
Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myofibrillar myopathies refer to a heterogeneous group of genetic disorders associated with disintegration of myofibrils and accumulation of Z-disc related proteins, with variable age of onset and disease progression. Patients present with lower limb muscle weakness, which slowly spreads to involve the upper limbs, truncal, neck-flexor, facial, bulbar, and respiratory muscles. Distribution of the weakness may be distal or both proximal and distal. Skeletal myopathy may be combined with or preceded by cardiomyopathy, conduction blocks, and arrhythmias that can result in sudden death. Respiratory muscle weakness can also be a major complication in some patients. In this updated article, the author highlights advances in the diagnosis and management of myofibrillar myopathies.
|
• Myofibrillar myopathies are a subgroup of hereditary protein aggregate myopathies. | |
|
• Muscle biopsy reveals characteristic amorphous or granular material, with focal areas of myofibrillar disintegration, reduced oxidative enzyme activity, and vacuolar changes. | |
|
• The clinical phenotype is highly variable; most commonly it presents as a distal myopathy involving the hands and feet, with progressive muscle weakness, respiratory dysfunction, and cardiomyopathy. | |
|
• Treatment remains primarily supportive; those with significant cardiac complications may require pacemaker implantation or cardiac transplant. |
Myofibrillar myopathies refer to a heterogeneous group of rare inherited primary chronic noninflammatory myopathies characterized by abnormal accumulation of cytoplasmic inclusion bodies and myofibrillar disarray in skeletal or cardiac muscles (47; 06; 41). The disease was originally described more than 30 years ago based on common muscle histopathological features (46). The onset usually occurs in adult life (between the second and fourth decade) but may be congenital or present in early childhood. A combination of distal and proximal weakness, cardiomyopathy, peripheral neuropathy, and autosomal dominant inheritance should suggest this disorder (87). However, sporadic, autosomal recessive, or X-linked inheritance have also been described (40; 119).
The first description of cytoplasmic body as a structural anomaly of the Z-disc was in 1969 (81). Cytoplasmic bodies occur in a number of unrelated neuromuscular disorders, but their relationship to desmin-related neuromuscular diseases was only recognized in the 1980s (35; 136; 95).
Sporadic cases of adult-onset myopathy with spheroid inclusions, cytoplasmic bodies, and myofibrillar aggregates were initially reported in the 1970s (88; 69; 65). An autosomal dominantly inherited form of proximal myopathy was also described with spheroid bodies (49), granulofilamentous aggregates (37; 20), and myofibrillar inclusions (20). Intermediate filaments were identified in association with sarcoplasmic bodies in families with a severe progressive late-onset autosomal dominant distal myopathy (35), an adult-onset limb-girdle myopathy (34), and a distal myopathy (60). Subsequent cases had cardiomyopathy with large proteinaceous inclusions identified in the cardiac muscle consisting of intermediate filaments that were immunofluorescent desmin-positive (104; 136). Desmin body myofibrillar myopathy was also associated with a congenital form of myopathy with or without cardiomyopathy (50; 152). The cardiomyopathy could precede the myopathy (11; 51).
In a typical presentation, the disease is characterized by bilateral weakness in the distal leg muscles spreading proximally, or in proximal muscles spreading distally and eventually leading to wheelchair dependence. There can also be slowly progressive proximal muscle weakness, significant distal and proximal weakness, predominant distal weakness, or generalized muscle weakness with dysphonia and dysphagia (37; 49; 35). The cranial nerves are usually spared. Subgroups are characterized according to clinical presentation (04).
Cardiac involvement is frequent. The cardiomyopathy may be hypertrophic (37; 104), congestive (152; 51), or restrictive (114); it may precede the myopathy by a number of years (140; 64). There are also reports of patients with cardiomyopathy and clinically silent or only mild skeletal muscle disease (104; 13; 132). The cardiomyopathy is associated with frequent cardiac arrhythmias and either ventricular conduction blocks or atrioventricular conduction blocks requiring a pacemaker, often with progression to congestive cardiac failure and premature demise (104; 58; 60; 64). Neither the severity of skeletal muscle weakness nor the type of DES mutations was found to correlate significantly with the extent of cardiac involvement (146).
