Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
GLUT1 deficiency syndrome is a metabolic disorder due to defective transport of glucose across the blood-brain barrier. First described by De Vivo in 1991, GLUT1 deficiency in its classic form presents in infancy with drug-resistant seizures and developmental delay. Dietary treatment can provide dramatic improvement in seizure control and may improve developmental outcome. Milder phenotypes have been described, including patients presenting with only paroxysmal dystonia.
|
• GLUT1 deficiency syndrome is a metabolic disorder due to defective transport of glucose across the blood-brain barrier. | |
|
• The classic picture is that of a child with refractory seizures starting in infancy, developmental delay, acquired microcephaly, hypotonia, and a movement disorder typically consisting of ataxia, spasticity, and dystonia. | |
|
• Increasingly, milder phenotypes are being recognized, including patients with normal cognition, early-onset absence or other generalized epilepsy, paroxysmal dystonia, or other movement disorders with and without epilepsy. | |
|
• Treatment with a ketogenic diet or a modified Atkins diet can lead to rapid seizure control and should be initiated as early as possible to provide the best possible outcome. |
GLUT1 deficiency syndrome (GLUT1-DS) is a metabolic disorder first described by De Vivo and colleagues in 1991 and is, therefore, sometimes called De Vivo disease (22). They noted two children with seizures and developmental delay whose spinal fluid demonstrated persistent low glucose concentrations (hypoglycorrhachia) in the face of a normal peripheral glucose concentration. A defect in the transport of glucose across the blood-brain barrier was suspected, and the authors hypothesized that these children needed an alternative energy source for brain metabolism. Both children were started on a ketogenic diet, which provides energy in the form of ketones in place of glucose. The children stopped having seizures within a few days of starting the diet.
Seven years later, the same group described heterogeneous mutations leading to functional defects of the type 1 glucose transporter protein (GLUT1), which is the glucose transporter most widely distributed in human brain and erythrocytes (91). The gene coding for this protein (known as SLC2A1 for solute carrier 2 A, type 1) had previously been mapped to the short arm of chromosome 1 (65). More than 100 different mutations in SCL2A1 have since been described (42).
There is a wide range of phenotypes for this treatable disease (04; 76; 18). By 2005, the classic phenotype, including infantile seizures, acquired microcephaly, hypotonia, ataxia, and developmental delay, was well established, and treatment with the ketogenic diet had emerged as the gold standard therapy for these children. Since 2008, many reports of patients with GLUT1 deficiency syndrome who presented with “atypical” features, namely idiopathic generalized epilepsy and early-onset absence epilepsy (before 4 years of age) have appeared, often in individuals with normal or only mildly impaired cognition (84; 12). Patients with a broad spectrum of movement disorders have also been reported, with and without epilepsy, ranging from mild tremor to dystonia and chorea (78).
|
• The classic presentation of GLUT1 deficiency syndrome is that of a child born with relatively normal birth history and without perinatal complications, who develops normally during the first few months of life, but then develops seizures, typically between 1 and 8 months of age. | |
|
• Seizures can be of various types and often are not immediately recognized as such because they are often brief and subtle. | |
|
• Infants with GLUT1 deficiency syndrome have a normal head size at birth, but the growth of the brain and skull is slow, in severe cases resulting in an abnormally small head size. | |
|
• Children with GLUT1 deficiency syndrome who have severe developmental delay also tend to have acquired microcephaly. | |
|
• Abnormal movements tend to become more prominent in adolescence, and patients are increasingly being recognized who have movement disorders of varying severity without epilepsy. | |
|
• The patients typically have an abnormal gait due to ataxia and spasticity, and they may have dyspraxia, dystonic posturing or generalized dystonia, intention tremor, chorea, dysarthria, myoclonus, and oculogyric crises. | |
|
• Paroxysmal exertional dyskinesia (also known as DYT18 in the catalogue of genetic dystonia syndromes) and the syndrome of paroxysmal choreoathetosis/spasticity (DYT9) are due to mutations in the GLUT1 gene. |
GLUT1 deficiency syndrome is caused by a defect in the function of the type 1 glucose transporter protein and is usually inherited in an autosomal dominant trait (although rare cases of autosomal recessive inheritance have also been reported).
GLUT1 deficiency syndrome causes a wide range of manifestations that include infantile-onset chronic encephalopathy, infantile seizures refractory to anticonvulsive drugs, developmental delay, acquired microcephaly, facial dysmorphism, and neurologic manifestations including intellectual disability, spasticity (and specifically spastic paraparesis), hypotonia, ataxia, nonepileptic paroxysmal movement disorders, and various other movement disorders (22; 100; 28; 31; 18; 19; 96; 02). There is a wide phenotypic spectrum and considerable intrafamilial variation (99; 19; 13; 48). The clinical manifestations of GLUT1 deficiency syndrome primarily include seizures, movement disorders, and developmental delay (111). Clinical manifestations may be continuous, paroxysmal, or continual with fluctuating severity. Even within the same family there may be considerable phenotypic variability among affected members with the same mutation in the SCL2A1 gene (68; 109).
