






Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this article, the author explains the clinical and genetic background of hepatorenal tyrosinemia. Hepatorenal tyrosinemia is an inborn metabolic disease caused by a defective fumarylacetoacetate hydrolase enzyme, the last enzyme of the tyrosine degradation pathway. Untreated, hepatorenal tyrosinemia can lead to hepatocellular carcinoma.
|
• Hepatorenal tyrosinemia, or tyrosinemia type 1, is the most severe form of genetic tyrosinemia. | |
|
• Hepatorenal tyrosinemia is a devastating disorder of childhood that causes liver failure, painful porphyria-like neurologic crises, Fanconi syndrome with secondary hypophosphatemic rickets, and hepatocellular carcinoma. | |
|
• If untreated, death typically occurs before 2 years of age, although some milder forms allow longer survival. | |
|
• Hepatorenal tyrosinemia is caused by defective fumarylacetoacetate hydrolase. | |
|
• The pathognomonic metabolite for hepatorenal tyrosinemia is succinylacetone. | |
|
• Secondary enzyme deficiencies in hepatorenal tyrosinemia (ie, delta-aminolevulinic acid dehydratase, methionine adenosyl transferase) result from inhibition by metabolites (ie, succinylacetone and fumarylacetoacetate), accumulating as a result of a primary deficiency of fumarylacetoacetate hydrolase. The secondary enzyme deficiency in delta-aminolevulinic acid dehydratase links tyrosinemia with porphyrin synthesis and is responsible for the porphyria-like crises in tyrosinemia. | |
|
• Hepatorenal tyrosinemia is treated by nitisinone administration and restriction of dietary tyrosine and phenylalanine. Occasionally, orthotopic hepatic transplantation may be indicated. |
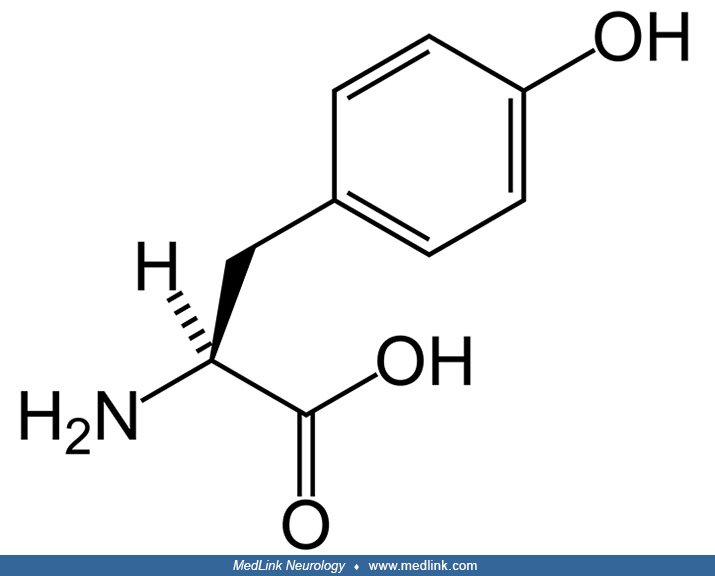
Tyrosine (from the Greek tyros, meaning cheese), or 4-hydroxy-phenylalanine, was discovered in 1846 by German chemist Justus Freiherr von Liebig (1803-1873) in the protein casein from cheese.
Tyrosine is a nonessential amino acid with a polar side group, one of the 22 amino acids that are used by cells to synthesize proteins. Tyrosine phosphorylation, mediated by protein kinases (so-called receptor tyrosine kinases), is one of the key steps in signal transduction and regulation of enzymatic activity. Tyrosine is also a precursor to neurotransmitters (ie, catecholamines) and hormones (ie, thyroxine and melatonin), and, in particular, in dopaminergic cells in the brain, tyrosine is converted to L-DOPA by the enzyme tyrosine hydroxylase, and L-DOPA can in turn be converted to dopamine, noradrenaline (norepinephrine), and adrenaline (epinephrine).
In 1932, American biochemist Grace Medes (1886-1967), at the University of Minnesota Medical School in Minneapolis, first described “a new disorder of tyrosine metabolism” and called it “tyrosinosis” after observing 4-hydroxyphenylpyruvate in the urine of a 49-year-old man with myasthenia gravis (61). She proposed that the metabolic defect in this patient was a deficiency of 4-hydroxyphenylpyruvate dioxygenase, but the case remains puzzling and has since been assigned a separate OMIM number (76800) (49).