Respiratory muscle involvement is common. In adult-onset cases, respiratory muscle weakness and acute respiratory incapacity may be the initial presenting features (65; 34; 60; 12). Respiratory failure also occurs in congenital cases (07).
A peripheral neuropathy can be present. Both a sensorimotor polyneuropathy and a motor or sensory predominant neuropathy have been described (114; 87; 63; 21; 24). The Achilles reflexes may initially be absent, followed by loss of knee reflexes and preservation of arm reflexes until late in the disease (60).
Rarely, homozygous variants in desmin gene (recessive desminopathies) may present as a congenital myasthenic syndrome with ocular, bulbar, and limb-girdle muscle weakness, confirmed by positive electrodecremental response to repetitive nerve stimulation and partial responsiveness to treatment with pyridostigmine and salbutamol (103).
Skeletal abnormalities, such as thoracispinal kyphoscoliosis (152; 114; 86) and lumbar lordosis, may occur (58; 43). Cranial acromegaly with large frontal sinus, high-arched palate (152; 43), micrognathia, and cleft palate has been described (69). Bilateral pes cavus and camptocormia can occur (114; 58; 110). Extramuscular manifestations including mental retardation and late onset cerebellar ataxia have been reported as well (86; 106).
Most cases of myofibrillar myopathies present after the first decade or later in life. Early onset disease with homozygous alpha-B-crystallin (CRYAB) mutations were described in Canadian aboriginal infants of Cree ancestry with hypertonia, muscle rigidity, and early respiratory insufficiency shortly after birth (73; 25). Extensive fibrosis and absence of alpha-B-crystallin immunoreactivity were important diagnostic clues for this entity (25; 84; 79). Other congenital forms of myofibrillar myopathies may be benign (43), severe (07), or associated with progressive muscle weakness (38; 43; 71).
Myofibrillar myopathy may be slowly progressive, remain relatively static, or become severe, with some patients becoming wheelchair-bound within 10 years (35). The prognosis also depends on the presence or absence of an accompanying cardiomyopathy with or without cardiorespiratory failure. Age of disease onset in patients with autosomal dominant disease is between 14 and 48 years of age, whereas patients with recessive mutations may present during childhood or adolescence. Disease progression may be faster in patients with autosomal recessive DES mutations and cardiomyopathy (52).
A 13-year-old girl was referred for initial assessment. Her past medical history was unremarkable apart from a recent history of intermittent dizziness and shortness of breath during intense physical activity. She denied any chest pain, palpitations, syncope, seizures, weakness, or numbness. Her exam was normal apart from mild distal muscle weakness and atrophy in her hands and feet, plus a mild stocking and glove distribution of reduced pinprick, temperature, vibration, and light touch up to her wrists and ankles in a symmetrical fashion.
Her family history was significant for cardiomyopathy in her father; he required a cardiac transplantation at 20 years of age. He noticed a gradual decline in strength during his early 30s, initially with distal lower extremities weakness, followed by gradual progression to involve his proximal lower and then upper limbs. His recent quadriceps biopsy revealed a myofibrillary myopathy with increased variation in fiber size, occasional atrophic myofibers, and multiple hyaline inclusions that reacted positively to desmin immunostaining.
Initial laboratory tests including serum creatine kinase, electrolytes, glucose, complete blood count, serum B12, and thyroid function were normal. Her electrocardiogram showed a first-degree atrioventricular block and a right bundle branch block. An exercise stress test showed a normal heart rate and blood pressure response, but the test was terminated early due to subjective sensation of chest tightness. A 24-hour Holter monitoring showed no other significant rhythm disturbance. Her echocardiogram, cardiac MRI, nerve conduction study, and pulmonary function tests were within normal limits. Molecular genetic testing confirmed a pathologic DES gene mutation in both the father and daughter. She had implantation of a MRI-compatible VVIR (demand ventricular pacing with physiologic response to exercise) pacemaker for persistent trifascicular conduction block. She remained otherwise asymptomatic, with stable cardiac, pulmonary, and neurologic assessments a year later.