In a series of 37 genetically confirmed cases, the most common presenting clinical manifestations were epilepsy (84%), intellectual disabilities (78%), and movement disorders (76%) (96). Paroxysmal dyskinesias occurred in 27% but were more frequently seen in familial cases (55%) than in sporadic cases (15%) (96). Glut1 deficiency syndrome patients with intellectual disabilities typically had either epilepsy or movement disorders.
Seizures. The classic presentation of GLUT1 deficiency syndrome is that of a child born with relatively normal birth history and without perinatal complications, who develops normally during the first few months of life, but then develops seizures, typically between 1 and 8 months of age. Seizures can be of various types and often are not immediately recognized as such because they are often brief and subtle. They may include abnormal eye movements, sudden pallor, limpness, and head nodding. They then tend to evolve into other seizure types, most commonly generalized tonic-clonic, absence (typical and atypical), myoclonic, or, less commonly, partial-onset seizures (95; 76; 110; 52; 54; 34; 39; 87). One case has been reported with infantile spasms (54). SLC2A1 mutations are common in individuals with absence epilepsy of early onset, being detected in 5 of 50 patients (10%) in one population-based study in Denmark (52); the greater the number of atypical characteristics in affected patients with absence seizures, the more likely they are to have an SLC2A1 mutation (93). Seizures may worsen with fasting (79). The seizures are often resistant to standard anticonvulsants, and phenobarbital in particular may worsen the seizures (47).
Milder and atypical phenotypes are being recognized with increasing frequency (07; 79; 13). One study found that one in 10 children diagnosed with early-onset absence epilepsy had GLUT1 deficiency syndrome, as did 1% of patients with other idiopathic generalized epilepsies; some of these patients may have good seizure control with standard anticonvulsants (07). Seizures that worsen with fasting or improve with steroid treatment may also be suggestive of GLUT1 deficiency (11). About 10% of affected individuals have nonepileptic GLUT1 deficiency syndrome, which is usually less severe than the common form, though still often manifesting developmental delay and intellectual disability.
Developmental delay and intellectual disability. Infants with GLUT1 deficiency syndrome have a normal head size at birth, but the growth of the brain and skull is slow, in severe cases resulting in an abnormally small head size (100). In children with the classic phenotype, global developmental delay with intellectual disability becomes apparent by 1 year of age (10). Although most of the children learn to walk, they tend to be ataxic and remain nonverbal (76). In a series of 57 affected children, all were found to have intellectual disability, with 36% in the mild range, 44% in the moderate range, and 17% in the severe range (55). However, affected patients with normal or near-normal cognition are being identified with increasing frequency.
Children with GLUT1 deficiency syndrome who have severe developmental delay also tend to have acquired microcephaly (100).
In a Dutch cross-sectional study of 54 patients, 6.5% had microcephaly (more than 2 standard deviations below the normal mean), and head circumference was below the normal mean in 76% of cases (101).
Movement disorder. Abnormal movements tend to become more prominent in adolescence, and patients are increasingly being recognized who have movement disorders of varying severity without epilepsy. The patients typically have an abnormal gait due to ataxia and spasticity, and may have dyspraxia, dystonic posturing or generalized dystonia, intention tremor, chorea, dysarthria, myoclonus, and oculogyric crises (19; 33; 10). The gait pattern of GLUT1 deficiency syndrome patients is marked by reduced fluidity, stability, and smoothness (16).
Paroxysmal exertional dyskinesia (also known as DYT18 in the catalogue of genetic dystonia syndromes) and the syndrome of paroxysmal choreoathetosis/spasticity (DYT9) are due to mutations in the GLUT1 gene (95; 106; 105; 13; 06). Paroxysmal exertional dyskinesia can occur in isolation or in association with epilepsy (95; 19; 13; 06). Paroxysmal exertional dyskinesia may manifest with choreoathetosis, dystonia, or both, and affects mainly the legs (95; 06). Adult-onset nonkinesigenic paroxysmal dyskinesia in GLUT1 deficiency syndrome has also been reported (02).
Episodic ataxia secondary to GLUT1 deficiency may occur with or without associated seizures or other neurologic defects (97). Episodic ataxia secondary to GLUT1 deficiency may respond to acetazolamide therapy (97).
Nearly 90% of patients with GLUT1 deficiency have paroxysmal or constant gait abnormalities, including ataxic, spastic, ataxic-spastic, and dystonic gaits (78). A paroxysmal gait disorder triggered by exertion or fasting, named “criss-cross gait” has also been described, which is characterized by lower-body choreo-dyskinesias, causing the legs to intersect repeatedly, producing irregular steps combined with intermittent loss of balance and compensatory upper-body movements (59).
Most patients, with or without seizures, have exacerbation of symptoms with fasting and improvement with a ketogenic diet (50).
Hereditary spastic paraparesis. An Italian family has been described with spastic paraplegia, often accompanied by epilepsy and intellectual disability, inherited as an autosomal dominant trait (24). Although a novel variant of hereditary spastic paraplegia was initially suspected, linkage studies of known hereditary spastic paraplegia genes did not identify the genetic cause. An exome-sequencing study identified a p.Arg126Cys mutation in the SLC2A1 gene, encoding GLUT1, which segregated with the paraplegia trait (24). The diagnosis of GLUT1 deficiency syndrome was confirmed by cerebrospinal fluid analysis, demonstrating hypoglycorrhachia (24).
