Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Familial Alzheimer disease is a type of Alzheimer disease that is inherited in an autosomal dominant pattern (15). This means that if someone receives a mutated gene from one parent, they will develop the disease. Familial Alzheimer disease accounts for only slightly more than 5% of all cases of Alzheimer disease (33). Although the symptoms of familial Alzheimer disease are similar to those of Alzheimer disease, they usually tend to appear in life in a person’s 30s, 40s, or 50s (136). In this article, the author provides an overview of the genetics and clinical characteristics of familial Alzheimer disease and explores variations in how it presents among different ethnic groups. The author also discusses the role of variations in causing Alzheimer disease and touches on the factors in and available genetic testing options for diagnosing Alzheimer disease. Finally, the author discusses the genetic aspects of familial Alzheimer disease and addresses new evidence of clinical heterogeneity in the disease and new loci of genetic risk factors for Alzheimer disease: TOMM40, the clusterin (CLU) or APOJ gene, and the PICALM gene.
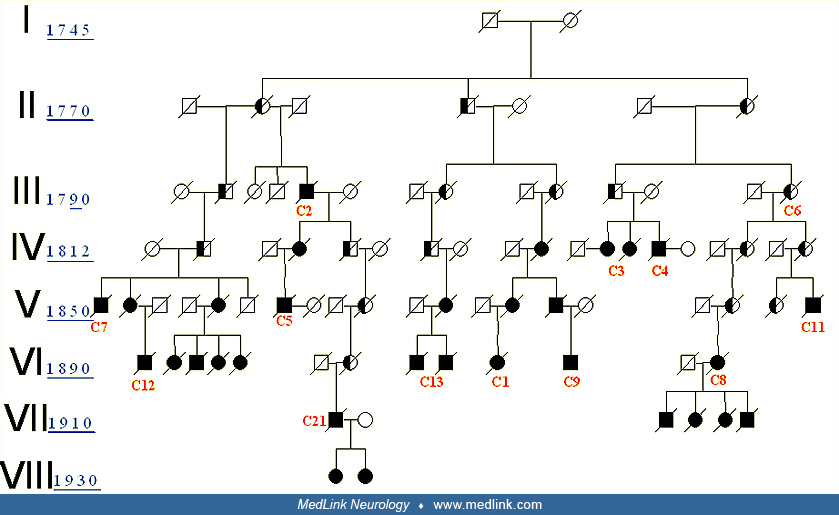
The term “familial Alzheimer disease” means that two or more persons in the same family are affected by Alzheimer disease. The first case of familial Alzheimer disease with neuropathological studies was reported in 1932 (132). Since then, more than 500 families have been described with a multigenerational autosomal dominant pattern of Alzheimer disease transmission. In 1979 Cook and colleagues reported three new families, bringing the total to 50 described in the medical literature, and suggested a link between familial Alzheimer disease and transmissible dementia. Among those families, there was a patient with histologically confirmed Alzheimer disease whose sister had proved spongiform encephalopathy (27). Ten years later, Bird and colleagues suggested genetic heterogeneity by describing the variation of clinical and neuropathological characteristics in 180 demented individuals from 24 kindreds with familial Alzheimer disease, with at least two affected generations and neuropathological confirmation in one case (18).
Masters and colleagues have purified and characterized the cerebral amyloid protein that forms the plaque core in Alzheimer disease and in aged individuals with Down syndrome. This protein consists of multimeric aggregates of a polypeptide of about 40 residues (4 kDa), but there are no similarities between its amino acid sequence and those of scrapie polypeptides (92). Two years later, amyloid fibrils were purified from the leptomeningeal vessels of a patient who had no clinical signs of dementia, whose family had hereditary cerebral hemorrhage, with amyloidosis restricted to the leptomeninges and cerebral cortex. This protein showed homology to the beta-protein of Alzheimer disease and Down syndrome, which suggested that hereditary cerebral hemorrhage with amyloidosis of Dutch origin was pathogenetically related to Alzheimer disease (151).
As in sporadic forms of Alzheimer disease, there are two kinds of familial Alzheimer disease, namely: The early and late-onset varieties. Men and women in early-onset families had equivalent risk of dementia. The lifetime risk of dementia in early-onset familial Alzheimer disease kindreds is consistent with an autosomal dominant inheritance model. Late-onset familial Alzheimer disease has at least two etiologies: Alzheimer disease in some families may be transmitted as a dominant trait, whereas in others, it may be caused by other genetic or shared environmental factors (41).
In familial Alzheimer disease, the inheritance pattern follows Mendelian genetics, specifically autosomal dominant inheritance (11; 93; 146). This means that if a parent has a mutation in one of the genes associated with familial Alzheimer disease, each child has a 50% chance of inheriting the same mutation (146). There are three genes that have been associated with early-onset familial Alzheimer disease: presenilin 1 (PS1) on chromosome 14, presenilin 2 (PS2) on chromosome 1, and amyloid precursor protein (APP) on chromosome 21. Mutations in any of these three genes can lead to the development of familial Alzheimer disease (146).
It is important to note that familial Alzheimer disease accounts for less than 5% of all cases of Alzheimer disease (141). In contrast, the majority of Alzheimer cases are sporadic and not directly caused by genetic mutations (04). However, having a family history of Alzheimer disease does increase the risk of developing the disease (14). Individuals who have a parent or sibling with Alzheimer disease are more likely to develop the disease compared to those without a first-degree relative with Alzheimer disease (63). The risk is even higher for individuals who have more than one first-degree relative with Alzheimer disease (63).
Genetic testing is available to detect mutations in the genes associated with familial Alzheimer disease (141). However, the Alzheimer's Association cautions against routine genetic testing for Alzheimer disease risk without proper counseling and an understanding of the implications (63). Genetic testing can help identify individuals who are at a higher risk of developing familial Alzheimer disease, but it is not a definitive predictor of whether or not someone will develop the disease (39).
Familial Alzheimer disease follows Mendelian inheritance with an autosomal dominant pattern. Mutations in genes, such as PS1, PS2, and APP, can lead to the development of familial Alzheimer disease. Although having a family history of Alzheimer disease increases the risk of developing the disease, it is important to approach genetic testing with caution and to seek proper counseling.
The initial symptoms of familial Alzheimer disease are usually similar to those of typical Alzheimer disease, primarily memory loss (117). However, the clinical presentation of familial Alzheimer disease can vary among different families (117). The majority of individuals with familial Alzheimer disease have similar clinical presentations to those with sporadic Alzheimer disease, comprising an early impairment of episodic memory, which is the ability to remember recent events (130). Other symptoms of familial Alzheimer disease can include difficulty with language, changes in mood and behavior, and difficulty with motor coordination. Some studies have noted that the pathologic hallmarks (protein deposits called plaques and tangles) are more severe in familial Alzheimer disease than in late-onset Alzheimer disease and may be concentrated in different regions (02). These differences in underlying mechanisms suggest that treatments for familial Alzheimer disease may overlap with, but not always be identical to, treatments being developed for late-onset Alzheimer disease (03).
Although progressive memory loss is the most frequent early finding, language disturbances, slow cognitive processing, attention problems, tremor, parkinsonism, extrapyramidal features, myoclonus, and seizures are also seen (74; 67). Whatever the case might be, memory loss is the most important symptom of familial Alzheimer disease, and it is under the strongest genetic influence, particularly declarative memory. None of the other cognitive symptoms showed heritability estimates as high as those observed for memory loss (78).
In familial Alzheimer disease clinical manifestations, temporal progression, and neuropathological features are not different from those of the more common sporadic form of Alzheimer disease (46). In occasional patients, myoclonus may be an early neurologic symptom in early-onset familial Alzheimer disease. Early-onset dementia, early myoclonus, and a familial history of dementia should suggest the possibility of a genetic form of Alzheimer disease (64).