Myofibrillar myopathies are considered a subgroup of protein aggregate myopathies (47; 06; 17). Myofibrillar myopathies (MFM) are characterized by a broad spectrum of pathologic changes found in muscle, including focal disintegration of myofibrils, mitochondrial dysfunction, and accumulation of multiple sarcoplasmic proteins including desmin (23; 87; 122; 123; 123). Desmin was initially identified as a key molecule associated with a diverse group of disorders. Molecular studies have demonstrated mutations in desmin and alpha-B-crystallin genes in patients with a clinical phenotype of desminopathy. Other proteins besides desmin were subsequently found to accumulate in the abnormal myofibrils.
Approximately half of the myofibrillar myopathies are caused by mutations in genes encoding sarcomeric and extra-sarcomeric proteins, including desmin (53; 85; 22; 89), alpha-B-crystallin (142; 125), myotilin (126), LIM-domain-binding protein 3 (LDB3, also known as ZASP) (127), filamin C (145; 113; 139; 75), Bcl2-associated athanogene 3 (Bag3) (130; 63), and DNAJ homolog subfamily B member 6 (DNAJB6) (150; 116). In addition, mutations involving four and a half LIM domain 1 (FHL1) and titin (TTN) were found in a subgroup of patients with myofibrillar myopathies, reducing body myopathy, or hereditary myopathy with early respiratory failure (120; 93; 40; 102). Defects in the genes encoding human plectin (PLEC), alpha-actin (ACTA1), small heat shock protein 22 (HSPB8) (90; 109), lamin A/C (LMNA) (27), kyphoscoliosis peptidase (KY) (33), pyridine nucleotide-disulfide oxidoreductase domain-containing protein 1 (PYROXDI) (92), sequestosome 1 (SQSTMI) in combination with a modifying variant in cytotoxic granule-associated RNA-bind protein (TIA1) (91; 97), and tripartite motif-containing protein 32 (TRIM32) (96) can also give rise to myofibrillar aggregations with vacuolar changes. Many of the above genes are associated with other types of myopathies.
Myopathic manifestations caused by desmin (DES) or alpha-B-crystallin (CRYAB) mutations are identical but are termed “desminopathy” when desmin is involved and “alpha-B-crystallinopathy” when alpha-B-crystallin is involved (142; 22; 125; 122; 123; 80). Similarly, mutations involving myotilin (MYOT), Z-band alternately spliced PDZ motif-containing protein (ZASP), filamin C (FLNC), or bcl-2-associated athanogene 3 (Bag3) result in myotilinopathy, zaspopathy, filaminopathy, or Bag3opathy, respectively (126; 127; 145; 130; 63; 113; 139; 72).
Muscle biopsy changes in myofibrillar myopathy are those of focal dissolution of myofibrils followed by accumulation of the products of myofibrillar degradation. Each process is associated with an anomaly of the Z-disc, indicating that this plays a role in the pathogenesis of the disease (87). A review suggested that myofibrillar myopathy should be classified as a “Z-disk-opathy” caused by mutations in genes directly involved in encoding Z-disk structural proteins or their interacting chaperones (62).
The main muscle intermediate filament of desmin, formally called "skeletin," is a 53-kd protein and a class 3 muscle-specific intermediate filament measuring 8 to 10 nm in diameter and occurring in skeletal, cardiac, and smooth muscle. Desmin is encoded by a single copy gene (DES), and the human desmin gene is localized to chromosome 2q35 (108; 144). It encompasses nine exons and codes for 476 amino acids. In mature skeletal muscle, desmin filaments encircle and interlink the myofibrils at the level of the Z-disc and connect to the plasma membrane and nuclear lamina, thus, aligning the myofibrils. Desmin is enhanced at the myotendinous and neuromuscular junctions (46). Desmin occurs in high concentration in developing striated myofibers, especially fetal myotubes, and then diminishes in mature muscle where it functions in maintaining the alignment of Z-bands of adjacent myofibrils (117; 118).