Other neurologic manifestations. A 4-generation Norwegian family with a mild phenotype of GLUT1 deficiency has been reported in which all affected members had early-onset epilepsy and paroxysmal exercise-induced dyskinesias, most had mild learning disability, and some had a distal neuropathy, a reduced sense of orientation, and excessive daytime sleep (80). Some may have a sleep disorder as part of their presentation, including excessive daytime sleepiness, insomnia, and restless sleep (06). Other manifestations include dysarthria (15) and hemiplegic migraine (10).
Adults and children with GLUT1 deficiency may experience stroke or stroke-like events with abrupt onset of neurologic symptoms and signs. In 42 patients with GLUT1 deficiency, eight individuals between the ages of 6 and 38 presented with an acute onset of neurologic disturbances: dysarthria/aphasia, oral dyskinesia, swallowing difficulties, paresthesia, facial palsy, hemi/monoplegia, vomiting, headache, and behavioral disturbances (71). When performed, MRI typically showed signs of venous congestion and hypoperfusion, although MRI demonstrated the presence of an ischemic brain lesion in two patients. Electroencephalography showed focal contralateral slowing. Deficits were transient in all patients except one. Four patients (50%) were on a ketogenic diet, but two had lower than usual ketonemia levels during the episode.
Non-neurologic features. At least four patients with GLUT1 deficiency have been described with hemolytic anemia associated with erythrocyte cation leak (106; 25; 08; 19). One of these patients also had paroxysmal exertional dyskinesia and epilepsy; the others had a multisystem disorder with hemolysis, hepatomegaly, and cataracts in addition to seizures and developmental delay.
Pseudohyperkalemia due to cryohydrocytosis. Cryohydrocytosis is a form of stomatocytosis, a rare condition of red blood cells (in which a mouth-like or slit-like pattern replaces the normal central zone of pallor). Cryohydrocytosis is characterized by a temperature-dependent anomaly in red cell membrane permeability causing the leakage of sodium and potassium from red blood cells at low temperatures, leading to high in vitro potassium levels in blood samples stored below 37°C, ie, pseudohyperkalemia.
Stomatin (human erythrocyte integral membrane protein band 7) is an integral membrane protein that localizes to the cell membrane of red blood cells, where it regulates ion channels and transporters.
Stomatin-deficient cryohydrocytosis is an extremely rare variant that is associated with pathogenic variants in the SLC2A1 gene, encoding GLUT1. Consequently, GLUT1 deficiency syndrome may present with stomatin-deficient cryohydrocytosis (27).
Other clinically significant associations of GLUT1. GLUTs are often deregulated in cancer (37; 58; 01). Increased glucose transport in malignant cells is associated with increased and deregulated expression of glucose transporter proteins, characteristically with overexpression of GLUT1 and/or GLUT3 (92; 58). GLUT1 expression is seen in most cases of lung carcinoma, particularly in squamous cell lung carcinoma (37). GLUT1 expression in adenocarcinoma of the lung is associated with reduced cell differentiation, larger tumor size, and lymph node metastases (37). High levels of GLUT1 expression in tumors are associated with poor survival (58). The identification and targeting of tumor-specific GLUTs may lead to therapies that impair glucose-regulated metabolism and signaling in cancer cells.
GLUT1 is also a receptor used by the human T cell leukemia virus (HTLV) virus to enter target cells (60). The receptor-binding domains of both HTLV-1 and HTLV-2 envelope glycoproteins inhibit glucose transport by interacting with GLUT-1 (60). Receptor binding and HTLV infection are selectively inhibited when glucose transport or GLUT-1 expression are blocked (60). Neurologic manifestations of HTLV infection are likely, at least in part, the result of disturbed CNS glucose metabolism resulting from interactions of HTLV envelope glycoproteins with GLUT-1.
GLUT1 is also a powerful histochemical marker for juvenile hemangiomas (70).
Children with GLUT1 deficiency and early-onset seizures typically have severe developmental delay that leads to intellectual disability. Most eventually learn to walk but tend to remain nonverbal. This has been the experience of children with the classic phenotype. However, there is often a long delay between the onset of seizures and diagnosis (28; 77); an average 6.6-year delay was reported in a large series (77), and some individuals are not diagnosed until adulthood, despite severe clinical manifestations (28). Earlier diagnosis and treatment may improve the developmental outcome (81; 34).
The long-term outcome is not yet known in those with milder phenotypes. However, most have an excellent response to the ketogenic diet or the modified Atkins diet.
An infant born at term with normal prenatal history and without complications developed seizures at 5 months of age that consisted of “strange eye movements, darting back and forth,” lasting for 2 to 3 minutes, one to two times per week. At 9 months of age, these features were replaced by new spells consisting of lip smacking associated with myoclonic jerking of all extremities without loss of consciousness. Treatment with phenobarbital was ineffective. The myoclonic jerks disappeared at 12 months of age, when they were replaced by generalized tonic-clonic seizures lasting for up to 10 minutes, occurring daily to several times per week. Phenobarbital was switched to carbamazepine, again without success, and the patient was then changed to levetiracetam at the age of 2. Valproate was added a year later, and levetiracetam was eventually replaced by topiramate, but he continued to have frequent seizures.
Developmentally, he crawled at 12 months and walked at 3 years. At the age of 8, he finally spoke a few words, including “Mom.”