The first typical patient with hepatorenal tyrosinemia was described in 1956 by Margaret D Baber at Edgware General Hospital in Middlesex, England (06). Starting the following year, Kiyoshi Sakai and colleagues, at the Jikei University School of Medicine in Tokyo, published three reports describing the clinical, biochemical, and pathological findings of a 2-year-old boy with hepatorenal tyrosinemia who was then thought to have an “atypical” case of tyrosinosis (“atypical” because it differed from the supposedly prototypical case reported by Medes) (78; 79; 80; 49).
Then, between 1963 and 1965, Swedish pediatrician Rolf Zetterström (1920-2011) and associates at the Karolinska Institutet in Sweden published the first detailed descriptions of hepatorenal tyrosinemia and its variants, a disorder then hypothesized to be caused by a defective 4-hydroxyphenylpyruvate dioxygenase enzyme (106; 36; 46; 37; 40; 94). Shortly thereafter, a Canadian group also described the clinical and laboratory findings of hepatorenal tyrosinemia (86). Both the Scandinavian and Canadian groups suggested that the Japanese patients described earlier by Sakai and colleagues had the same disorder, ie, hepatorenal tyrosinemia (49).
From 1965, doubts arose over the hypothesis that hepatorenal tyrosinemia is caused by a defective 4-hydroxyphenylpyruvate dioxygenase enzyme, but it was not until 1977 that Bengt Lindblad and colleagues at the University of Gothenburg in Sweden showed that the primary enzyme defect in hepatorenal tyrosinemia involved the fumarylacetoacetate hydrolase enzyme (57). This was subsequently confirmed with a direct enzyme assay.
Untreated, most patients with hepatorenal tyrosinemia become symptomatic within the first few months of life, but patients can present as late as 6 months of age (47; 85). Failure to thrive usually precedes the appearance of more severe findings. Clinical presentations vary from early-onset acute liver failure with severe coagulopathy, to slowly progressing cirrhosis with multiple nodules and variable renal dysfunction, to normal liver function with renal failure (56; 69; 95). One third of cases develop hepatocellular carcinoma (69; 47). Untreated patients have a peculiar “cabbage-like” odor to the skin or urine. Other manifestations can include porphyria-like neurologic crises, Fanconi syndrome with urine loss of phosphate and secondary hypophosphatemic rickets, and cardiomyopathy (39; 38; 57; 47; 65; 95).
In a study of 22 affected children from Finland, the median age at diagnosis was 5 months, with an approximately equal gender distribution (55% were girls) (01). Four patients were detected through screening and 18 clinically. Thirteen (59%) had a homozygous Finnish type c.786G>A, (p.Trp262X) mutation. The main findings were liver failure, ascites, renal tubulopathy, hypophosphatemic rickets, growth failure, thrombocytopenia, and anemia. Three patients needed transplantation later. Kidney dysfunction, hypertension, and osteopenia/osteoporosis were more frequent in transplanted than in nitisinone-treated patients.
Due to the effect of succinylacetone on heme-metabolism, porphyria-like neurologic crises can also occur (39; 38; 57; 85). However, treatment of patients with nitisinone prevents the development of acute hepatic and neurologic crises.
Historically, patients with hepatorenal tyrosinemia were classified as “acute” or “chronic,” with patients classified as “chronic” when they survived more than 2 years on medical treatment. Classification of patients with hepatorenal tyrosinemia in this manner may be deceptive because patients classified as acute in the first year of life may survive with a chronic course. Conversely, patients classified as chronic may still experience life-threatening neurologic and hepatic crises.
Van Spronsen and colleagues proposed the classification of hepatorenal tyrosinemia as very early-, early-, and late-presenting based on the time of the onset of symptoms (less than 2 months, 2-6 months, and more than 6 months, respectively) (99). Similarly, Demers and colleagues classified hepatorenal tyrosinemia as acute, subacute, and chronic based on the time of liver transplantation. With the Demers classification, patients undergoing liver transplantation before 2 years of age are classified as acute, between the ages of 2 and 6 as subacute, and after 6 years of age as chronic (30).