Genetic influences on Alzheimer disease include genes linked to familial Alzheimer disease and sporadic Alzheimer disease. Key genes implicated in familial Alzheimer disease are APP (amyloid precursor protein), PSEN1 (presenilin 1), and PSEN2 (presenilin 2), whereas the APOE gene is associated with sporadic and late-onset familial Alzheimer disease (38). Additional molecular factors involve immunological elements, such as TREM2, and disorders related to lipid metabolism (ABCA1, ABCA7) or biothiol metabolism (MTHFD1), as well as the transport of metabolites (BIN1). Research suggests that APOE is a risk factor for both sporadic Alzheimer disease and late-onset familial Alzheimer disease. The amyloid cascade theory is the predominant explanation for the pathomechanism of Alzheimer disease, primarily associated with familial Alzheimer disease, but it has also been noted in cases of sporadic Alzheimer disease. Excessive accumulation of beta-amyloid (Abeta) peptides, along with intracellular neurofibrillary tangles of hyperphosphorylated tau protein, appears to lead to damage of DNA and RNA. Furthermore, RNA interference may influence the levels of pathological proteins (Abeta and tau) and affects the onset and progression of Alzheimer disease (148).
Sometimes patients with familial Alzheimer disease have an atypical clinical picture. For instance, a novel Leu174Arg PS1 mutation was found in two members of a Bavarian family, initially diagnosed with frontotemporal dementia (75). In an African-American family with autosomal dominant rapidly progressive dementia and psychosis, occurring early in the 5th decade of life, two brothers developed frontotemporal dementia with personality change, auditory and visual hallucinations, delusions, memory impairment, word-finding difficulties, and, subsequently, rigidity, dystonia, myoclonus, and mutism. In one of them, neuropsychometric testing results suggested frontotemporal dementia, but neuroimaging studies revealed diffuse cortical involvement. A clinical diagnosis of familial non-Alzheimer dementia was made. However, autopsy results confirmed the diagnosis of Alzheimer disease, and gene sequencing revealed a PS1 point mutation (M139V) co-segregating with the disease (124). PS1 mutations can cause a form of Pick disease without evidence of Alzheimer disease. A family with a PS1 M146L mutation had both Pick bodies and Alzheimer disease. M146L mutant PS1 may predispose to both Pick disease and Alzheimer disease by affecting multiple intracellular pathways involving tau phosphorylation and amyloid metabolism. Language impairment with relative preservation of memory was the initial sign in two members of a kindred with the PS1 mutation R278I (51). A 27-year-old man who developed early-onset dementia with spastic paraparesis had the mutation (Leu85Pro) in PS1. This was the first report of early-onset Alzheimer disease with spastic paraparesis and the visual variant form of the disease, which is associated with visuospatial cognitive disorder (09). Another unusual presentation was reported in members of an Irish family with familial Alzheimer disease due to an E280G mutation in exon 8 of the presenilin-1 gene. One member had spastic paraparesis and white matter abnormalities on cranial MRI. A sibling had an internuclear ophthalmoplegia, spastic-ataxic quadriparesis, and "cotton-wool plaques" with amyloid angiopathy in the brain biopsy. In another affected sibling, MRI also revealed white matter abnormalities consistent with an ischemic leukoencephalopathy due to amyloid angiopathy affecting meningocortical vessels (103). A novel variant of Alzheimer disease was described in a Finnish pedigree with 17 affected individuals across three generations. Their disease was characterized by progressive dementia, in most cases preceded by spastic paraparesis. Neuropathological studies revealed numerous, distinct, large, round, and eosinophilic plaques as well as neurofibrillary tangles and amyloid angiopathy throughout the cerebral cortex. The predominant plaques resembled cotton wool balls and were immunoreactive for A-beta. However, they lacked a congophilic dense core or marked plaque-related neuritic pathology. Molecular genetic analyses revealed that the disease was caused by a deletion of exon 9 (delta9) of the PS1 gene (30). In another family, exhibiting early-onset autosomal dominant Alzheimer disease with spastic paraplegia, dystonia, and dysarthria, a novel 6-nucleotide insertional mutation in exon 3 of the PS1 gene was described (96). In a single Greek family with early-onset familial Alzheimer disease owing to the N135S PSEN1 mutation, three individuals had disease onset in their 30s, with a clinical phenotype that was remarkable for spastic dysarthria, limb spasticity, and seizures in addition to more typical features of Alzheimer disease. At autopsy both the mother and her daughter had pathologic findings of Alzheimer disease and histologic evidence of corticospinal tract degeneration (129). In an Italian family with hereditary dementia associated with the novel mutation PSEN2 A85V in the presenilin 2 gene, the proband showed a clinical phenotype indicative of Lewy body dementia, and the neuropathologic examination demonstrated the presence of unusually abundant and widespread cortical Lewy bodies in addition to the hallmark lesions of Alzheimer disease (110). In an Iranian family with the APP Thr714Ala mutation, the phenotype was atypical with a long prodromal phase, autonomic failure, and seizures. The proband had epilepsy with complex partial seizures and central degenerative autonomic failure (84). This is another example of clinical heterogeneity in familial Alzheimer disease.
There is heterogeneity in the age of dementia onset. In a large pedigree linked to chromosome 14q, including 34 subjects with early-onset progressive dementia, the mean age at onset was 46 ± 3.5 years, and the mean age at death was 52.6 ± 5.7 years. Myoclonus and extrapyramidal signs were common, and seizures were present in all affected individuals, which suggest that epilepsy could represent a particular feature linked to chromosome 14q in Alzheimer disease families (22). In another study of 180 demented individuals from 24 kindreds with familial Alzheimer disease, phenotypic heterogeneity was also found. The mean age at onset for the total group was 54.7 ± 11.5 years, with a wide range of 30 to 84 years. The mean age at death was 63.5 ± 12.2 years (range 46 to 85). Five out of these families represented an early-onset group with the mean age of 42 years at the beginning of the disease. Eight more families represented a late-onset group with the mean age of 68 years when the disease began (18). In nine kindreds of German ancestry from the Volga River region, with 89 known demented persons (53 male, 36 female), the mean age at onset was 57.6 ± 8.4 years (range 40 to 84), and the mean age at death was 66.5 ± 7.6 years (range 50 to 80). Out of these patients, 24% had seizures; 72%, language disturbance; 36%, rigidity; 16%, tremor; and 12%, myoclonus (17).
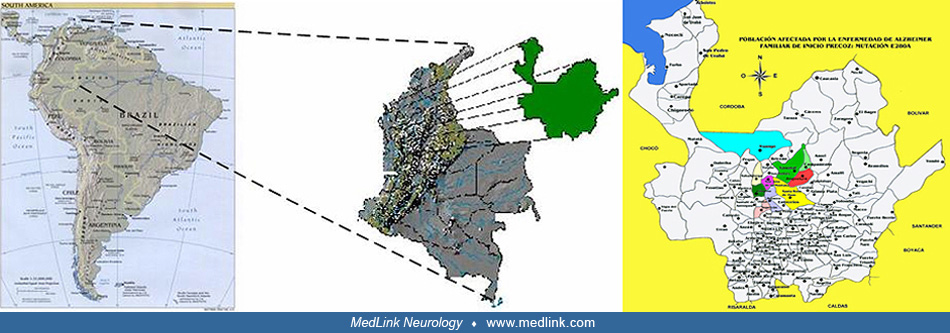
Presenilin 1 (PS1) mutations are the most common cause of early-onset familial Alzheimer disease (EOFAD), showing a common phenotypic profile characterized by early age of onset, severe dementia, and distinct neurodegeneration. The largest population of EOFAD carries the E280A mutation in PS1 and resides in Antioquia, Colombia, which comprises around 5,000 individuals. Carriers start showing memory impairment in the third decade of life, followed by progressive impairment of language and other cognitive processes. They reach mild cognitive impairment around 45 years of age and dementia around 50 years of age. There is some phenotypic variability among the carriers of this single PS1 mutation. Some patients present with epilepsy, verbal impairment, and cerebellar ataxia. Neuropathologically, PS1 E280A cases show pronounced brain atrophy, severe amyloid-beta pathology, distinct hyperphosphorylated tau-related pathology, and cerebellar damage (135).