Work-up included normal MRI of the brain. Initial EEG showed only mild background slowing. Subsequent video-EEG monitoring demonstrated diffuse background slowing and intermittent high-amplitude bursts of generalized spikes lasting 3 to 5 seconds. Urinary organic acid and plasma amino acid analyses, carnitine/acylcarnitine profile, plasma lactate, and ammonia concentrations were normal. The following tests were all negative: single-nucleotide microarray; fragile X syndrome testing; methylation studies for Angelman syndrome; and UBE3A, SCN1-A, 22q11, and SLC9A6 testing for Pitt-Hopkins syndrome.
A lumbar puncture was performed at 7 years of age, which demonstrated normal neurotransmitter metabolites and pterins, but CSF glucose concentration was 24 and plasma glucose concentration was 88. GLUT1 deficiency syndrome was confirmed by the presence of a missense mutation at c376 in the SLC2A1 gene.
The child was started on the ketogenic diet and became seizure-free and had improvements in both speech and gait.
|
• The GLUT gene family of transporter proteins facilitates passive diffusion of glucose across tissue barriers by energy-independent mechanisms. | |
|
• GLUT1 deficiency syndrome is caused by a defect in the function of the type 1 glucose transporter protein. In 70% to 80% of cases, a mutation of the SLC2A1 gene on chromosome 1p34.2 has been identified. |
Glucose transporter 1 (or GLUT1), also known as "solute carrier family 2, facilitated glucose transporter member 1" (SLC2A1), is a uniporter protein—a membrane transport protein that transports a single species of substrate across a cell membrane—that is encoded by the SLC2A1 gene. GLUT1 facilitates the transport of glucose across the plasma membranes of mammalian cells and in particular is a major glucose transporter across the mammalian blood-brain barrier. GLUT1 is responsible both for the transport of glucose from the bloodstream into the extracellular cleft and from the extracellular cleft into astrocytes (103). The encoded protein is found primarily in the cell membrane and on the cell surface. GLUT1 expression occurs in almost all tissues, but the degree of expression varies with the rate of cellular glucose metabolism.
Collectively, the GLUT gene family of transporter proteins facilitates passive diffusion of glucose across tissue barriers by energy-independent mechanisms (72). The GLUT gene family is part of a superfamily of 12 transport facilitators designated SLC2A (solute carrier 2A). GLUT1 encoded by the SLC2A1 gene on chromosome 1p34.2 is widely expressed in fetal tissues, and in adults it is highly expressed in erythrocytes and also in the endothelial cells of barrier tissues such as the blood–brain barrier (74).
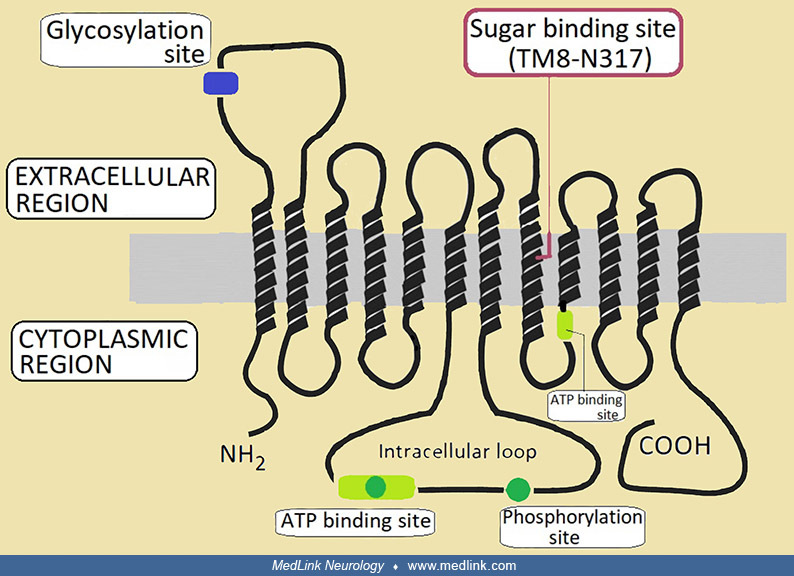
GLUT1 has 12 hydrophobic segments, each of which forms a membrane-spanning alpha helix. The side-by-side assembly of several helices produces a transmembrane channel lined with hydrophilic residues that can hydrogen bond with glucose as it moves through the channel (36; 23; 103). The hydrophobic regions remain on the outside of the channel adjacent to the fatty acid tails of the membrane (103).
Glucose transporters (GLUT): GLUT1 (green) is expressed in endothelial cells (part of the blood-brain barrier) and in the glial cell membrane; GLUT3 (yellow) is expressed in the neuron membrane. Monocarboxylate transporter fami...
Levels of GLUT1 in cell membranes are increased by reduced glucose levels and decreased by increased glucose levels.
As part of a vitamin C recycling process, GLUT1 is also a major receptor for uptake of vitamin C, especially in humans and other mammals that cannot synthesize vitamin C (64).
Classically, GLUT1 deficiency syndrome is caused by a defect in the function of the type 1 glucose transporter protein. In 70% to 80% of cases, a mutation of the SLC2A1 gene on chromosome 1p34.2 has been identified. Most cases (approximately 90%) are due to a de novo mutation, whereas about 10% are due to autosomal dominant mutations with the parent only mildly affected. The disorder is usually inherited in an autosomal dominant trait (95), but rare cases of autosomal recessive inheritance have also been reported (45; 83).