Earlier research suggests that desmin plays a role in the functional and spatial relationship between the nucleus and plasma membrane, linked to the nuclear intermediate filament laminin B (46). In the heart, desmin is increased at the intercalated discs and in the Purkinje fibers (107). Like other intermediate filament proteins, desmin is composed of three major structural domains (59). N-terminal end is a nonconserved non-alpha-helical head domain; middle is a conserved alpha-helical coiled-coil rod domain that contains three linker regions; and C-terminal end is a nonconserved tail domain (45). Partial knockdown of the desmin genes resulted in smaller organisms with diminished active tension and force generation as well as increased vulnerability to acute stretch injury (77; 66).
DES mutations cause loss of function of both desmin and its binding partners, as well as toxic accumulation of aggregates containing misfolded desmin and debris from other ectopically expressed proteins within the myofiber (14; 68; 01). Mutant desmin also interacts with other cytoskeletal proteins. The pathologic process starts with disintegration of Z-disc, which is the functionally important site for tension transmission between the sarcomeres. The amount of desmin in affected muscle fibers of desminopathy patients increases over time (03). In cultured satellite cells taken from a patient with p.Leu345Pro mutation in the DES gene, desmin created a fully normal network in the early passages; however, in further passages an increasing number of cells showed abnormal accumulation of desmin-positive material in the form of perinuclear, spot-like, or subsarcolemmal pattern (15). Animal models and experiments in desmin knockout mice suggest that desmin is essential for the structural integrity, adhesion, and migration of muscle cells (99; 55). Mutated desmin led to impaired myocyte structure and function (151; 28; 44). Mitochondrial abnormalities also contribute to the development of cardiomyopathy in desmin-related myopathies (61).
Alpha-B-crystallin is a ubiquitous small heat shock protein that has multiple cellular functions, including chaperone-like antiaggregation properties. It normally stabilizes proteins including desmin and prevents their irreversible aggregation (30); one study suggests that alpha-B-crystallin may function as a sensor for proper assembly of intermediate filaments (131). It is found in the lens, skeletal, and cardiac muscle, and to a lesser extent in the skin, brain, and kidney. The human alpha-B-crystallin gene (CRYAB) was first mapped to the 26-cM interval in chromosome 11q21-23, a region containing the CRYAB gene, in a large French pedigree (143; 142). Hyperphosphorylation of alpha-B-crystallin is postulated to be responsible for the deleterious change in various cellular processes (02). Expression of mutant alpha-B crystallin leads to formation of abnormal aggregates that contain both desmin and alpha-B-crystallin (25). Aggregation of amyloid-like material has been described as a typical feature of many human cardiomyopathies, especially those caused by CRYAB mutations (115).
Some studies suggest an important role of autophagy in protection against alpha-B-crystallin or desmin-related myopathies (148). In both mice and in vitro studies, p62 mRNA and protein expression were significantly upregulated in cardiomyocytes by transgenic overexpression of mutant desmin or alpha-B-crystallin (153). The p62 upregulation in mouse proteinopathic hearts promoted aggresome formation and autophagy activation (153).