There was no significant difference in the age at onset between men and women. The mean age of death, based on 54 observations, was 54.8 years (range 38 to 65), with no significant differences between men and women (87). In a retrospective study with this same population, Acosta-Baena and colleagues found that clinical deterioration can be detected as measurable cognitive impairment around 2 decades before dementia onset (01). They assessed descendants of individuals with the E280A (PSEN1) mutation, with the aim of identifying distinct stages of clinical progression to Alzheimer disease-related dementia. A total of 1784 patients were initially evaluated, 449 of whom were carriers. Median age at onset was 35 years (95% CI 30 to 36) for asymptomatic pre-mild cognitive impairment (MCI), 38 years (37 to 40) for symptomatic pre-MCI, 44 years (43 to 45) for MCI, and 49 years (49 to 50) for dementia. The median age at death was 59 years (95% CI 58 to 61). The median time of progression from asymptomatic to symptomatic pre-MCI was 4 years (95% CI 2 to 8), from symptomatic pre-MCI to MCI was 6 years (4 to 7), from MCI to dementia was 5 years (4 to 6), and from dementia to death was 10 years (9 to 12).
Jimenez and colleagues suggested that a novel gene, not linked to a previously known genetic locus, is related to late-onset familial Alzheimer disease in four families with autosomal dominant Alzheimer disease, from a genetically isolated population. Twelve affected individuals who were examined had progressive memory loss with onset between 57 and 74 years of age, along with seizures, myoclonus, and parkinsonism in advanced stages. The brain of the case examined postmortem showed widespread neocortical neuritic plaques and neurofibrillary tangles, amyloid angiopathy, and Lewy bodies restricted to limbic areas (67).
The outlook and potential complications of familial Alzheimer disease can vary depending on the individual and the specific genetic mutation involved (118). The following are some important points to note regarding prognosis and expected complications.
Onset. Familial Alzheimer disease symptoms typically become evident before the age of 65, most commonly in persons in their 40s or 50s. Some PSEN1 mutations produce onset as early as the 30s, whereas certain APP and PSEN2 variants may manifest later (at approximately 50 to 60 years of age). In Familial Alzheimer disease cohorts, both parental and variant-derived onset ages predict the carrier’s onset (131).
Progressive nature. Like other Alzheimer variants, Familial Alzheimer disease is a relentlessly progressive neurodegenerative disorder. Recent multi-omics analyses across APP, PSEN1, and PSEN2 mutations identify shared molecular endotypes--synaptic dysfunction, mitochondrial stress, and neuroinflammation--that contribute to accelerated decline (149).
Decline in cognitive abilities. Initially, familial Alzheimer disease exhibits symptoms similar to those seen in Alzheimer disease, namely, memory loss being prominently observed (117). As the disease progresses further, individuals may encounter difficulties with language skills, problem-solving capabilities, decision-making processes, and other cognitive functions. Average survival after symptom onset remains about 8 to 12 years, with the mutation site influencing course length (133).
Psychological symptoms. Familial Alzheimer disease can also be accompanied by various behavioral and psychological symptoms, such as shifts in mood patterns or alterations in personality traits or behaviors (118). These symptoms may include restlessness or agitation, episodes of aggression, feelings of depression or anxiety, as well as withdrawal from social interactions.
Motor skills and tasks. As familial Alzheimer disease progresses, individuals might encounter challenges in performing tasks like getting dressed, taking a bath, and eating (118). They may also experience difficulties with motor skills.
Impact on quality of life. Familial Alzheimer disease can have a significant impact on the quality of life for both individuals with the disease and their caregivers (118). The progressive nature of the disease and the cognitive and functional decline can lead to increased dependency and the need for extensive care and support.
Lack of effective treatment. There are no medications or treatments that can prevent or cure familial Alzheimer disease. Anti-amyloid monoclonal antibodies (lecanemab, donanemab) are now under evaluation in presymptomatic APP/PSEN mutation carriers within DIAN trials (85).
The prognosis of mutation carriers in APP, PS1, and PS2 genes is not favorable, and they will usually develop Alzheimer disease in a similar age range as other affected members of their families because these are dominant mutations with 100% penetrance. Concerning APOE e4, gene dose was shown to have an effect on the risk of developing Alzheimer disease, the age at onset, the accumulation of senile plaques in the brain, and a reduction of choline-acetyltransferase (ChAT) activity in the hippocampus of Alzheimer disease subjects. APOE e4 plays a crucial role in the cholinergic dysfunction associated with Alzheimer disease and may be a prognostic indicator of poor response to therapy with acetylcholinesterase inhibitors in patients with Alzheimer disease (111). Bleeding complications, particularly amyloid-related imaging abnormalities with hemorrhage (ARIA-H), occur more frequently in Alzheimer disease patients carrying the APOE4 allele who are treated with anti-amyloid monoclonal antibodies, and this risk appears accentuated in older, later-onset cases (116; 43).
Mutations in these three genes are seldom silent. Although mutation A713T in APP is a silent mutation or polymorphism, a carrier has been identified with clinical and neuropathological evidence of Alzheimer disease (08).
The population with single-domain amnesic MCI has a higher risk of conversion to dementia by Alzheimer disease than other kinds of MCI. A total of 881 MCI subjects recruited from 20 memory clinics and followed for up to 5 years shows that one cluster (aMCIsingle) had a significantly higher conversion rate (19%), compared to subjective cognitive impairment (SCI, p < 0.0001) and non-amnestic MCI (naMCI, p = 0.012) (34). This cluster was the only one showing a significantly different biomarker profile (Aβ42, t-tau, APOE ε4, and medial temporal atrophy), compared to SCI or naMCI. In subjects with mild MCI, the single-domain amnestic MCI profile was associated with the highest risk of conversion. A cognitive profile characterized by isolated memory deficits may be sufficient to warrant applying prevention strategies in MCI.
A 49-year-old farmer from the village of Belmira consulted the neurologic service of the San Vicente de Paul University Hospital in Medellín, Colombia. Since the age of 45 years, he had been gradually losing his memory and the ability to understand the meaning of words. He was crying or laughing out of context. His eyes were unable to concentrate on any person or object. Physically, he was weak. The family wanted palliative measures for their relative’s disease, with which they were familiar because over the years it had killed many other members of the family. It appeared that the symptoms of progressive cognitive impairment, hallucinations, and delusions were similar to those described by Alois Alzheimer in 1906 in the case of a 51-year-old woman who died shortly afterwards in a hospital in Frankfurt, Germany. Nine members of the family of the original patient had died around the age of 47 years.
In 1990, a second person, this one from the village of Yarumal, Colombia, was studied with similar symptoms of dementia. This person’s occupation was to sell lottery tickets, and periodic losses of memory affected his business. Another patient, a woman from Angostura, was examined with a complete loss of her memory. Fieldwork in these villages confirmed that other members of these families had been affected by this precocious form of Alzheimer disease, which was inherited by 50% of the children when one of the parents was affected.
The brain autopsy of one of these affected individuals contained large amyloid deposits similar to those found in the case of Alzheimer disease. Neurofibrillary tangles were seen widely throughout the brain. Meanwhile, a fourth family with the same illness was identified, living in the village of San Jos‚ de la Montana. A link to chromosome 14 was established by Goate and colleagues at Washington University in the United States (50). This gene was eventually identified in another family by a Canadian research group (137) as the Presenilin-1 gene (PS1). The Antioquia gene was found to consist of a substitution error in the codon 280 (glutamic acid x alanine); today it is known as the Paisa mutation or mutation E280A of the PS1 (06). These families affected by genetically determined Alzheimer disease due to the E280A mutation in the PS1 gene on chromosome 14, were found in 26 families distributed all over Antioquia. The following elements were found that suggest all of these pedigrees are linked: (1) all the disabled individuals have the same phenotype; (2) all of them carry the E280A mutation in the PS1; (3) all of them live in the same geographical area; (4) several families share the same family name; (5) several families have the same infrequent haplotype (genetic label); this suggests that they are part of the same hereditary trunk (79); (6) it was possible to interconnect the common trunk in 13 of the 26 families and to classify them in a common genealogy (21). This is the biggest family known with hereditary early-onset Alzheimer disease.
There are two different types of Alzheimer disease, namely: (1) 5% to 10% of the cases are due to mutations in either one of three genes resulting in the increased generation of beta amyloid proteins; (2) the great majority of Alzheimer disease cases appear sporadic, with old age being the main risk factor.
Although several loci have been identified as contributors to Alzheimer disease, only three of them are sufficient to produce it, namely, the amyloid precursor protein gene (AD1), the presenilin 1 gene (AD3), and the presenilin 2 gene (AD4). More than 200 disease-associated mutations have been identified at these three loci.