Fanconi-Debré-de Toni syndrome (also known as Fanconi renotubular syndrome, or FRST) often presents as a secondary feature of tyrosinemia and other systemic disorders that impair energy supply (eg, Lowe syndrome, Dent disease, cystinosis, hereditary fructose intolerance, galactosemia, Alport syndrome, and Wilson disease) (11; 63; 60; 33; 35; 68; 02). Fanconi syndrome is a defect of the proximal renal tubule leading to malabsorption of various electrolytes and substances that are usually absorbed by the proximal tubule (eg, water, calcium, bicarbonate, phosphate, potassium, glucose, uric acid, amino acids, etc.). Various animal models of tyrosinemia have been employed to elaborate the mechanism of Fanconi syndrome development in tyrosinemia (90; 89; 77; 76; 75; 74; 103; 102; 92; 66). Succinylacetone, a metabolite of tyrosine excreted in excess by infants and children with hereditary tyrosinemia and the renal Fanconi syndrome, was found to impair sugar and amino acid uptake by both newborn renal tubules and 7-day renal brush-border membrane vesicles (BBMV) (90; 76). Succinylacetone administration in experimental models was also associated with glucosuria, polyuria, and reduced renal phosphate reabsorption (102).
Although the introduction of newborn screening programs and nitisinone treatment has altered the clinical course of hepatorenal tyrosinemia by making early detection and treatment possible (29), natural disease progression is still seen in many parts of the world where early screening and treatment are not available (35). In addition, even with appropriate treatment, patients develop several behavior problems and have a lower-than-normal health-related quality of life (100).
The prognosis of type 1 tyrosinemia has improved with the advent of nitisinone, particularly when combined with newborn screening to identify cases early (01; 29). Indeed, newborn screening and early treatment are essential for a good outcome (29). Nevertheless, there is a long-term risk for complications, particularly with late diagnosis or insufficient nitisinone levels.
In 1955, a 2-year-old boy was admitted with marked hepatosplenomegaly and failure to thrive (78; 79; 80; 49). He was the second child of a consanguineous marriage and was a normal term delivery and at birth weighed 3900 grams. Although he grew and developed normally as an infant, during his second year his parents noticed a loss of appetite and gradually increasing abdominal distension. Routine laboratory tests were normal, except for low serum inorganic phosphorus and slightly elevated liver function test results. His urine contained a large amount of 4-hydroxyphenyllactate (4-HPL) and small amounts of 4-hydroxyphenylpyruvate (4-HPP), 4-hydroxyphenylacetate (4-HPA), and tyrosine.
His weight gradually decreased, and he developed rachitic changes, including genu valgum, evident clinically and confirmed by x-rays. From clinical observations and persistent hypophosphatemia, Fanconi syndrome (nephrotic-glucosuric dwarfism with hypophosphatemic rickets) was suspected. He had a series of infections and ultimately developed hepatic coma and died in November 1957. At autopsy, the liver showed portal cirrhosis with malignant hepatoma. The tyrosine oxidation activity of a liver homogenate (obtained from a liver biopsy 1 hour after death) was only about 20% of normal.
|
• Hepatorenal tyrosinemia is a monogenic disorder with an autosomal recessive pattern of inheritance. | |
|
• Compound heterozygotes for c.1021C > T (p.Arg341Trp) and a severely deficient FAH allele had only mild hypersuccinylacetonemia and remained asymptomatic and clinically normal without nitisinone or dietary treatment for ages 9 to 15 years. |
Hepatorenal tyrosinemia is a monogenic disorder with an autosomal recessive pattern of inheritance. The disease is caused by reduced activity of fumarylacetoacetate hydrolase (FAH, E.C.3.7.1.2), the last enzyme of the tyrosine degradation pathway (57).
In some series, a high proportion of cases occur as a result of consanguineous unions; for example, in an Egyptian report of 76 cases from 70 families, consanguinity was identified in 61 affected families (32).
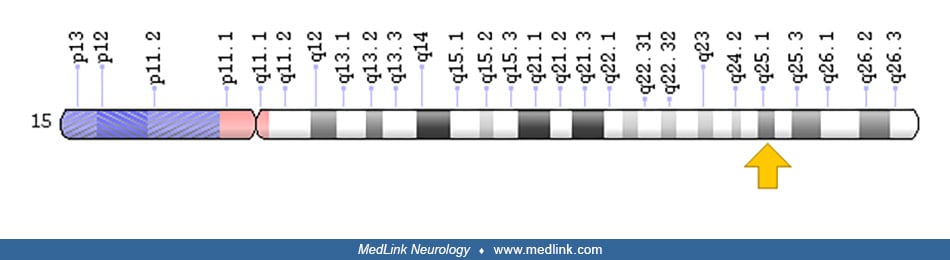
The FAH gene is located on chromosome 15q25.1. The gene spans over 35 kb of DNA and has five protein-coding transcripts.