A variety of different mutations have been identified (46; 95).
A genetic diagnosis cannot be made in 10% of patients with GLUT1 deficiency syndrome (108). Using whole-genome sequencing, a de novo 5'-UTR variant has been identified in SLC2A1 that generates a novel translation initiation codon, severely compromising SLC2A1 function (108). Thus, impaired upstream SLC2A1 translation initiation can cause GLUT1 deficiency syndrome (108).
In GLUT1 deficiency syndrome, GLUT1 expression is reduced in red blood cells of affected patients (30).
Glucose, after entering the brain, can follow three metabolic pathways: (1) it may be converted to pyruvate and energy via glycolysis; (2) the pyruvate may be converted further via the Krebs citric acid cycle to much larger amounts of energy; or (3) the glucose may enter the pentose phosphate shunt pathway, which is important for amino acid and nucleotide synthesis, fatty acid synthesis, and glycogen storage. Decreased entry of glucose into the brain limits the activity in these metabolic pathways and was, therefore, thought to constitute the major pathophysiology of GLUT1 deficiency syndrome (74). Functional imaging studies have identified alterations in glucose metabolism in the corticostriate pathways in affected GLUT1 deficiency syndrome patients with paroxysmal exercise-induced dyskinesia and in the frontal lobe cortex in those with epileptic seizures (95).
Metabolic adaptations and epigenetic reprogramming are involved in macrophage plasticity and phenotype change from monocyte to activated monocytes and then the macrophages (103). Glut1 is necessary for the increased glucose uptake, increased glycolysis, and increased synthesis of lipids, proteins, and nucleotides (103). Glut1 is also required for the homeostasis of T and B lymphocytes. Glut1 deficiency selectively impairs thymocyte and T effector functions, and serum levels of antibodies are significantly lower, favoring the development of severe infections (103).
The transition of monocytes to macrophages is characterized by increased GLUT1 expression and glycolysis. Naive (unactivated) monocytes are metabolically quiescent, with low basal metabolic activity and ATP derived primarily vi...
Animal models of GLUT1 deficiency. In creating the Glut1-deficient mouse, a transgenic antisense mouse model for GLUT1 haploinsufficiency, mouse embryos with homozygous mutations developed a severe embryopathy with various neurologic malformations, including microcephaly, anencephaly, anophthalmia, and caudal regression, generally incompatible with life (35). Mice with hemizygous mutations demonstrated both ataxia and CNS hyperexcitability brought on by fasting (104).
In a heterozygous Glut1-deficient mouse model, expression of the GLUT1 protein was reduced by 31% to 50% in the thalamus and cortex compared to control animals (61); deoxyglucose deposition was also greatly reduced in cortex. On EEG, Glut1-deficient mice had very frequent epileptic spikes and spike-wave activity. Contrary to expectations, concentrations of Krebs cycle intermediates and neurotransmitters were similar to those of control animals whereas concentrations of fatty acids and acetyl-coenzyme A were lower than normal in the Glut1-deficient animals. GLUT1-deficient brain appears to use up available acetyl-CoA with a negative impact on myelin synthesis and deposition, while preserving whole-brain Krebs cycle contents and neurotransmitters. Thus, energy failure may not be the critical element it was previously thought to be GLUT1 deficiency syndrome.
|
• GLUT1 deficiency syndrome is a rare condition. |
GLUT1 deficiency syndrome is a rare condition with only about 500 cases reported. In 2006, even before milder phenotypes were recognized, an Australian study suggested a prevalence of 1:90,000 (14). A study in Denmark found a similar prevalence of SLC2A1 mutations of approximately 1:83,000 (52). Milder presentations may still be significantly underrecognized.
|
• Because most cases are due to spontaneous mutations, genetic counseling typically does not help prevent the disorder. | |
|
• For families in which a parent has an identified disease-causing mutation, prenatal testing is available via amniocentesis or chorionic villus sampling. | |
|
• Early diagnosis and treatment with a ketogenic or a modified Atkins diet effectively controls epilepsy, improves movement disorders, and may help to improve long-term outcome in affected individuals. |
Because most cases are due to spontaneous mutations, genetic counseling typically does not help prevent the disorder. However, for families in which a parent has an identified disease-causing mutation, prenatal testing is available via amniocentesis or chorionic villus sampling. Preimplantation genetic diagnosis may be an option for some families.
Early diagnosis and treatment with a ketogenic or a modified Atkins diet effectively controls epilepsy, improves movement disorders, and may help to improve long-term outcome in affected individuals (81).
The differential diagnosis for early-onset epileptic encephalopathy, developmental delay, and movement disorders is vast and includes a wide variety of metabolic and genetic disorders.