APP mutations. Over 30 coding mutations in the APP gene have been found. About 25 of these are pathogenic, resulting in autosomal dominant Alzheimer disease with an early onset. However, in a set of whole-genome sequence data from 1,795 Icelanders, Jonsson and colleagues found a coding mutation (A673T) in the APP gene that protects against Alzheimer disease and age-related cognitive decline (70). The strong protective effect of the A673T substitution against Alzheimer disease provides proof of principle for the hypothesis that reducing the b-cleavage of APP may protect against the disease and may also protect against cognitive decline in the elderly without Alzheimer disease, by the same or similar mechanisms. The authors provided indirect support for the hypothesis that the pathogenesis of Alzheimer disease and normal cognitive decline of the elderly may be shared, at least in part. They, therefore, propose that Alzheimer disease may represent the extreme of the age-related decline in cognitive function.
Most mutations in the amyloid precursor protein (APP) gene have been associated with early-onset familial Alzheimer disease; however, some mutations within the Abeta-coding sequence have been described in families with other phenotypes such as leukoencephalopathy, schizophrenia, amyloidosis, late-onset familial Alzheimer disease, or recurrent cerebral hemorrhage (see Table 1). For example, hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D) is an autosomal dominant disease characterized by amyloid deposition in cerebral blood vessels, which results in cerebral hemorrhages, leukoencephalopathy, dementia, and death (58). Almost all of the mutations in APP are autosomal dominant; however, the APP mutation A673V causes Alzheimer disease in only the homozygous state, whereas heterozygous carriers are unaffected, which is consistent with a recessive Mendelian trait of inheritance (37). The neuropathological picture of the proband of this family presents distinctive characteristics compared to sporadic Alzheimer disease or familial Alzheimer disease inherited as a dominant trait. This fact has implications for genetic screening of Alzheimer disease (48).
|
Phenotype |
APP mutation |
Author |
|
Amyloidosis, with cerebral hemorrhage, hereditary, Dutch type (HCHWA-D) |
GLU693GLN |
(80) |
|
Familial Alzheimer disease |
VAL717ILE |
(50) |
|
Familial Alzheimer disease |
VAL717PHE |
(99) |
|
Familial Alzheimer disease |
VAL717GLY |
(25) |
|
Familial Alzheimer disease |
VAL717LEU |
(100) |
|
Dementia, presenile, and cerebroarterial amyloidosis |
ALA692GLY |
(61) |
|
Schizophrenia |
ALA713VAL |
(69) |
|
Polymorphism |
NT2124, C-T |
(10) |
|
Familial Alzheimer disease |
LYS670ASN |
(98) |
|
Familial Alzheimer disease |
ALA713THR |
(23) |
|
Familial Alzheimer disease, late-onset |
GLU665ASP |
(108) |
|
Familial Alzheimer disease |
ILE716VAL |
(40) |
|
Familial Alzheimer disease |
VAL715MET |
(05) |
|
Familial Alzheimer disease |
GLU693GLY |
(72) |
|
Amyloidosis, cerebroarterial, Italian type |
GLU693LYS |
(95) |
|
Familial Alzheimer disease |
THR714ILE |
(76) |
|
Familial Alzheimer disease, leukoencephalopathy |
ASN23ASP |
(56) |
|
Familial Alzheimer disease |
THR714ALA |
(106) |
|
Cerebroarterial amyloidosis, recurrent intracerebral hemorrhages |
LEU705VAL |
(102) |
|
Alzheimer disease, early-onset, with cerebral amyloid angiopathy |
APP duplication |
(20; 128) |
|
Alzheimer disease, primarily caused by soluble Abeta oligomers |
E693Delta |
(145) |
|
Recessive Mendelian familial Alzheimer disease |
A673V |
(48) |
|
Protection against Alzheimer disease and age-related cognitive decline |
A673T |
(70) |
PS1 and PS2 mutations. PS1 (S182) is the gene linked to chromosomal locus 14q24.3, related to early-onset familial Alzheimer disease type AD3, which encodes a 467-amino acid protein. This protein has at least seven transmembrane helical domains. The first missense mutations that cosegregate with early-onset familial Alzheimer disease type AD3 were identified in this gene. Because these changes occurred in conserved domains of this gene but were not present in normal controls, they were considered to be causative of disease (06; 137). Since then, 177 mutations have been described in this gene and account for more than approximately 80% of all familial Alzheimer disease mutations. More than 200 fully penetrant mutations in the amyloid beta-protein precursor (APP), presenilin 1 (or PSEN1), and presenilin 2 (PSEN2) have been linked to early-onset familial Alzheimer disease.
Li and colleagues reported the existence of a gene (PS2), localized on chromosome 1, that encodes a 7 transmembrane domain protein, and is similar in structure and sequence to that encoded by AD3 (83). This gene is expressed in a variety of tissues, including the brain, and causes familial Alzheimer disease (AD4) (83). Mutations in presenilin genes are the major cause of familial Alzheimer disease, but they account for less than 5% of the cases. PS1 and PS2 are important participating components of the gamma-secretase activity that is responsible for the proteolytic cleavage of the amyloid precursor protein and the NOTCH receptor proteins. PS1 is involved in the intracellular trafficking of selected membrane proteins (ie, APP, nicastrin, trkB, telencephalin) and also in the trafficking of N-cadherin from the endoplasmic reticulum to the plasma membrane via the microtubule network. Alterations in protein transport caused by a dysfunction in PS1 could lead to a disturbance in synaptic transmission and eventually to neurodegeneration (147).
During development, presenilins play crucial roles in the maintenance of neural progenitor cell proliferation, the temporal control of neuronal differentiation, and the survival and proper neuronal migration in the developing cerebral cortex. Analysis of presenilin function in the adult cerebral cortex has revealed essential roles for presenilins in synaptic plasticity, long-term memory, and neuronal survival. The molecular mechanisms through which presenilins may mediate these functions include the Notch, CREB, and NMDA receptor-mediated signaling pathways. These diverse functions of presenilins in cortical development and function and in neuronal survival have important implications for the pathogenesis of neurodegenerative dementia (155).
PS1 expression is essential for neurogenesis during embryonic development and may influence neurogenesis in the adult brain as well. However, the functions of PS1 and how mutations in its gene cause familial Alzheimer disease are incompletely understood. PS1 mutations impair new neuron production in the adult hippocampus by decreasing neural progenitor survival (154). Expression of STM2 (PS2) was high in the skeletal muscle and the pancreas, with comparatively low levels observed in the brain. This expression pattern is intriguing because pathology and degeneration in Alzheimer disease are observed only in the central nervous system (81).
The presenilins are expressed in the heart and are critical to cardiac development. Mutations in presenilins may also be associated with dilated cardiomyopathy. A novel PSEN1 missense mutation (Asp333Gly) and a single PSEN2 missense mutation (Ser130Leu) segregated with dilated cardiomyopathy and heart failure (82).
Mutations in presenilin 2 are a rare cause of early-onset familial Alzheimer disease. Eighteen presenilin 2 mutations have been reported, although not all have been confirmed to be pathogenic. The best studied, the N141I mutation, produces an Alzheimer disease phenotype with a wide range of onset ages overlapping both early- and late-onset Alzheimer disease; it is often associated with seizures, high penetrance, and typical Alzheimer disease neuropathology (65).
In patients with Alzheimer disease linked to chromosome 1 and with the PS2 gene mutation, among the Volga German families, Abeta42(43) was the predominant peptide species present, although the total amount of Abeta40 and Abeta42(43) deposited in plaques did not differ from that seen in sporadic Alzheimer disease and was significantly lower than that occurring in Alzheimer disease due to PS1 gene mutations. It seems that the effect of the PS2 gene mutation on the brain, related to Abeta deposition, is much less severe than that of the PS1 mutation, which seems to lead to a much earlier and much more aggressive development of Alzheimer disease (89).
Mutations of the presenilin 1 and 2 genes more severely affect the lysosomal system in humans than does sporadic Alzheimer disease. The neuronal lysosomal system is a major degradative pathway, induced by cell stress and linked to neurodegenerative diseases. Cathepsin D and B levels are higher in PS-familial Alzheimer disease neocortexes than in those of sporadic Alzheimer disease. Accumulation of hydrolases in dystrophic neurites in senile plaques suggests that amyloid deposition may be a stimulus for local mobilization of the lysosomal system (24).