Numerous mutations responsible for hepatorenal tyrosinemia have been identified in FAH, including nucleotide substitutions (missense and nonsense), splicing mutations, gross deletions, and small indels (ie, insertions or deletions of bases) (71; 53; 52; 73; 72; 14; 31; 30; 18; 21; 45; 22; 24). Mutations are referenced at the Human Genome Mutation Database (HGMD®).
Reversions to wild-type allele have been reported for some hepatorenal tyrosinemia mutations, including the following: NM_000137.2:c.1062+5G>A, NM_000137.2:c.192G>T, NM_000137.2:c.1009G>A, and NM_000137.2:c.836A>G (53; 52; 72; 31; 30; 18; 21). This reversion of one allele to the wild-type is sufficient to re-establish a normal metabolic milieu, giving such cells a growth advantage over cells with non-corrected mutations. FAH-immunopositive nodules are found in the livers of a large majority (88%) of affected patients (52; 30), thereby creating a liver mosaic.
FAH is a soluble cytosolic homodimer of 46.3 kDa subunits (54). It is predominantly expressed in the liver but is also found in a wide range of tissue and cell types, including brain and skeletal muscle (12; 15).
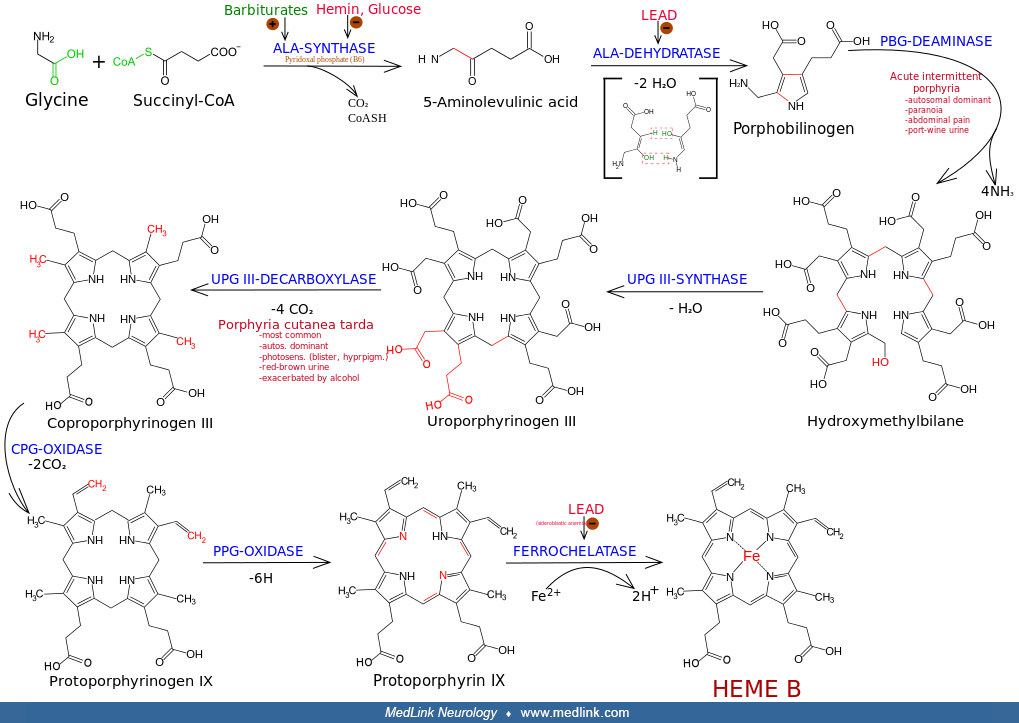
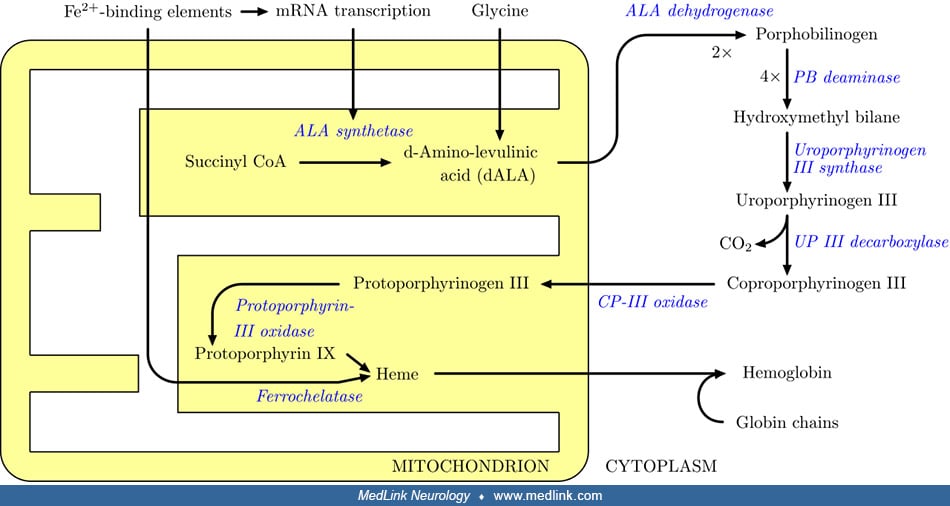
In the absence of FAH, various metabolites accumulate, including fumarylacetoacetate, maleylacetoacetate, homogentisate, p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, p-hydroxyphenylacetate, and succinylacetone (47). In addition, the absence of FAH activity leads to oxidative damage in the liver that progresses to liver failure and hepatocarcinoma, renal injury, and neurologic crises (43).
