (23%) had a potentially treatable neurologic disease that included immune-mediated disorders (26) (primary CNS angiitis, acute disseminated encephalomyelitis, limbic encephalitis, neurosarcoidosis, paraneoplastic cerebellar degeneration, and Wegener granulomatosis), neoplasms, infections (fungal, viral, and parasitic), and toxic encephalopathies (6). As noted above, one disease that can masquerade as Creutzfeldt-Jakob disease is Alzheimer disease, especially familial Alzheimer disease (76). The pathologic features of these two diseases may sometimes also be confused; in these cases, the use of antisera directed against beta peptide or prion protein may be critical for differentiating Alzheimer disease amyloid plaques from those of Creutzfeldt-Jakob disease (or Gerstmann-Sträussler-Scheinker syndrome).
Some subacute spongiform encephalopathies result from an environmental source. For example, kuru is caused by endocannabalism involving ingestion of prion-infected tissue by individuals in the New Guinea Highlands. Variant Creutzfeldt-Jakob disease is caused by ingestion of bovine spongiform encephalopathy-contaminated beef. In some cases, there is infection as a result of iatrogenic exposure, such as via corneal transplants.
Patients or animals with familial prion diseases have a mutation in PRNP. At times, the mutation is specific for a particular prion disease. Non-PRNP host genetics likely influence the age of onset and duration of the prion disease; however, these genes are still being investigated (13).
The most important diagnostic assay in diagnosing Creutzfeldt-Jakob disease is a misfolding cyclic amplification test called the RT-QuIC (97). MRI may also be helpful, as can be the case with the EEG (27).
As one awaits the results of these tests and in order to rule out other causes of rapidly progressive disease, the following studies should be performed: complete blood count, comprehensive metabolic profile, sedimentation rate, collagen vascular screen, HIV-1 antibody, paraneoplastic antibodies, an autoimmune encephalitis panel (including NMDA receptor antibody), a toxic screen, antithyroid antibody and consideration for CSF analysis with culture of the fluid, and tests for paraneoplastic antibodies and autoimmune encephalitis. The CSF is noninflammatory, such that detection of a pleocytosis strongly mitigates against the diagnosis. A brain MRI study with gadolinium infusion should be performed to look for hyperintensities in the striatum, thalamus, or cortex and to rule out the possibility of a mass lesion or inflammatory focus. If vasculitis is strongly suspected, an arteriogram should be considered. An EEG is important to identify the typical pattern seen in Creutzfeldt-Jakob disease and to rule out seizures.
The emergence of RT-QuIC has provided a much-needed test for Creutzfeldt-Jakob disease. The results of RT-QuIC on CSF from 126 patients who were suspected to have Creutzfeldt-Jakob disease (111 sporadic disease and 15 genetic) and had prion disease confirmed at autopsy and also from 67 patients who had other diseases at autopsy had a specificity of 98.5% and sensitivity of 92.1% (24). A subsequently published review notes that the sensitivity of RT-QuIC for CSF in Creutzfeldt-Jakob disease CSF is 96%, and the specificity is 100% (96). Modifications of the amplification test have been used to detect variant Creutzfeldt-Jakob prions in blood (16) and urine (55).
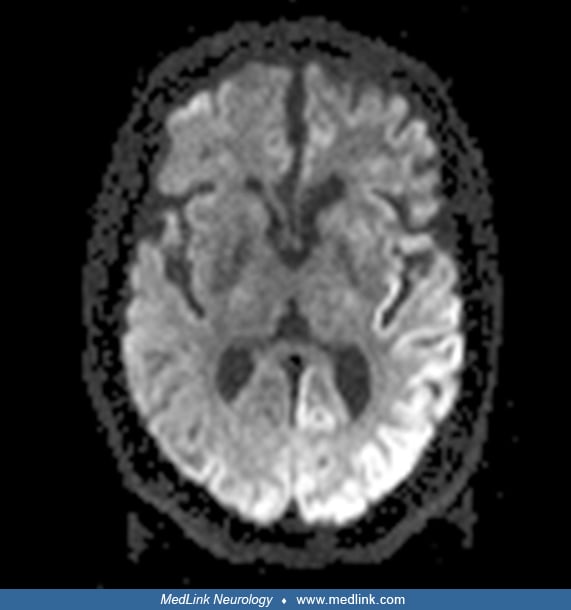
MRI is also frequently helpful in making the diagnosis of Creutzfeldt-Jakob disease. A consensus review of 48 cases of Creutzfeldt-Jakob disease by two neuroradiologists blinded to the diagnosis found that MRI had a sensitivity of 96% and 93%, respectively, and that the pattern of FLAIR/DWI can effectively differentiate Creutzfeldt-Jakob disease from other rapidly progressive dementias (88). There were gray matter hyperintensities in DWI sequences of the MRI in all the 48 cases, with certain regions preferentially involved, as found in this study and others, especially the cortex (“cortical ribboning”), basal ganglia, and thalamus and to a lesser degree the occipital lobe.