A low CSF glucose concentration (hypoglycorrhachia) in the presence of normal peripheral glucose concentration is much less common, but may be seen in cases of the following:
|
Bacterial or fungal meningitis |
|
• GLUT1 deficiency syndrome is likely underdiagnosed due to its complex and pleiotropic phenotype. | |
|
• GLUT1 deficiency should be considered in patients with early-onset generalized epilepsy or paroxysmal movement disorders, particularly in those with early-onset seizures of any type combined with developmental delay; drug-resistant seizures; early-onset absence seizures; absence seizures associated with irregular ictal EEG discharges, mild intellectual disability, migraine, or microcephaly; exercise-induced dyskinesias; or fasting-induced worsening of seizures of movement disorders. | |
|
• Diagnosis of GLUT1 deficiency syndrome is confirmed by the presence of characteristic clinical signs in association with hypoglycorrhachia documented by lumbar puncture, and genetic analysis showing pathogenic SLC2A1 variants. | |
|
• A CSF-to-blood glucose concentration ratio less than 0.5 (and often as low as 0.33 to 0.37) and a CSF glucose concentration of less than 40 mg/dl (2.2 mmol) are strongly suggestive of GLUT1 deficiency syndrome. A ratio less than 0.4 is considered diagnostic, but this cutoff is not very sensitive (59). | |
|
• Approximately 70% to 80% of patients have mutations in the SLC2A1 gene. | |
|
• An abnormally low erythrocyte 3-O-methyl-D-glucose uptake assay confirms the clinical diagnosis of GLUT1 deficiency syndrome. | |
|
• The EEG may be normal interictally, or it may demonstrate mild to moderate background slowing, or generalized spike-wave complexes. | |
|
• Brain MRI is usually normal but may show mild atrophy, delayed myelination, or other white matter abnormalities. |
GLUT1 deficiency syndrome is likely underdiagnosed due to its complex and pleiotropic phenotype (ie, one gene influences two or more seemingly unrelated phenotypic traits) (31). It should be considered in patients with early-onset generalized epilepsy or paroxysmal movement disorders (31), particularly in those with early-onset seizures of any type combined with developmental delay; drug-resistant seizures; early-onset absence seizures; absence seizures associated with irregular ictal EEG discharges, mild intellectual disability, migraine, or microcephaly; exercise-induced dyskinesias; or fasting-induced worsening of seizures of movement disorders (31; 79; 82).
Diagnosis of GLUT1 deficiency syndrome is confirmed by the presence of characteristic clinical signs in association with hypoglycorrhachia documented by lumbar puncture, and genetic analysis showing pathogenic SLC2A1 variants (43; 103). Lumbar puncture is important when investigating a child with epileptic seizures and abnormal gait or developmental delay to avoid missing treatable neurometabolic conditions, such as Glut1 deficiency syndrome (05).
Lumbar puncture. Most affected children had CSF glucose levels below 40 mg/dl (2.2 mmol/L) (111). CSF-to-blood glucose concentration ratio of less than 0.5 (and often as low as 0.33 to 0.37) and a CSF glucose concentration of less than 40 mg/dl (2.2 mmol) are strongly suggestive of GLUT1 deficiency syndrome (95; 20; 62). With increasing recognition of milder variants, higher values are noted, with ratios up to 0.60 and CSF glucose concentrations of up to 64 mg/dl (3.6 mmol) (95), but in most classic cases, the ratio is below 0.37 (20).
CSF lactate is generally low or low normal, never high (42).
Genetic testing. Approximately 70% to 80% of patients have mutations in the SLC2A1 gene (42). SLC2A1 molecular analysis should be performed in patients with a family history of Glut1 deficiency syndrome or with a low CSF glucose level and at least one of the following: epilepsy and movement disorder (including paroxysmal dyskinesia) (96). Genetic testing is commercially available and should include sequencing of all 10 exons, splice site, and the promoter region. If this is negative, deletions/duplications within the SCL2A1 gene can be detected by multiplex ligation-dependent probe amplification (MLPA) (56). Extending genetic screening to noncoding regions will enable diagnosis of some additional patients with GLUT1 deficiency because pathogenic deep intronic SLC2A1 variants have been identified (57).
GLUT1 deficiency syndrome may be caused by mutations in genes other than SLC2A1 in patients with a compatible phenotype, low CSF glucose level, and good response to the ketogenic diet (40; 86). These other genes include SLC6A1, SLC9A6, KCNQ2, and NALCN. Patients with SLC2A1 mutations have marked phenotypic variability that overlaps with the phenotypes of patients without SLC2A1 mutations to the point of behaving as phenocopies (40).
Massive sequencing identified a presumably severe novel hemizygous mutation in the SLC9A6 gene (c.803+1G> A), which encodes sodium-proton exchanger SLC9A6; alterations in the gene may cause Christianson syndrome, an X-linked disorder (Xq26.3) characterized by severe intellectual disability, absent language development, autistic spectrum disorder, epilepsy, microcephaly, late-onset ataxia, muscle weakness, and dystonia. A genetic study detected the same mutation in his mother.