In hepatorenal tyrosinemia, secondary enzymes are inhibited (ie, porphobilinogen synthase and methionine adenosyl transferase) by accumulating metabolites (ie, succinylacetone and fumarylacetoacetate) resulting from the primary deficiency of fumarylacetoacetate hydrolase (57; 26; 82; 13; 83; 81; 69). For example, a secondary deficiency of porphobilinogen synthase (ALA dehydratase, aminolevulinate dehydratase; EC 4.2.1.24), the second enzymatic step in the synthesis of porphyrin, results mainly from inhibition by succinylacetone (4,6-dioxoheptanoic acid), and to a lesser degree by fumarylacetoacetate (57; 26; 82; 13; 81; 69).
Succinylacetone is formed from a buildup in the metabolites maleylacetoacetate and fumarylacetoacetate (57; 26; 82; 81; 69). The formation and accumulation of toxic metabolites such as succinylacetone results in kidney and liver damage (59).
Because of the inhibition of the cytosolic enzyme porphobilinogen synthase by succinylacetone, patients with hepatorenal tyrosinemia build up and excrete 5-aminolevulinic acid, as do patients in whom the activity of this enzyme is disturbed for other reasons (eg, acute intermittent porphyria, porphyria variegata, hereditary coproporphyria, and lead intoxication) (50). This process of secondary enzyme inhibition effectively links tyrosinemia with porphyrin synthesis and is responsible for the porphyria-like neurologic crises in patients with hepatorenal tyrosinemia (13). Similarly, the secondary enzyme deficiency in methionine adenosyl transferase (by succinylacetone) results in elevated methionine levels (13).
Compound heterozygotes for c.1021C > T (p.Arg341Trp) and a severely deficient FAH allele had only mild hypersuccinylacetonemia and remained asymptomatic and clinically normal without nitisinone or dietary treatment for ages 9 to 15 years (105).
|
• Hepatorenal tyrosinemia generally affects about one in 100,000 newborns. | |
|
• Due to a complex founder effect, hepatorenal tyrosinemia is much more common in French Canadians. |
Hepatorenal tyrosinemia generally affects about one in 100,000 newborns (43; 05; 20; 01), but it differs somewhat in different populations. For example, the incidence is estimated at one in 74,800 births among Norwegians (18) and between 2.2 and 4.9 per 100,000 births among Emiratis (04). Due to a complex founder effect, hepatorenal tyrosinemia is much more common in French Canadians (55; 16; 44), clustering particularly in the Saguenay-Lac-St.-Jean area of Quebec, where the incidence is more than one in 2000 newborns, and where one of every 22 persons is a carrier (44). It is also relatively common in Tunisia, where the incidence is approximately one in 15,000 births (67).
Hypertyrosinemia may be caused by several different conditions, including severe hepatocellular dysfunction, transient tyrosinemia of the newborn, inborn errors of tyrosine catabolism (hepatorenal tyrosinemia, oculocutaneous tyrosinemia, 4-HPPD deficiency), nitisinone treatment, scurvy, hyperthyroidism, and the postprandial state. In cirrhosis and acute liver failure, plasma tyrosine and methionine may both be nonspecifically elevated. Concurrent hepatorenal failure and renal tubular dysfunction may also be seen in other inherited diseases, (eg, galactosemia, hereditary fructose intolerance, lactic acidosis, and glycogen storage disease type IV).