Accumulation of misfolded proteins is a common pathological pattern in all myofibrillar myopathies (112; 150). The effect of different disease-associated mutations at the molecular level was evaluated by confocal single-particle fluorescence spectroscopy (76). De novo aggregation properties of desmin in vitro and divergent assembly patterns for three different desmin missense mutations were detected. R350P-desmin showed a strong inhibition of assembly formation with a reduced level of tetramers and an increase in dimers in native cell extracts. It interacted with wild-type protein resulting in a dominant negative effect on desmin assembly. E413K-desmin formed hyperstable tetramers whereas R454W-desmin had subtle effects at the dimer and tetramer levels. The study of cultured human myoblasts suggests that increased stiffness as expressed by desmin mutation may contribute to the progressive muscle damage due to excessive mechanical stress (08). Some studies suggest that desmin plays a key role in the rheological properties of the cytoplasm in myoblasts (36; 18). In a study, pretreatment with N-acetyl-L-cysteine (NAC) decreased stress-induced protein aggregation in desmin mutant cell lines; however, NAC did not reduce protein aggregation in AAV-injected mouse models of desminopathies (26).
The location of DES mutations can exert a significant influence on the clinical phenotype (52; 141). Pathologic DES variants located in the 1B segment of the alpha-helical rod domain and the tail domain were more likely to present with early-onset severe cardiomyopathy compared to patients with variants in other segments (135). Myopathy, cardiomyopathy, smooth muscle myopathy, neuropathy, respiratory dysfunction, facial paralysis, or cataracts may be present in some desminopathy patients and may be absent in others. There was a tendency for the age of onset to be 4 to 10 years earlier, and disease progression to be faster, in patients with cardiomyopathy or autosomal recessive disease (52; 94). Recessive desmin mutations with complete loss of function were found to be associated with infantile-onset generalized weakness, cardiomyopathy, as well as a congenital myasthenic syndrome in two children attributed to impaired neuromuscular endplates formation, in addition to the myopathy (32).
The incidence and prevalence of desmin body myofibrillar myopathy in the general population is unknown. Among 63 patients in Mayo Clinic’s cohort of myofibrillar myopathies, only 6% showed mutations in DES, whereas 3% had sequence alterations in CRYAB (128). The genetic basis for the majority cases of myofibrillar myopathies remained unclear (123). Pathogenic variants in DES, MYOT, or DNAJB6 were the most common disease-causing genes among 132 patients with genetically confirmed myofibrillar myopathy followed in the Italian myology neuromuscular centers (09), whereas DES followed by FLNC, BAG3, and TTN accounted for majority of 39 patients with myofibrillar myopathy followed in a single center in China (147).
No means of prevention are known.
Neuromuscular diseases with early distal involvement suspected of having a neurogenic etiology are mainly in the differential diagnosis (101). Distal myopathies, myotonic dystrophy, limb girdle or distal muscular dystrophy, and scapuloperoneal syndrome should also be considered (29; 78). Unlike many inheritable distal myopathies, the pattern of weakness in desmin body myofibrillar myopathies is not specific. The arm flexors are more involved than the arm extensors; this contrasts with Welander disease and myotonic dystrophy. There is mild involvement of the neck and facial muscles, so it can be differentiated from the dominantly inherited facioscapulohumeral dystrophy (35). In sporadic cases, the differential diagnosis is wider and includes inclusion-body myositis, vacuolar myopathies, and congenital myopathies (83). The pathological changes of myofibrillar disarray and ectopic expression of multiple proteins can be seen in congenital myopathies related to selenoprotein (SEPN1) and skeletal muscle alpha-actin (ACTA1) mutations (138; 124), biallelic variants in the myosin chaperone UNC45B (31; 133), homozygous loss-of-function mutations in supervillin (SV2) (57), as well as hereditary myopathy due to recessive variants in DNAJB4 (149). Synemin (SYNM) is another intermediate filament that copolymerizes with desmin and vimentin; it is a potential candidate for cardiac and skeletal myopathies of unknown etiology (98).