The hyperintensity is usually greater on DWI than FLAIR, and the ADC in these regions is hypointense, indicating restricted diffusion. MRI criteria for the diagnosis of sporadic Creutzfeldt-Jakob disease are available (82). MRI abnormalities have also been found preclinically in at-risk patients who carry a mutation that is highly penetrant for familial Creutzfeldt-Jakob disease with diffusion significantly reduced in thalamic-striatal structures, including the putamen and mediodorsal, ventrolateral, and pulvinar thalamic nuclei (41). Patients with variant Creutzfeldt-Jakob disease have a distinctive finding with increased signal in the posterior thalamus on T2-weighted images (the “pulvinar sign”). The EEG characteristically shows positive sharp wave complexes. In a large, well-controlled study, 58% of sporadic Creutzfeldt-Jakob disease patients (1261 of 2083 cases) showed this typical EEG, although the chances of finding it were more likely with a patient over 50 years of age and a disease duration under 6 months (15); the typical EEG was more likely seen with patients with a particular subtype of PrP: the MM1 subtype and less likely with the MV1, MV2, and VV2 subtypes. If the characteristic EEG findings are not initially seen, serial EEGs should be performed because these abnormalities frequently appear as the clinical syndrome evolves.
The following details the past European criteria for diagnosing sporadic Creutzfeldt-Jakob disease (82). The availability of RT-QuIC will play a major role in future diagnostic criteria. The prior criteria are listed below:
|
Rapidly progressive cognitive impairment with two of the following clinical features: | |
|
(a) myoclonus | |
|
In combination with either: | |
|
(a) periodic EEG discharges or | |
Other criteria for the diagnosis of sporadic Creutzfeldt-Jakob are also available (82), as well as criteria for the diagnosis of iatrogenic and genetic transmissible spongiform encephalopathy and variant Creutzfeldt-Jakob disease and can be accessed at the following site:www.cjd.ed.ac.uk.
As a result of difficulties in obtaining a reliable family history, RT-QuIC is important for all individuals suspected of having Creutzfeldt-Jakob disease. Patients with a positive test should have prion protein gene sequencing on white blood cell DNA to look for evidence of a pathogenic mutation.
The performance of a brain biopsy is usually unnecessary with the availability of RT-QuIC. In addition, there are difficulties in carrying out the biopsy because of biohazard concerns. The CNS tissue usually shows the typical spongiform changes as well as evidence of protease-resistant PrPSc. Affected tissue generally transmits disease in experimental animals.
The fear of an epidemic of variant Creutzfeldt-Jakob disease, the poor prognosis of classical Creutzfeldt-Jakob disease, and the presence of individuals who carry a PrP pathogenic mutation but are still asymptomatic have challenged the scientific community to identify potential treatments. A number of drugs have been tested in mice carrying PrPSc; however, there remains no confirmed effective treatment for any prion disease (95). Of interest is a publication showing that treatment of a scrapie-infected mouse with a drug that inhibits a key kinase in the unfolded protein response (triggered by the aggregation of PrPSc) abrogates the development of clinical disease and decreases the neuropathological abnormalities (56). A number of subsequent studies that used different strategies to target this pathway again found an amelioration of disease (34; 79).
A promising path for treating prion diseases is related to knocking down or knocking out PrP. As noted above, PrP knock-out mice do not develop prion disease following inoculation of (mouse-adapted) PrPSc, and reintroduction of PrPC as a transgene into the knock-out mouse leads to a return in susceptibility of these mice to prion disease (10). Furthermore, turning off the expression of PrPC in prion-infected mice rescues them from disease and leads to recovery from cognitive and behavioral symptoms (45; 46). The latter results suggest that the disease process is not inexorably fatal after it starts. Other experiments have shown that knockdown of PrPC following lentiviral delivery of RNAi into the hippocampus of scrapie-infected mice led to a prolongation in survival, with a decrease in behavioral defects and neuropathology (91). A promising avenue for treatment is intracerebroventricular injection of antisense oligonucleotides directed against the prion protein, which led to a significant extension in the survival of scrapie-infected mice (73). There have also been attempts to knockdown PrP by means of immunotherapy, primarily with antibodies directed to all or part of PrP (75; 52). These antibodies have been shown to increase lifespan and, in some cases, prolong the incubation period of scrapie mice. One could also deliver shRNA that knocks down PrP via virus vector delivery. There may be problems, however, in knocking down PrP expression because this knockdown or knockout may cause toxicities.
Unfortunately, as of this date, there is still no treatment recommended to slow the course of Creutzfeldt-Jakob disease, and there is no way to prevent disease from developing in a clinically asymptomatic individual who carries a mutant PrP gene associated with familial Creutzfeldt-Jakob disease. Furthermore, it is clear that there is significant damage to the brain when most Creutzfeldt-Jakob disease patients are diagnosed, so treatments may sadly have little benefit in symptomatic individuals. Despite this outlook, there are investigations using screens to identify interventions that might limit prion propagation (02).
No information is available regarding the effect of pregnancy on the course of Creutzfeldt-Jakob disease. To date, there is no evidence that children born to pregnant women with Creutzfeldt-Jakob disease develop the disease (94). An investigation of a child born from a pregnant woman with Creutzfeldt-Jakob disease found no evidence of proteinase K-resistant protein in gestational tissues, including the placenta and amniotic fluid (94); however, transmission studies or amplification-based detection assays were not performed. Of concern is the detection of chronic wasting disease prions in fetal tissues of white-tailed deer using an amplification method, demonstrating vertical transmission of prions (07; 57).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Raymond P Roos MD
Dr. Roos of University of Chicago Medical Center has no relevant financial relationships to disclose.
See Profile
John E Greenlee MD
Dr. Greenlee of the University of Utah School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Infectious Disorders
May. 15, 2026
General Neurology
May. 13, 2026
Infectious Disorders
May. 12, 2026
Infectious Disorders
May. 05, 2026
Infectious Disorders
May. 01, 2026
Infectious Disorders
Apr. 30, 2026
Headache & Pain
Apr. 10, 2026
Infectious Disorders
Apr. 08, 2026