All of the genetic tyrosinemias are characterized by accumulation of tyrosine in body fluids and tissues, and excretion of characteristic tyrosine metabolites in urine (85). Oculocutaneous tyrosinemia (type II), caused by deficiency of tyrosine aminotransferase (TAT) encoded by the TAT gene on chromosome 16q22.1, presents with photophobia (resulting from deposition of tyrosine crystals in the cornea) and hyperkeratotic plaques on the hands and soles of the feet.
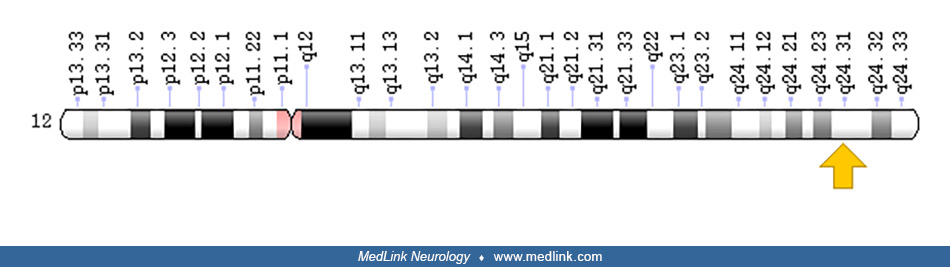
Tyrosinemia type III, a very rare condition caused by a deficiency of 4-hydroxyphenylpyruvic dioxygenase (4-HPD) encoded by the HPD gene on chromosome 12q24.31, manifests with ataxia and mild intellectual disability.
Urine organic acids show elevated p-hydroxy-phenyl organic acids in each type of tyrosinemia, and pathognomic succinylacetone in hepatorenal tyrosinemia (tyrosinemia type I) (85). Methionine is also elevated in hepatorenal tyrosinemia, reflecting impaired hepatocellular function (85).
|
• Clinical features of hepatorenal tyrosinemia include severe liver dysfunction in infancy. | |
|
• Porphyria-like neurologic crises, hypophosphatemic rickets, and other renal tubular diseases may also suggest the diagnosis of hepatorenal tyrosinemia, especially if found in combination with liver dysfunction. | |
|
• A high level of succinylacetone in blood, plasma, or urine is characteristic of hepatorenal tyrosinemia. | |
|
• Diagnosis of hepatorenal tyrosinemia has to be confirmed with enzyme or mutation assays of FAH. These assays can be performed on liver biopsies, lymphocytes, fibroblasts, or dried blood spots. | |
|
• Targeted mutation analysis or sequence analysis may be performed to confirm hepatorenal tyrosinemia. | |
|
• Prenatal diagnosis of hepatorenal tyrosinemia may be obtained by either determination of succinylacetone in amniotic fluid, an FAH assay, or molecular analysis in amniocytes of chorionic villi. | |
|
• Neonatal screening for hepatorenal tyrosinemia is performed by one of two approaches: screening for elevated blood tyrosine and methionine levels or screening for more specific indicators, such as succinylacetone or delta-ALA-dehydratase (PBG synthase) enzyme activity. | |
|
• Hepatocellular carcinoma should be suspected in patients with hepatorenal tyrosinemia when there are increased alpha1-fetoprotein levels and characteristic imaging features, but a high index of suspicion is needed when imaging features change without a clear increase in alpha1-fetoprotein. |
Clinical features of hepatorenal tyrosinemia include severe liver dysfunction in infancy. Porphyria-like neurologic crises, hypophosphatemic rickets, and other renal tubular diseases may also suggest the diagnosis of hepatorenal tyrosinemia, especially if found in combination with liver dysfunction. A high level of succinylacetone in blood, plasma, or urine is characteristic of hepatorenal tyrosinemia (42) but can also occur with impaired function of maleylacetoacetate isomerase (ie, due to loss of function mutations in the GSTZ1 gene), the enzyme preceding fumarylacetoacetate hydrolase in tyrosine degradation (104). Elevated plasma levels of tyrosine, phenylalanine, and methionine, and elevated urinary levels of p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, p-hydroxyphenylacetate, and δ-aminolevulinic acid, may also be detected (47). Elevated urinary and plasma succinylacetone levels may persist even after orthotopic liver transplantation (09). Inconsistencies persist in the target ranges of biochemical markers (51).
Diagnosis of hepatorenal tyrosinemia has to be confirmed with enzyme or mutation assays of FAH. These assays can be performed on liver biopsies, lymphocytes, fibroblasts, or dried blood spots (34).