APOE e4 as a risk factor for Alzheimer disease. Apolipoprotein E is immunochemically localized to the senile plaques, vascular amyloid, and neurofibrillary tangles of Alzheimer disease. The apolipoprotein E gene is localized on chromosome 19q13.2, within the region previously associated with linkage of late-onset familial Alzheimer disease. Analysis of APOE alleles in patients with Alzheimer disease and control individuals demonstrated that there was a highly significant association of APOE e4 allele and late-onset familial Alzheimer disease (139). APOE e4 also has a strong relationship with sporadic Alzheimer disease. The risk for late-onset Alzheimer disease increased from 20% to 90%, and the mean age at onset decreased from 84 to 68 years with an increasing number of APOE e4 alleles (29), whereas possession of the APOE e2 allele may be associated with a protective effect (28). The age at onset is inversely related to the dose of APOE e4 alleles.
In late-onset familial Alzheimer disease, women have a significantly higher risk of developing the disease than men do. A direct comparison of APOE e4 heterozygous men and women revealed a significant twofold increased risk in women. These observations are consistent with the increased incidence of familial Alzheimer disease in women and may be a critical clue to the role of gender in the pathogenesis of Alzheimer disease (107).
Other genetic risk factors. The low-density lipoprotein receptor-related protein gene (LRP1) is often mentioned as a candidate gene for Alzheimer disease risk because of its role as a receptor for apolipoprotein E (apoE) but other authors have not found any evidence of linkage or association of LRP1 and Alzheimer disease (134). The risk for Alzheimer disease increases with certain polymorphisms in the genes that encode the alpha and beta isoforms of interleukin-1 (IL-1). IL-1 levels are elevated in the brains of patients with Alzheimer disease, and overexpression of IL-1 is associated with beta-amyloid plaque deposition. IL-1 interacts with the gene products of several other known or suspected genetic risk factors for Alzheimer disease, including ApoE, alpha1-antichymotrypsin, and alpha2-macroglobulin (97). Although linkage to chromosome 12p for familial Alzheimer disease has been inconsistent, it is possible that a gene acting as a risk factor for Alzheimer disease is localized on chromosome 12p (109; 73).
Inherited variants in the SORL1 neuronal sorting receptor have been reported to be associated with late-onset Alzheimer disease. The variants LR11 or SORLA may regulate tissue-specific expression of SORL1. The SORL1 directs trafficking of APP into recycling pathways, and when SORL1 is underexpressed, APP is sorted into Abeta-generating compartments. These data suggest that changes in SORL1 expression or function are mechanistically involved in causing Alzheimer disease (125).
Phylogenetic analysis has identified a polymorphic poly-T variant, rs10524523, in the translocase of outer mitochondrial membrane 40 homolog (TOMM40) gene, which provides greatly increased precision in the estimation of age of late-onset Alzheimer disease onset for apolipoprotein E are epsilon3 carriers (126). In two independent clinical cohorts, longer lengths of rs10524523 were associated with a higher risk of late-onset Alzheimer disease.
In a 2-stage genome-wide association study of Alzheimer disease involving over 16,000 individuals, a genome-wide significant association was observed with single-nucleotide polymorphisms at two loci not previously associated with Alzheimer disease: the clusterin (CLU, also known as APOJ) gene and the PICALM gene (59).
To determine whether risk for Alzheimer disease associated with novel genes CLU, PICALM, and CR1 is influenced by apolipoprotein E (APOE) genotypes, 7070 cases with Alzheimer disease, 3055 with autopsies, and 8169 elderly cognitively normal controls, 1092 with autopsies, from 12 different studies were included in a meta-analysis (71). Genotypes at PICALM confer risk predominantly in APOE e4-positive subjects. Thus, APOE and PICALM synergistically interact.
Genetic variants within the TREM2 gene are associated with increased risk of Alzheimer disease and FTD. The most commonly associated variant, rs75932628 (encoding R47H), showed a highly significant association with Alzheimer disease (57). Giraldo and colleagues investigated a consanguineous family segregating autosomal recessive behavioral variant FTLD from Antioquia, Colombia (49). Exome sequencing identified a nonsense mutation in TREM2 (p.Trp198X) segregating with disease. TREM2 is significantly overexpressed in the brain tissue from Alzheimer disease cases. In general, these data suggest that a mutational burden in TREM2 may serve as a risk factor for neurodegenerative disease and that potentially this class of TREM2 variant carriers with dementia should be considered as having a molecularly distinct form of neurodegenerative disease.
Eight pathogenic variants (one in APP, one in MAPT, two in PSEN1, and four in GRN) were found in 14 samples from 439 probands from late-onset Alzheimer disease families with a history of four or more affected individuals (31). Thirteen additional variants, present in 23 families, did not segregate with disease, but the frequency of these variants is higher in Alzheimer disease cases than in controls, indicating these variants may also modify risk for disease. The presence of variants in these genes in LOAD and early-onset Alzheimer disease demonstrates that factors other than the mutation can impact the age at onset and penetrance of at least some variants associated with Alzheimer disease.
Velez and colleagues used a novel sequential strategy that combined pooling of DNA and bootstrapping (pbGWAS) in patients with early onset Familial Alzheimer disease, carrying the PSEN1 p.Glu280Ala mutation (often referred to as E280A mutation), and they identified novel loci genome-wide significantly associated as modifiers of the age of onset of Alzheimer disease (CD44, rs187116, P=1.29 x 10(-12); NPHP1, rs10173717, P=1.74 x 10(-12); CADPS2, rs3757536, P=1.54 x 10(-10); GREM2, rs12129547, P=1.69 x 10(-13)), as well as other loci known to be associated with Alzheimer disease (152). Regions identified by pbGWAS were confirmed by subsequent individual genotyping. The pbGWAS methodology and the genes it targeted could provide important insights in determining the genetic causes of Alzheimer disease and other complex conditions.
Genetic protector factors in Alzheimer disease have also been described. People with major mitochondrial haplotypes H6A1A and H6A1B showed a reduced risk of Alzheimer disease (121). The protective haplotypes were defined by three variants: m.3915G>A, m.4727A>G, and m.9380G>A. These findings are the results of the largest study to date with complete mtDNA genome sequence data, but the functional significance of the associated haplotypes remains unknown.
Amyloidosis appears to be a stereotyped mode of central nervous system reaction to various types of genetic stimuli or injury, such as mutations involving different genes, trisomy 21 or repeated head trauma. Three lesions may be considered typical in Alzheimer disease, namely: neurofibrillary pathology, amyloid deposits, and neuronal and synaptic loss. According to the "amyloid cascade hypothesis", accumulation of amyloid beta (Abeta) peptides in the brain is a primary event in the pathogenesis of Alzheimer disease. One of the strongest pieces of evidence supporting this hypothesis was the identification of pathogenic mutations in APP, PS1, and PS2 genes, responsible for familial autosomal dominant Alzheimer disease. Mutational effects are the overproduction of long-tailed amyloid beta-peptides and alterations of beta-catenin signaling pathways (101).
Transgenic mouse models of Alzheimer disease expressing high levels of amyloid precursor protein (APP) with familial Alzheimer disease mutations have proven to be useful in understanding pathogenic processes of Alzheimer disease. These data support earlier observations of A beta accumulation in the endosomal-lysosomal pathway and the hypothesis that intraneuronal accumulation of A beta could be an important factor in the Alzheimer disease pathogenesis (150).
The relationship between beta-amyloid plaques and neurofibrillary tangles is unclear. However, injection of beta-amyloid Abeta42 fibrils into the brains of P301L mutant tau transgenic mice caused a 5-fold increase in the number of neurofibrillary tangles in cell bodies within the amygdala. This finding supports the hypothesis that Abeta42 fibrils can accelerate neurofibrillary tangles formation in vivo (54).
Parenchymal and vascular amyloid deposits are associated with a different group of Abeta peptide species. Round lesions lacking a central amyloid core and that have been observed in individuals affected by early-onset familial Alzheimer disease, associated with mutations in the PS1 gene, have been named “Cotton wool plaques.” Cotton wool plaques associated with the PS1 V261I mutation, and isolated by laser microdissection, were not found to be associated with vessels and did not contain Abeta1-40 peptides, which are the main component of vascular deposits (94).