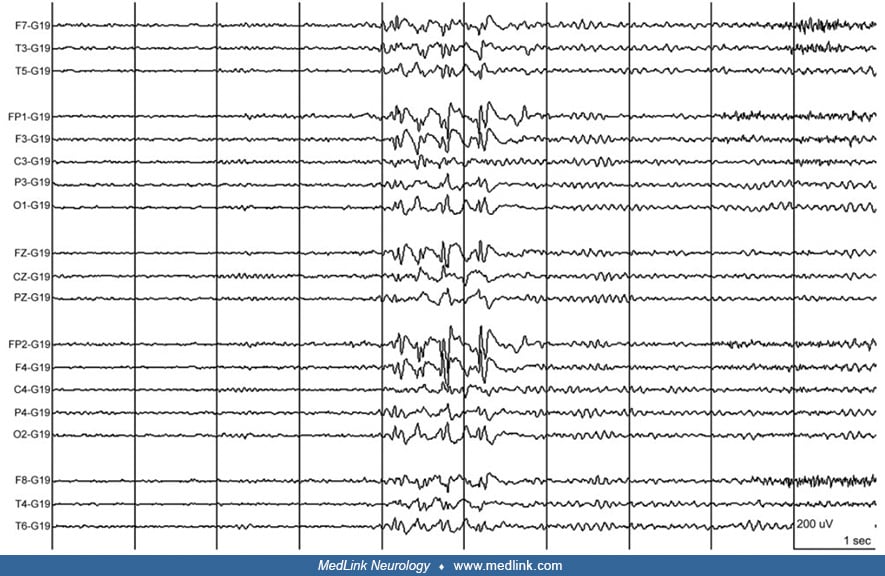
EEG. There is no one characteristic EEG pattern for GLUT1 deficiency syndrome. The EEG may be normal interictally, or it may demonstrate mild to moderate background slowing, or generalized spike-wave complexes (95).
Infants will often present with multifocal spike discharges on EEG; later, the EEG evolves into a pattern of generalized spike-wave discharges with a frequency of 2.5 to 3 Hz (53; 29). Hyperventilation may elicit absence seizures with high-voltage 3Hz generalized spike-wave discharges on EEG (95).
Some authors have reported improvement in EEG after eating or after intravenous glucose administration (102).
Brain imaging. Brain MRI is usually normal but may show mild atrophy, delayed myelination (44), or other white matter abnormalities (34).
The age-adjusted lenticular nuclei/thalami radioactivity ratio on PET could distinguish patients with GLUT1 deficiency syndrome from patients with epilepsy of unknown etiology with high accuracy in a small sample (eight patients and 45 controls) (69), but it remains to be established that these results are replicated in other samples and that this adds significantly to the other diagnostic methods available.
• The ketogenic diet, a high-fat, very-low carbohydrate regimen, is the gold standard of treatment for GLUT1 deficiency syndrome. | |
• Triheptanoin can favorably influence cardinal aspects of neural function in GLUT1 deficiency syndrome, although it may not reduce seizure frequency in affected individuals who are not on a ketogenic diet. | |
• Phenobarbital and valproic acid should be avoided because they may inhibit GLUT1 activity, and valproate also partially inhibits fatty acid oxidation. |
Ketogenic diet. The ketogenic diet, a high-fat, very-low carbohydrate regimen, has been the gold standard of treatment for GLUT1 deficiency syndrome since the condition was first described (22; 41; 31; 82; 89; 30; 97; 93; 107; 43; 90; 49; 51; 85; 75; 103; 98). Through severe restriction of carbohydrates, it mimics the metabolic state of fasting by forcing the consumption of fat and by providing ketones as a fuel source in place of glucose. The classic 4:1 ketogenic diet, which uses a ratio of 4 grams of fat for every 1 gram of protein and carbohydrate combined, is widely used for other forms of intractable epilepsy and is highly effective in GLUT1 deficiency syndrome. Many patients with GLUT1 deficiency syndrome have more than 90% reductions in seizure frequency and some are seizure-free on the ketogenic diet (41; 82; 88; 26), though the effects on neurodevelopment and on the movement disorder are fewer (41), with some studies suggesting only equivocal benefit on intellectual development (26).
Other forms of ketogenic diet have also been used, including the modified Atkins diet, which is a stricter version of the well-known weight-control diet, providing 10% carbohydrates, 30% protein, and 60% fats without restriction of calories or fluids (32; 82; 34; 85). Experience suggests that the modified Atkins diet may equal the classic ketogenic diet in controlling seizures and other symptoms of GLUT1 deficiency syndrome, with less severe dietary restriction and more palatability (38; 32; 82; 03; 26; 80; 85), particularly in cases having a mild phenotype (80). In some patients, a switch from the ketogenic diet to a modified Adkins diet produces improvements in physical abilities in growth without any exacerbation or recurrence of symptoms (03).
Ketogenic diets may also have a positive effect on GLUT1 deficiency-associated movement disorders (81; 82; 97). The impact on developmental delay is less well established, but studies suggest that early introduction of the ketogenic diet in the first years of life provides better cognitive outcomes (81; 82). Early diagnosis and dietary treatment are important to prevent developmental delay (82). Adults with GLUT1 deficiency syndrome may also benefit from dietary treatment by improving alertness (82).
In a study of 270 patients with GLUT1 deficiency syndrome with dietary treatment followed for a mean of 53 months, the ketogenic diet was generally well tolerated with minimal side effects, although 18% reported poor compliance (90). The results of treatment were excellent: epilepsy improved for 83% of 230 patients; movement disorders improved for 82% of 127 patients; and cognition improved for 59% of 58 patients (90). Effects of the ketogenic diet on epilepsy were seen within days/weeks and were most pronounced in patients with early treatment initiation. Effects of the ketogenic diet on epilepsy on movement disorders were noticed within months and were strongest in patients with higher CSF-to-blood glucose ratio. Among treated patients, symptoms worsened with low ketosis, poor compliance, or treatment discontinuation.