Targeted mutation analysis or sequence analysis may be performed to confirm hepatorenal tyrosinemia. Four common mutations (NM_000137.2:c.1062+5G> A, NM_000137.2:c.554-1G> T, NM_000137.2:c.607-6T> G, NM_000137.2:c.782C> T) account for 60% of hepatorenal tyrosinemia mutations in the United States, Europe, Morocco, and Turley. The NM_000137.2:c.782C>T mutation is found in almost 100% of patients with hepatorenal tyrosinemia in the Ashkenazi Jewish population, and the NM_000137.2:c.1062+5G>A accounts for 90% of mutations in the French-Canadian population. In Norway, 65% of patients with hepatorenal tyrosinemia are compound heterozygous for different mutations (18).
Prenatal diagnosis of hepatorenal tyrosinemia may be obtained by either determination of succinylacetone in amniotic fluid, an FAH assay, or molecular analysis in amniocytes of chorionic villi (93).
Neonatal screening for hepatorenal tyrosinemia is performed by one of two approaches: screening for elevated blood tyrosine and methionine levels or screening for more specific indicators, such as succinylacetone or delta-ALA-dehydratase (PBG synthase) enzyme activity (47). There is now strong consensus in favor of newborn screening for hepatorenal tyrosinemia (hereditary tyrosinemia type I; HT-1) using blood succinylacetone as a marker (29), followed by diagnostic confirmation and early treatment with nitisinone and diet.
Hepatocellular carcinoma should be suspected in patients with hepatorenal tyrosinemia when there are increased alpha1-fetoprotein levels and characteristic imaging features, but a high index of suspicion is needed when imaging features change without a clear increase in alpha1-fetoprotein (96).
|
• Standard treatment for hepatorenal tyrosinemia consists of protein-restricted diet with administration of nitisinone or NTBC. | |
|
• Initiating treatment of NTBC before 6 months of age significantly reduces the risk of development of hepatocellular carcinoma, improves renal tubular dysfunction, and preserves renal function even after orthotopic liver transplantation. |
Standard treatment for hepatorenal tyrosinemia consists of protein-restricted diet with administration of nitisinone or NTBC [2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione] (03; 25; 28; 64). Nevertheless, inconsistencies persist in the frequency and methods of follow-up, and the dosing of NTBC (51).
Diet. A low-protein diet is strongly recommended. International groups of nutrition specialists now advocate for a simplified low-protein diet and to allow more natural protein intake (07; 29). Preliminary data indicate that simplification of the diet with increased natural protein intake based on three categories of food may be implemented in the diet of patients with tyrosinemia type 1 without significantly altering metabolic control (29). Compliance with dietary recommendations was higher using the simplified diet in comparison to the stricter approach.
Casein glycomacropeptide (CGMP), a bioactive peptide, is an alternative protein source to traditional amino acids. CGMP contains residual tyrosine and phenylalanine and requires supplementation with tryptophan, histidine, methionine, leucine, cysteine, and arginine. In a 12-month study in children with hepatorenal tyrosinemia, CGMP was well tolerated, with no deterioration in metabolic control or growth (27).
Nitisinone (NTBC). Typically, the starting dose of NTBC is 1.0 mg/kg per day divided into two doses. However, Schlune and colleagues suggested that a single dose may be as effective as multiple daily doses (84). According to consensus group recommendations from the German-speaking countries, the dose of NTBC should be as low as possible without losing metabolic control (29). To be of therapeutic value, the level of NTBC in the blood should be between 40 and 60 µmol/L. If this level is maintained, succinylacetone levels in urine do not need to be monitored.
NTBC blocks the accumulation of toxic metabolites by inhibiting the 4-hydroxyphenylpyruvate dioxygenase enzyme (4HPPD), an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine: conversion of 4-hydroxyphenylpyruvate into homogentisate. Inhibition of 4-HPPD prevents tyrosine degradation and results in decreases of fumarylacetoacetate, maleylacetoacetate, succinylacetone, and δ-aminolevulinate concentrations (48) (step 3 in diagram).
Concentrations of p-hydroxyphenyllactate and tyrosine may increase after inhibition of 4HPPD. Therefore, dietary intake of phenylalanine and tyrosine should be restricted based on plasma levels. Factors such as growth rate, protein and energy intake, and health may influence the tyrosine and phenylalanine requirements of an individual. The concentration of plasma tyrosine and phenylalanine should be controlled with diet to levels of 200-500 µmol/L and 20-80 µmol/L, respectively.