An early-onset familial dementia with ataxia linked to the BRI2 gene was found in a Danish family. The main neuropathological features of their disease were: cerebral deposits of two unrelated amyloid and preamyloid molecules, Danish amyloid (ADan) and beta-amyloid (Abeta), absence of compact plaques, and bearing neurofibrillary degeneration indistinguishable from that observed in Alzheimer disease. In all cases, the presence of Abeta40 was negligible, a surprising finding in view of the prevalence of Abeta40 in vascular deposits observed in sporadic and familial Alzheimer disease, Down syndrome, and normal aging. Whether the presence of the two amyloid subunits is imperative for the disease phenotype remains to be elucidated. Extensive neurofibrillar degeneration in the absence of compact plaques strongly suggests that the latter, fundamental lesions for Alzheimer disease diagnosis, are not essential for the mechanism of dementia (144).
Studies in Alzheimer disease models have identified marked dysregulations in calcium signaling and related downstream pathways, which occur long before the diagnostic histopathological or cognitive changes. "Calcinists" think that Alzheimer disease pathogenesis reflects long-term calcium dysregulation. They do not reject “betaAptist” or “Tauist” doctrine, but rather believe that their genesis is associated with earlier calcium signaling dysregulations (140).
Alzheimer disease is hypothesized to be caused by an overproduction or reduced clearance of amyloid-beta (Aβ) peptide. Autosomal dominant Alzheimer disease (ADAD) caused by mutations in the presenilin (PSEN) gene has been postulated to result from increased production of Aβ42 compared to Aβ40 in the central nervous system (CNS). Central nervous system Aβ42 to Aβ40 production rates were 24% higher in mutation carriers compared to noncarriers, and this was independent of fibrillar amyloid deposits quantified by PET PIB imaging (113). The fractional turnover rate of soluble Aβ42 relative to Aβ40 was 65% faster in mutation carriers and correlated with amyloid deposition, and it was consistent with increased deposition of Aβ42 into plaques, leading to reduced recovery of Aβ42 in cerebrospinal fluid (CSF). These findings support the hypothesis that Aβ42 is overproduced in the central nervous system of humans with PSEN mutations that cause Alzheimer disease, and they demonstrate that soluble Aβ42 turnover and exchange processes are altered in the presence of amyloid plaques, causing a reduction in Aβ42 concentrations in the cerebrospinal fluid.
Presently, Alzheimer disease affects more than 60 million people worldwide, according to the World Alzheimer Report 2024 (Alzheimer’s Disease International 2024). Global prevalence is projected to exceed 150 million by 2050, driven largely by aging populations and improved survival. Early-onset familial Alzheimer disease represents less than 5% of all Alzheimer disease cases, though it accounts for as much as 10% to 15% of early-onset cases (onset earlier than 65 years) (149). Having a first-degree relative with Alzheimer disease roughly doubles lifetime risk (127). When two or more relatives are affected, risk increases up to fourfold, depending on APOE genotype and shared lifestyle factors (35). In families affected with autosomal-dominant inheritance, the risk of familial Alzheimer disease is 50%. This reflects the fully penetrant nature of mutations in PSEN1, PSEN2, or APP genes (131). Individuals carrying one or two APOE ε4 alleles have an elevated lifetime risk and earlier onset, though APOE ε4 is a susceptibility allele rather than a deterministic one (35). Protective alleles, such as APOE ε2 and variants in TREM2 and SORL1, modify risk and may influence age of onset in familial and sporadic forms alike (47). Current clinical practice already includes predictive genetic testing and counseling for at-risk relatives in known Familial Alzheimer disease families (85). Population-based polygenic risk scoring and blood-based biomarkers (Aβ42/40 ratio, p-tau217) are increasingly incorporated into preventive trials to refine early detection strategies (149).
Identification of the genes responsible for autosomal dominant early-onset familial Alzheimer disease has awakened considerable interest in the application of this genetic information to medical practice through genetic testing and counseling. A screening test for mutations in the APP, PS1, and PS2 genes is available to individuals with Alzheimer disease symptoms and to those at risk. In families with a confirmed pathogenic variant, each first-degree relative carries a 50% probability of inheritance due to autosomal-dominant transmission (131).
The goal of genetic testing is to identify at-risk individuals in order to enable accurate counseling, reproductive decision-making, and participation in preventive trials aimed at delaying symptom onset in presymptomatic carriers (85). APOE genotyping remains of limited predictive value for clinical use; it indicates risk modulation but not disease certainty (45). Guidelines recommend formal genetic counseling before and after any testing to address ethical, psychological, and insurance concerns (04).
Carriers of PSEN1, PSEN2, and APP mutations are now the primary participants in ongoing prevention studies, such as the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) and the Alzheimer’s Prevention Initiative (API). These efforts evaluate anti-amyloid monoclonal antibodies (eg, lecanemab, donanemab, and the discontinued crenezumab) and emerging anti-tau therapies in mutation carriers years before the expected onset (85). Carriers of PS1, PS2, and APP mutations are now important participants in clinical trials oriented to primary prevention therapy.
Updated API protocols incorporate multimodal biomarkers--amyloid/tau PET, plasma p-tau217, and neurofilament light chain--to track early pathophysiologic change and treatment response (149). Initial findings from a clinical trial supported by the National Institutes of Health suggest that crenezumab, an experimental anti-amyloid medication, did not exhibit a statistically significant therapeutic advantage in individuals without cognitive impairment who possess a rare genetic mutation responsible for the onset of autosomal dominant Alzheimer disease at an early stage (119).
The 2023 API-Colombia crenezumab trial showed safe amyloid reduction but no measurable cognitive benefit, reinforcing the need for earlier and combination therapies (45). Subsequent DIAN-TU open-label extensions report that sustained amyloid plaque clearance with gantenerumab or lecanemab may delay symptom onset by several years in presymptomatic carriers (12). Lifestyle-based interventions--including physical exercise, cardiovascular risk control, and cognitive training--may confer modest additive protection even in genetically determined Familial Alzheimer disease, though evidence remains limited (86).
Differential diagnosis must be done between Alzheimer disease and other dementias as tauopathies. Tau filament formation, in the absence of Abeta production, is a feature of several neurodegenerative diseases including progressive supranuclear palsy, corticobasal degeneration, Pick disease, and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). No mutations have been identified in the tau gene in Alzheimer disease to date. The identification of mutations in the tau gene that are linked to FTDP-17 established that dysfunction of tau can, as well as Abeta formation, lead to neurodegeneration and dementia (54).
Clinically, familial and sporadic forms of Alzheimer disease are recognized, with a further division into patients with presenile (early) and senile (late) onset. Clinical, neuroimaging, neuropathological, and neurochemical studies have attempted to identify differences between the early- and late-onset cases, but, so far, no categorical biological difference between the two groups has been identified.
Advances in the molecular genetics of familial Alzheimer disease and the discovery of defined genetic abnormalities have provided a promising approach for distinguishing between the early- and late-onset cases within the group of autosomal dominant familial Alzheimer disease. Future studies may be able to contrast early- versus late-onset familial Alzheimer disease cases, and early-onset familial versus early-onset sporadic forms of the disease (60).
Diagnostic markers. A probable Alzheimer disease diagnosis is based mostly on clinical grounds. However, for a definitive diagnosis, neuropathological studies are necessary. It is feasible to do a definitive in vivo Alzheimer disease diagnosis by means of molecular studies in people with the clinical diagnosis and mutations in the APP, PS1, and PS2 genes. Other studies based on markers that may be of aid in the diagnosis of probable Alzheimer disease are available.
Neuroimaging markers. Patients with dementia had significantly lower parietal metabolism, measured using PET, than did at-risk subjects with APOE e4. The inheritance of APOE e4 is associated with reduced cerebral parietal metabolism and increased asymmetry in nondemented relatives at risk for probable Alzheimer disease. Longitudinal studies will determine if glucose metabolic measures provide a means to monitor experimental treatment responses during the early phases of the disorder (138).
Regional cerebral perfusion abnormalities based on SPECT studies are detectable before the development of the clinical Alzheimer disease symptoms in carriers of the PS1 mutation (68). Asymptomatic subjects with the PS1 mutation demonstrated reduced perfusion, in comparison with the normal control individuals, in the hippocampal complex, the anterior and posterior cingulates, the posterior parietal lobe, and the anterior frontal lobe. Patients with Alzheimer disease demonstrated decreased perfusion in the posterior parietal and the superior frontal cortex in comparison with the normal control subjects.