As shown in the case illustration, the recommended investigations in patients with suspected myofibrillar myopathies include the following (52; 56):
|
(1) Electrophysiological investigations, including nerve conduction studies and EMG examination, to exclude neurogenic causes of weakness, such as motor neuron disease and peripheral neuropathy. | |
|
(2) ECG, to identify arrhythmias and cardiac conduction defects. | |
|
(3) Holter monitoring, especially if symptoms suggest an intermittent arrhythmia. | |
|
(4) Echocardiography to detect and diagnose the type of cardiomyopathy, even in patients with no cardiac symptoms. | |
|
(5) Cardiac MRI and MR delayed enhancement imaging if available, to detect subtle myocardial changes or early myocardial fibrosis. | |
|
(6) Respiratory function tests, even in patients with no respiratory symptoms. | |
|
(7) Muscle imaging studies and/or muscle biopsy, to differentiate from other myopathies. | |
|
(8) Genetic testing (including whole exome sequencing), which is essential in establishing an accurate diagnosis and to provide reliable genetic counseling. |
Cardiac MRI is superior to conventional echocardiography to identify patients at risk of cardiovascular complications (137; 19). Skeletal muscle MRI can also help confirm the patterns of muscle involvement in myofibrillar myopathies (90), with early involvement of the semitendinosus, sartorius, and gracilis in DES-related myopathies (67).
Electromyogram usually shows a myopathic pattern in the majority of the patients with myofibrillar myopathies; a minority of patients showed mixed neurogenic and myopathic features with myotonic discharges (49; 65; 87). Nerve conduction studies may be normal or show reduced conduction velocities and decreased motor and sensory amplitudes (114; 87). The serum creatine kinase level is usually normal or mildly elevated (less than 7-fold above the upper normal limit).
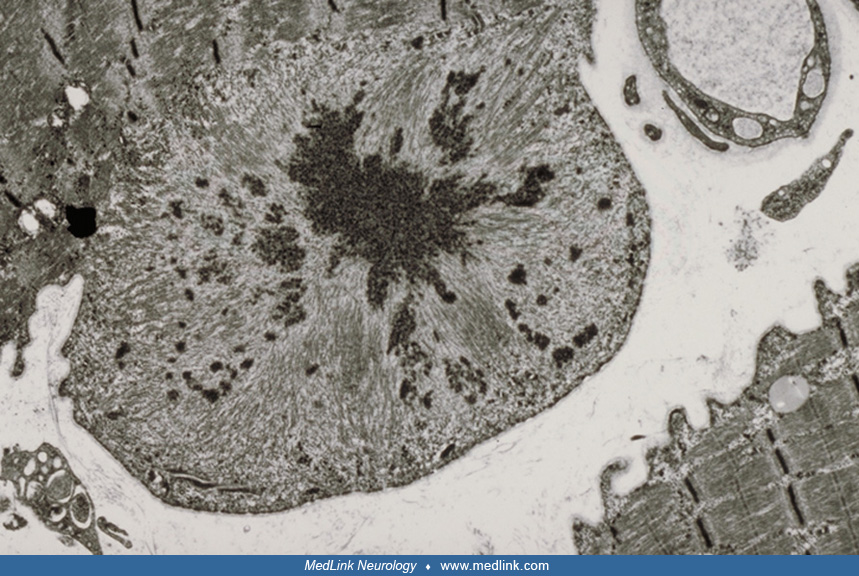
A muscle biopsy is required to make the diagnosis. The histology usually shows a myopathy. The main features are eosinophilic inclusions of amorphous, granular, or hyaline material in a subsarcolemmal distribution or throughout the sarcoplasm, which on the modified Gomori trichrome stain appear a blue-red-purple color. The NADH stain shows reduced oxidative enzyme activity corresponding to the abnormal hyaline structures. These structures have been termed "inclusion bodies" (88), "spheroid bodies" (49), "Mallory bodies" (42), and "vermiform inclusions" (11). They occur in normal, atrophic, and hypertrophic fibers, mainly in type 1 fibers but also in type 2 fibers (12). There is a variation in fiber size, with atrophy ranging from 6 to 25 µm and hypertrophy from 80 to 150 µm. Inflammatory infiltrates are usually absent. However, focal necrotic fibers with macrophages (11; 58; 60; 87) and regenerating fibers have been reported (87). Desmin is diffusely expressed in regenerating fibers (117). Other features include type 1 fiber predominance, fiber splitting, fibrosis, prominent internal nuclei, and lakes of para-aminosalicylic acid-positive material (87). There are also several vacuoles showing acid phosphatase positivity (11; 87).