In contradistinction to previous reports focusing on children with intractable epilepsy, limited clinical experience maintaining adults with GLUT1 deficiency syndrome on a ketogenic diet for more than 5 years suggests that this diet does not negatively affect body composition, bone mineral content, or bone mineral density (09).
EEG did not prove to be reliable tool for adjusting the ketogenic diet in these patients (98).
One 4-year-old boy with GLUT1 deficiency and episodic ataxia, but without seizures or other baseline abnormalities on neurologic examination, had a more than 75% improvement in frequency and severity of episodes with acetazolamide therapy (97); his symptoms subsequently resolved with initiation of the ketogenic diet despite discontinuation of acetazolamide (97).
Alpha-lipoic acid. Alpha-lipoic acid is an antioxidant that serves as a coenzyme in energy metabolism. It has been recommended in GLUT1 deficiency syndrome because it improves glucose transport in muscle cell cultures (21). However, there are currently no clinical data in humans to support the use of alpha-lipoic acid as a treatment for GLUT1 deficiency syndrome.
Gene therapy. Gene therapy in a mouse model suggests that this will be an effective human therapy in the future (67; 66). In heterozygous knock-out murine Glut1 mice, an adeno-associated virus vector in which the human SLC2A1 gene was expressed under the synapsin I promoter produced exogenous GLUT1 in neural cells and also improved CSF glucose levels (67). Intracerebroventricular injection of an adeno-associated virus vector, in which the human SLC2A1 gene encoding GLUT1 was expressed under the human endogenous GLUT1 promoter (AAV-GLUT1), produced strong expression of exogenous GLUT1 in the cerebral cortex, hippocampus, and thalamus in GLUT1-deficient mice (66). Exogenous GLUT1 was mainly expressed in endothelial cells and partially expressed in neural cells and oligodendrocytes. CSF glucose levels and motor function were significantly improved.
Unhelpful therapies.
Phenobarbital and valproic acid. Phenobarbital and valproic acid should be avoided because they may inhibit GLUT1 activity; valproate also partially inhibits fatty acid oxidation (45). Carbonic anhydrase inhibitors, such as acetazolamide or zonisamide, can be effective for seizure control (99), though some advocate avoiding these agents because they can worsen metabolic acidosis and promote kidney stone formation in patients on the ketogenic diet.
Deep brain stimulation. In one patient with GLUT1 deficiency and progressive generalized dystonia since age 25 who underwent deep brain stimulation of the globus pallidus internus at age 44 (prior to diagnosis of GLUT1 deficiency), there was marginal therapeutic response despite proper electrode localization and sufficient stimulation programming. Genetic testing at age 53 ultimately identified a novel heterozygous pathogenic SLC2A1 mutation as the cause of glucose transporter type 1 deficiency syndrome (33).
Triheptanoin. Triheptanoin is a triglyceride that is broken down into carbon compounds, which can easily cross the blood-brain barrier and may enhance the effect of regular ketones as an alternative fuel for the brain (42). Early open-label studies reported that triheptanoin can favorably influence cardinal aspects of neural function in GLUT1 deficiency syndrome (73). In a case series of 14 children and adults with GLUT1 deficiency syndrome who were not receiving a ketogenic diet, supplementation of the regular diet with triheptanoin reportedly decreased spike-waves by 70% in 13 patients (73). In addition, neuropsychological performance and cerebral metabolic rate increased in most patients. Triheptanoin was generally well tolerated: 11 patients (78%) had no adverse effects after prolonged use, whereas three patients (21%) experienced gastrointestinal symptoms, and one (7%) discontinued the use of triheptanoin. In another open-label study among eight patients who objected to or did not tolerate ketogenic diets, triheptanoin treatment produced a 90% clinical improvement in nonepileptic paroxysmal manifestations (63).
However, subsequent randomized, double-blind studies found that triheptanoin did not significantly reduce seizure frequency or disabling movement disorder events in patients with Glut1 deficiency syndrome who were not on the ketogenic diet (94; 17). Triheptanoin treatment was associated with mild-to-moderate gastrointestinal treatment–related events, but most of these resolved following dose reduction or interruption (94; 17). Triheptanoin was not associated with any long-term safety concerns when administered at dose levels up to 35% of total daily caloric intake for up to 1 year (94).
Children with GLUT1 deficiency-associated early-onset seizures typically have severe developmental delay that leads to intellectual disability. Most eventually learn to walk but tend to remain nonverbal. This has been the experience of children with the classic phenotype. However, in the past there has often been a long delay between onset of seizures and diagnosis (28; 77); an average 6.6-year delay was reported in a large series (77), and some individuals are not diagnosed until adulthood, despite severe clinical manifestations (28). Earlier diagnosis and treatment may improve the developmental outcome (81).
The long-term outcome is not yet known in those with milder phenotypes. However, most have an excellent response to the ketogenic diet or the modified Atkins diet.
Barbiturates may need to be used with care. In vitro studies suggest that barbiturates may aggravate the GLUT1 transport defect in erythrocytes, and some parents have reported worsening of seizures with phenobarbital (45).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026