Longitudinal measures of atrophy rates with serial MRI scanning of autosomal dominant mutation carriers for Alzheimer disease can identify differences 2 to 3 years earlier than cross-sectional volumetric measures. Differences in hippocampal and whole-brain atrophy rates between controls and mutation carriers were evident 5.5 and 3.5 years, respectively, before diagnosis of Alzheimer disease (122).
Another study found that cortical atrophy is present in preclinical Alzheimer disease more than 5 years prior to symptom onset (115). Forty cognitively normal volunteer members of the cohort from the Colombian Alzheimer's Prevention Initiative Registry (18 E280A mutation carriers and 22 noncarriers) were involved in a study of biomarkers for Alzheimer disease. Compared to noncarriers, presymptomatic presenilin 1 mutation carriers exhibited a thinner cortex within the Alzheimer disease-signature summary measure. Analyses of individual regions demonstrated a thinner angular gyrus, precuneus, and superior parietal lobule in carriers compared to noncarriers.
Using tensor-based morphometry (TBM), Lee and colleagues examined brain volume differences between presymptomatic and symptomatic familial Alzheimer disease mutation carriers and noncarrier (NC) relatives (77). Twenty-five mutation carriers and 10 noncarrier relatives underwent brain MRI and clinical assessment. Cognitively intact familial Alzheimer disease mutation carriers had lower thalamic, caudate, and putamen volume. These regions may be affected earliest during prodromal stages of familial Alzheimer disease, whereas cortical atrophy may occur in later stages when carriers show cognitive deficits.
Using magnetic resonance spectroscopy, we can see metabolic changes in presymptomatic mutation carriers years before the expected onset of Alzheimer disease. Their magnitude is related to proximity of expected age at onset (52).
Seven presenilin 1 (PSEN1) mutation carriers, one amyloid precursor protein (APP) mutation carrier, 30 healthy control subjects, and 30 subjects with probable sporadic Alzheimer disease were participants in a study to evaluate the pattern of Pittsburgh Compound B (PiB) retention (153). All mutation carriers had high PiB retention in the striatum. The striatal pattern of PiB retention was similar in the PSEN1 and APP mutation carriers. The pattern of Abeta deposition in familial Alzheimer disease differed from that in sporadic Alzheimer disease, with higher striatal and somewhat lower cortical PiB retention in familial Alzheimer disease. The pattern and degree of Abeta deposition were not associated with mutation type nor cognitive status.
Fibrillar amyloid-β (Aβ) is thought to begin accumulating in the brain many years before the onset of clinical impairment in patients with Alzheimer disease and can be detected with florbetapir. Fifty members of the cohort from the Colombian Alzheimer's Prevention Initiative Registry, including 11 symptomatic individuals, 19 presymptomatic E280A mutation carriers, and 20 asymptomatic noncarriers, ranging in age from 20 to 56 years old, were studied to characterize the age-related accumulation of Aβ deposition using PET-Amiloide with florbetapir (42). There was greater florbetapir binding in asymptomatic PSEN1 E280A mutation carriers than in age-matched noncarriers. Fibrillar Aβ began to accumulate in PSEN 1E280A mutation carriers at a mean age of 28.2 years, which was about 16 and 21 years before the predicted median ages at mild cognitive impairment and dementia onset, respectively. Prominent florbetapir binding was seen in the anterior and posterior cingulate, precuneus, and parietotemporal and frontal grey matter, as well as in the basal ganglia.
A highly selective positron emission tomography (PET) imaging agent targeting PHF-tau in human Alzheimer disease brains has been developed. In-vitro autoradiography results show that [18F] T807 exhibits strong binding to PHF-tau-positive human brain sections. A comparison of autoradiography and double immunohistochemical staining of PHF-tau and Aβ on adjacent sections demonstrated that [18F] T807 binding localized with immunoreactive PHF-tau pathology but did not highlight Aβ plaques (157). [18F]T807 demonstrates high affinity and selectivity to PHF-tau, as well as favorable in vivo properties, making this a promising candidate as an imaging agent for Alzheimer disease. There have been few reports on imaging agents that selectively target tau aggregates. [18F]-T808 showed rapid uptake and washout in rodent brains, and preclinical in vivo studies suggest that [18F]-T808 possesses suitable properties and characteristics to be a specific and selective PET probe for imaging of paired helical filament tau in human brains. Amyloid positron emission tomography (PET) imaging has demonstrated that individuals with familial Alzheimer disease who possess mutations in the presenilin genes exhibit elevated levels of amyloid deposition in the frontal cortex, parietal cortex, and striatum in comparison to those with mutations in the amyloid precursor protein gene (16; 26). Additionally, fluorodeoxyglucose (FDG)-PET imaging has revealed that patients with familial Alzheimer disease with presenilin mutations tend to display reduced glucose metabolism in the posterior cingulate cortex, precuneus, and parietal cortex, as opposed to those with amyloid precursor protein mutations (16; 26). Furthermore, PET imaging can be employed to monitor disease progression and assess treatment response in specific brain regions among patients with familial Alzheimer disease (91; 62).
In summary, PET imaging offers valuable insights for the diagnosis and management of familial Alzheimer disease, whereas regional findings contribute to a deeper understanding of the underlying mechanisms of the disease within specific brain regions.
Electrophysiological markers. Event-related potentials (ERPs) were obtained during a semantic-matching task from 24 presymptomatic carriers and 25 symptomatic carriers of the E280A presenilin-1 (PS1) mutation, as well as 27 noncarriers (from the same families) (19). As expected, the symptomatic carrier group performed worse in the matching task and had lower N400 amplitudes than both asymptomatic groups, which did not differ from each other on these variables. However, N400 topography differed in mutation carrier groups with respect to the noncarriers. Intracranial source analysis evinced that the presymptomatic carriers presented a decrease of N400 generator strength in right inferior-temporal and medial cingulate areas and increased generator strength in the left hippocampus and parahippocampus as compared to the controls. This represents alterations in neural function without translation into behavioral impairments and may be an electrophysiological marker for Alzheimer disease. In these same families, Quiroz and colleagues used high-density ERPs to examine brain physiology in 10 presymptomatic subjects positive for the E280A PSEN1 mutation (carriers) and 11 siblings without the mutation (controls) (114). Subjects performed a visual recognition memory test while 128-channel ERPs were recorded. Despite identical behavioral performance, PSEN1 mutation carriers showed less positivity in frontal regions and more positivity in occipital regions compared to controls. These differences were more pronounced during the 200- to 300-msec period. Discriminant analysis at this time interval showed promising sensitivity (72.7%) and specificity (81.8%) of the ERP measures to predict the presence of Alzheimer disease pathology. These findings indicate that control subjects may use frontally mediated processes to distinguish between studied and unstudied visual items, whereas carriers appear to rely more on perceptual details of the items to distinguish between them. These findings also demonstrate the potential usefulness of ERP brain correlates as preclinical markers of Alzheimer disease.
Cognitive preclinical markers. Changes in lexical-semantic tasks and verbal expression could be cognitive markers in the preclinical phase of familial Alzheimer disease. Carriers of the E280A mutation in PS1 scored significantly lower than noncarriers on naming of famous faces and produced fewer semantic categories than noncarriers during the preclinical phase of Alzheimer disease describing the scene of the Cookie Theft Picture Card from the Boston Diagnostic Aphasia Examination (07; 32). Intrusive errors in verbal memory tests could be also considered as a preclinical marker of familial Alzheimer disease: nondemented carriers of E280A PS1 mutation presented more intrusive errors than asymptomatic noncarriers in the first and second trials and in the delay recall of the CERAD verbal memory task; they also had more intrusive errors than the demented carriers in the first trial and delay recall of the same task (143). Visual short-term memory binding deficits may be a preclinical marker for familial Alzheimer disease. Twenty-two patients with familial Alzheimer disease caused by the E280A single presenilin-1 mutation, 30 carriers of the mutation who did not meet Alzheimer disease criteria (asymptomatic carriers), and 30 healthy relatives (noncarrier healthy controls) were assessed with a visual short-term memory task and a neuropsychological battery (104). The short-term memory task assessed the recognition of shapes, colors, or shape-color bindings presented in two consecutive arrays (ie, study and test). Patients with Alzheimer disease and asymptomatic carriers performed significantly worse than healthy controls in the feature binding condition only. Classification and area under the curve analyses confirmed that the binding task combines more sensitivity and specificity for patients with Alzheimer disease, and most notably for asymptomatic carriers of the mutation, than other traditional neuropsychological measures.