Both immunofluorescence and immunoperoxidase staining have demonstrated the association of the granulofilamentous inclusions with desmin accumulation. A large number of proteins, which fall into different categories, have been found overexpressed (23). These proteins include cytoskeletal proteins such as dystrophin (51; 58; 16; 23) and vimentin (58), myofibril-associated proteins such as alpha-actin and actin (11; 34; 121), and proteins seen in relation to amyloid formation such as gelsolin, beta-A4 protein, and the beta-A4 amyloid precursor protein (23). Other proteins include ubiquitin (11; 140), alpha-B-crystallin (51), and antichymotrypsin, a marker of histiocytes (23). Accumulation of RNA polymerase II-associated proteins (RPAPS) was found in the cytoplasm of muscle fibers in familial and sporadic cases of myofibrillar myopathies; RPAP2 immunostaining may be a useful tool to identify abnormal protein aggregation (54). As well, proteomic analysis of the ratio and order of different proteins may help in the differential diagnosis of protein aggregate myopathies (82).
The ultrastructural patterns of the inclusion bodies are heterogeneous. The commonest inclusion is granulofilamentous, segregated into filamentous and electron-dense, finely granular components in various arrangements. The increased use of laser microdissection has permitted quantitative proteomic analysis and novel understanding of the pathogenesis of desminopathy (39; 70). Foci of myofibrillar disruption contain small deposits of irregularly shaped Z-disc material and accumulations of degraded myofibrillar components (87). The filaments described differ and include short fine filaments 6 to 8 nm in diameter representing fragmented actin. The filaments in spheroid bodies range in size from 12 to 15 nm (49). Although intermediate filaments 7 to 10 nm in diameter occur in association with the granular masses (35; 104; 95; 121), they are not usually present in desmin body myofibrillar myopathy. Membrane-bound vacuoles contain degenerating membranous organelles and myeloid structures, and the surrounding myofiber may show abnormal sarcotubular systems, scattered glycogen granules, and mitochondria.
The other inclusions seen are the classic cytoplasmic body (88; 65; 95; 121) and filamentous bodies without electron-dense material (104). Immunogold labeling has demonstrated different localizations of desmin positivity involving the intermediate filaments, the filamentous component (100), or the granulo-amorphous material (12). Desmin, alpha-B-crystallin, and dystrophin occur in the granulofilamentous inclusions (51). The filamentous component is also actin positive (12).
In summary, muscle histology reveals characteristic amorphous or granular material in a variable proportion of the muscle fibers with Gomori trichrome staining; sharply circumscribed decreases of oxidative enzyme activity in many abnormal fiber regions; small vacuoles in a variable number of fibers and abnormal ectopic expression of desmin, alpha-B-crystallin, dystrophin, and other proteins in immunocytochemical studies (52).
Next generation sequencing is essential to identify the disease-causing mutations (105).
The management is mainly related to the cardiorespiratory abnormalities that may be detected. Implantation of a pacemaker or an implantable cardioverter defibrillator (ICD) can be lifesaving in patients with cardiac arrhythmias and conduction defects. Patients with progressive or life-threatening cardiomyopathy are candidates for cardiac transplantation (132). Support with noninvasive positive pressure ventilation and prevention of recurrent lung infections are essential for those with respiratory involvement. Emerging treatments including gene and stem cell therapies are active areas of research (10; 05; 111; 134).
Preoperative cardiac and pulmonary assessments should be performed due to potential for cardiorespiratory complications. Volatile anesthetics should be used cautiously in patients with myofibrillar myopathies. The risk of malignant hyperthermia or anesthesia-induced rhabdomyolysis is presently unknown (74).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Jean K Mah MD
Dr. Mah of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neuroimmunology
Apr. 26, 2026