Initiating treatment of NTBC before 6 months of age significantly reduces the risk of development of hepatocellular carcinoma (34), prevents life-limiting hepatic disease (91), improves renal tubular dysfunction (58), and preserves renal function even after orthotopic liver transplantation (08). However, the long-term effects of NTBC treatment are unknown. In patients who do not respond to NTBC treatment, orthotopic liver transplantation may be necessary (34).
Poor compliance with dietary and pharmacological therapy in hepatorenal tyrosinemia is a major issue that adversely impacts clinical outcomes in this disease. In a Finnish study of 22 patients, low mean nitisinone concentration (ie, typically indicating poor medication compliance) was associated with a higher risk of severe complications (01). Treatment compliance worsens with age (91). Nevertheless, despite initial poor adherence to dietary and pharmacological treatment, positive reinforcement at medical consultations can produce marked improvement in NTBC levels, clearly demonstrating the importance and utility of systematic positive reinforcement at medical visits (41).
Orthotopic liver transplantation. Because orthotopic liver transplantation is associated with significant risks and complications, it should only be considered for patients who do not respond to NTBC treatment (eg, persistent neurologic crises), and for patients who develop severe liver or hepatocellular carcinoma (69; 47; 87; 17). Treatment with NTBC prior to orthotopic liver transplantation may, however, preserve renal function (08). Combined liver and kidney transplantations have also been performed in some cases with both hepatic and renal failure (69).
Some have, nevertheless, argued for use of liver transplantation as a "safe and definitive alternative to lifelong nitisinone [therapy]," especially when nitisinone is unavailable or prohibitively expensive (93; 62). Because orthotopic liver transplantation can be curative for hepatorenal tyrosinemia, and because excellent results can be obtained in experienced centers, liver transplantation is "especially favorable in countries with limited resources where the cost of medical therapy is highly prohibitive, with lifelong diet restrictions and unclear long-term risk of [hepatocellular carcinoma]" (62).
Gene therapy. Preclinical studies for viral-mediated gene transfer for correction of hepatorenal tyrosinemia have shown limited success (70; 101; 88). Animal studies have also provided support for the use of dendrimer-based lipid nanoparticles to deliver therapeutic fumarylacetoacetate hydrolase mRNA to normalize liver function and extend survival (23).
If untreated, hepatorenal tyrosinemia has a high mortality rate. Patients with early-onset hepatorenal tyrosinemia who do not receive treatment often die of chronic hepatic failure by 2 years of age. Similarly, in untreated patients with later-onset hepatorenal tyrosinemia, death may occur in childhood as the result of hepatic failure or hepatocellular carcinoma.
Treatment of patients with a phenylalanine-and-tyrosine-restricted diet reversed renal tubular dysfunction, and restored erythrocyte ALA dehydratase activity toward normal, but there was a less clear effect on liver function, even when the diet was started at an early age (19; 82; 83). Phenylalanine supplementation of 20 mg/kg/day in combination with phenylalanine-tyrosine free L-amino acid supplements can prevent most low phenylalanine concentrations without increasing tyrosine to concentrations above the target range or influencing NTBC and succinylacetone concentrations whereas 40 mg/kg/day increases tyrosine concentrations to values above the targeted range (98).
NTBC treatment appears to protect against inflammation, as well as DNA and protein oxidative damage, by decreasing succinylacetone levels (59).
Treatment with NTBC should start as soon as possible after diagnosis. Metabolic and neurologic decompensations and the need for orthotopic liver transplantation are markedly diminished or abolished by treatment with NTBC (08). If started early, NTBC can prevent liver failure, renal problems, and neurologic attacks and decrease the risk for hepatocellular carcinoma (97). However, NTBC treatment and a protein-restricted diet may not prevent impaired cognitive function (10).
As demonstrated in a noninterventional, noncomparative, multicenter study in 77 sites across 17 countries in Europe, long-term NTBC treatment is well tolerated (91).
Treatment with NTBC and dietary phenylalanine and tyrosine restriction improves physical health and life expectancy in tyrosinemia type 1 but the neurocognitive outcome continues to be suboptimal (100).
Limited information is available on the treatment of women with hepatorenal tyrosinemia during pregnancy and the puerperium. Nitisinone therapy during pregnancy and the short breastfeeding period did not result in adverse events in a 20-year-old woman with hepatorenal tyrosinemia or her children (107).
Regular assessments of tyrosine, succinylacetone, and nitisinone should be made during pregnancy and the breastfeeding period in both the mother and the infant (107). Additionally, even though experience with breastfeeding is limited for women taking nitisinone, there is no reason to consider breastfeeding unsafe or to not recommend it.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026