A deficit in the coordination mechanism of the central executive may be another cognitive preclinical marker for the early detection of Alzheimer disease due to the E280A presenilin-1 gene mutation (88). Thirty-nine carriers of the E280A PS1 mutation who did not meet the criteria for Alzheimer disease and 29 noncarrier healthy controls were asked to perform digit recall accompanied by a secondary tracking task. Individuals who were carriers of the genetic mutation demonstrated significantly higher dual-task costs than healthy noncarriers. Dual task performance was found to be more sensitive to this very early stage of familial Alzheimer disease than episodic memory measures.
Preclinical biomarkers. Persons at risk for familial Alzheimer disease provide a model in which biomarkers can be studied in presymptomatic carriers. Ringman and colleagues found that Abeta(42) is elevated in plasma in familial Alzheimer disease mutation carriers, and this level may decrease with disease progression prior to the development of overt dementia (123). The ratio of Abeta(42) to Abeta(40) is reduced in the CSF of nondemented carriers who have elevations of t-tau and p-tau(181), likely sensitive indicators of presymptomatic disease. Ringman and colleagues also found elevated F(2)-isoprostane levels in the CSF of preclinical carriers (123). The FDA has cleared blood-based tests for Alzheimer disease that incorporate tau measurements, including assays of phosphorylated tau such as pTau217 (as a pTau217/β-amyloid 1-42 plasma ratio) and pTau181 in plasma, to aid in detecting or ruling out Alzheimer disease-related amyloid pathology in symptomatic adults (see FDA Press Announcement).
In another study that studied the preclinical phase of Alzheimer disease, 44 participants of a cohort from the Colombian Alzheimer's Prevention Initiative Registry, who were between 18 to 26 years of age and who carried the presenilin 1 (PSEN1) E280A mutation that causes early-onset Alzheimer disease (20 carriers and 24 noncarriers) were studied with structural and functional MRI (120). Compared with noncarriers, carriers had greater right hippocampal and parahippocampal activation, less precuneus and posterior cingulate deactivation, and less grey matter in several parietal regions. In the 20 participants (10 PSEN1 E280A mutation carriers and 10 noncarriers) who had lumbar punctures and venipunctures, mutation carriers had higher CSF Aβ(1-42) concentrations and plasma Aβ(1-42) concentrations than noncarriers. Young adults at genetic risk for autosomal dominant Alzheimer disease have functional and structural MRI findings and CSF and plasma biomarker findings consistent with Aβ(1-42) overproduction. In one prospective, longitudinal study using 128 participants, it was found that autosomal dominant Alzheimer disease was associated with a series of pathophysiological changes over decades in CSF biochemical markers of Alzheimer disease, brain amyloid deposition, and brain metabolism, as well as progressive cognitive impairment (13). Concentrations of amyloid-beta (Aβ)(42) in the CSF appeared to decline 25 years before expected symptom onset. Aβ deposition, as measured by positron-emission tomography with the use of Pittsburgh compound B, was detected 15 years before expected symptom onset. Increased concentrations of tau protein in the CSF and an increase in brain atrophy were also detected 15 years before expected symptom onset. Cerebral hypometabolism and impaired episodic memory were observed 10 years before expected symptom onset. Global cognitive impairment, as measured by the Mini-Mental State Examination and the Clinical Dementia Rating scale, was detected 5 years before expected symptom onset, and patients met diagnostic criteria for dementia at an average of 3 years after expected symptom onset.
PET and SPECT have revealed a typical pattern of metabolic deficits in the temporal and parietal lobes in Alzheimer disease. Additionally, numerous studies indicate that a similar deficit pattern may be observed in nondemented subjects at risk of developing the disease, among them, those with recognized genetic traits such as mutations associated with familial Alzheimer disease on chromosomes 21 and 14, subjects with the epsilon4 allele of the apolipoprotein E gene, and individuals with mild cognitive impairment. These findings might have implications when selecting patients for clinical trials (66). Regional cerebral perfusion abnormalities are detectable by means of SPECT studies before the development of clinical symptoms of Alzheimer disease in carriers of the E280A PS1 mutation (68). This is an important issue to be considered for future therapies in the preclinical stage of Alzheimer disease.
Presently, patients with familial Alzheimer disease and sporadic Alzheimer disease are similarly managed. However, it may become feasible to treat patients with the familial forms of the disease when they have memory complaints but not dementia, or those who are carriers of mutations of risk genes when they are still in the preclinical stage.
As in sporadic Alzheimer disease, no treatments can prevent or cure familial Alzheimer disease. When prescribing medications for the management of familial Alzheimer disease, clinicians should consider the individual's specific needs and medical history. Cholinesterase inhibitors, such as donepezil, galantamine, and rivastigmine, can improve quality of life and help prolong independence in patients with familial Alzheimer disease (156; 44; 105; 53). Memantine, an NMDA receptor modulator, can also be used to treat moderate to severe Alzheimer disease (105; 53). Combination therapy involving the administration of one cholinergic drug and one NMDA receptor modulator, such as memantine, may also be considered (156; 105). It is important to note that medications do not work for everyone, and they may lose effectiveness over time (156; 44). Clinicians should consider the individual's specific needs and medical history when prescribing medications for the management of familial Alzheimer disease.
Inhibition of production of amyloid peptides by inhibitors of beta and gamma secretases has been suggested as a possible future rational and more specific therapeutic approach. Additionally, the activation of alpha secretase would increase nonamyloidogenic processing of APP (36). Secretase inhibitors, including both beta- and gamma-secretase inhibitors, have so far failed to demonstrate convincing clinical efficacy and have often been limited by safety concerns in Alzheimer disease trials, whereas anti-amyloid monoclonal antibodies such as aducanumab, lecanemab, and donanemab have shown modest but statistically significant benefits on cognitive and functional decline in early symptomatic Alzheimer disease (159).
A future approach in therapy for many autosomal dominant diseases, such as familial Alzheimer disease, is to develop a method to specifically eliminate the aberrant protein. Duplex of 21-nt RNA, known as siRNA, is another possible tool to silence genes for neurodegenerative diseases of autosomal dominant inheritance (158).
These days, prenatal diagnosis of genetic conditions is available. Abortion is a choice open to women in some countries, such as the United States, though this now differs from state to state. They can decide on the termination of pregnancy when their fetuses are carriers of a known mutation for familial Alzheimer disease. However, it is desirable to discuss pregnancy termination on purely ethical grounds in the case of late-onset but severe diseases (112).
In a large Cuban family with early-onset familial Alzheimer disease, 56 first-degree relatives of familial cases of Alzheimer disease were interviewed concerning their attitudes toward the possibility of using presymptomatic genetic testing for Alzheimer disease. All of them would use such testing to know their own risk of Alzheimer disease. Fifty percent would choose not to have children if they themselves had the mutation. A positive prenatal test would lead 48.2% of the participants to have an abortion, whereas 19.7% would carry on their pregnancy regardless of the positive test result (90).
In another paper, the acceptability of presymptomatic testing was assessed in people at 50% risk for the APP-692 mutation causing early-onset familial Alzheimer disease. Thirty-nine people (64%) in the whole group would request presymptomatic testing if it were clinically available, although two thirds did not yet feel ready to take it. Most respondents believed that an unfavorable result would not adversely affect their personal mood or relationships (142).
Apparently, pregnancy poses no risk for the mother if her fetus carries a mutation in a gene that causes familial Alzheimer disease. Nevertheless, prenatal diagnosis will become more common in families with familial Alzheimer disease residing in countries where termination of pregnancy is accepted, and intrauterine therapies are feasible.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Fardin Nabizadeh MD
Mr. Nabizadeh of Iran University of Medical Sciences has no relevant financial relationships to disclose.
See Profile
Howard S Kirshner MD
Dr. Kirshner of Vanderbilt University School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026
General Child Neurology
Apr. 29, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Infectious Disorders
Apr. 08, 2026
Neurogenetic Disorders
Apr. 01, 2026