Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Kearns-Sayre syndrome is a multisystem mitochondrial disease characterized by the obligate triad of onset before the age of 20, progressive external ophthalmoplegia, and pigmentary retinopathy, plus at least one of the following: cardiac conduction block, cerebrospinal fluid protein greater than 100 mg/dl, and cerebellar ataxia. The disorder is usually caused by a single large-scale deletion of mitochondrial DNA (mtDNA). The authors review the clinical features and molecular pathogenesis of this unusual disease, with a particular emphasis on reports regarding promising potential new therapeutic approaches targeting mitochondria and cells.
|
• Kearns-Sayre syndrome is a multisystemic disorder defined by the triad of chronic progressive external ophthalmoplegia, retinitis pigmentosa, and onset before 20 years of age. | |
|
• Kearns-Sayre syndrome is usually a result of single, large-scale deletions (1.1 to 10 kb) of mitochondrial DNA. | |
|
• Although Kearns-Sayre syndrome is typically sporadic, affected women with a large-scale deletion of mitochondrial DNA have a 4% to 11% risk of transmitting the mutation to a child. | |
|
• In Kearns-Sayre syndrome, progressive cardiac conduction block is common and can be fatal; therefore, timely placement of a cardiac pacemaker can extend lifespan. |
In 1958, Kearns and Sayre reported two patients with the clinical triad of "retinitis pigmentosa, external ophthalmoplegia, and complete heart block" (37). For years, many physicians did not accept the existence of Kearns-Sayre syndrome; in the late 1960s, Dr. David Drachman lumped neurodegenerative disorders with progressive external ophthalmoplegia as "ophthalmoplegia plus" (18). In the late 1970s, Berenberg and colleagues reported five new patients and reviewed 30 literature cases with the triad of clinical features described by Kearns and Sayre. Berenberg and his colleagues further noted that the syndrome was sporadic, began before the age of 20 years, and was frequently accompanied by elevated cerebrospinal fluid protein (08). In 1983, Rowland and colleagues proposed a clinical definition that included these features (67). In 1988, large-scale deletions of mitochondrial DNA were discovered as the underlying gene defect in patients with Kearns-Sayre syndrome (31; 90; 32).
Multiple organ systems are affected in Kearns-Sayre syndrome; the predominant clinical features are found in the central nervous system, skeletal muscle, and heart (29). The syndrome is defined by the obligatory triad of (1) onset before the age of 20, (2) pigmentary retinopathy, and (3) progressive external ophthalmoplegia. In addition, at least one of the following must be present: (1) cardiac conduction block, (2) cerebrospinal fluid protein greater than 100 mg/dL, or (3) cerebellar ataxia (67).
Other clinical manifestations seen in the majority of patients with Kearns-Sayre syndrome include short stature, sensorineural hearing loss, impaired intellect (intellectual disability, dementia, or both), and limb weakness. Neuropsychological testing of 15 patients with Kearns-Sayre syndrome revealed specific cognitive defects, particularly in visual construction, attention, and abstraction/flexibility, suggesting dysfunction of parietooccipital lobes and prefrontal cortex (10). A 2012 review of the psychiatric manifestations of mitochondrial disorders included four cases of Kearns-Sayre syndrome with bipolar disorder, cognitive impairment, psychotic disorder, or psychosomatic disorder (06). Facial dysmorphia has also been reported in Kearns-Sayre syndrome, attributed to abnormal neural crest cell development caused by mtDNA deletion (09).
Multiple endocrinopathies have been reported in Kearns-Sayre syndrome, including diabetes mellitus, hypoparathyroidism, hypothyroidism, hypogonadotropic hypogonadism, growth hormone deficiency, and adrenal insufficiency (02). Diabetes mellitus is not due to an insulin receptor defect but, rather, to insufficient insulin secretion. Atypical presentations in early childhood with severe, autoantibody-negative insulin-dependent diabetic ketoacidosis long before the onset of classic neurologic and ocular symptoms have been reported (14). In a diabetic patient, autopsy revealed small pancreatic islets with the absence of beta cells (59). Short stature in these patients has been attributed to a lack of growth hormone. Cortisol deficiency resulting from primary adrenal insufficiency is a significant aspect of the clinical spectrum in single large-scale mitochondrial DNA deletions. Early and routine evaluation of the adrenocortical axis, both at baseline and following stimulation, is essential for detecting any level of dysfunction (76). Renal dysfunctions so far reported in Kearns-Sayre syndrome include renal failure, renal tubular acidosis, Bartter-like syndrome, and DeToni-Fanconi-Debré syndrome (21). Impaired mitochondrial function can also be associated with hypomagnesemia, most probably due to impairment of energy-dependent reabsorption of the magnesium cation in the kidney (83). Sensorimotor neuropathy is occasionally found in Kearns-Sayre syndrome. Heart involvement in Kearns-Sayre syndrome is variable, including atrioventricular block, dilated cardiomyopathy, bradycardia, myocardial scarring, ventricular arrhythmias, and sudden cardiac death (20). The most common cardiac involvement in Kearns-Sayre syndrome is atrioventricular block, which may develop at any time. Cardiomyopathy is less common than cardiac conduction block, but using cardiovascular magnetic resonance imaging, a pattern of diffuse intramural late-gadolinium-enhancement in the inferolateral segments of the left ventricle was identified in 10 out of 24 (42%) patients with chronic progressive external ophthalmoplegia or Kearns-Sayre syndrome (87; 23). Seizures and strokes are two major clinical features of myoclonus epilepsy with ragged-red fibers (MERFF) and mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), but these features are uncommon in patients with Kearns-Sayre syndrome (93). Strokes were attributed to cardiac emboli in at least two patients with Kearns-Sayre syndrome (60).
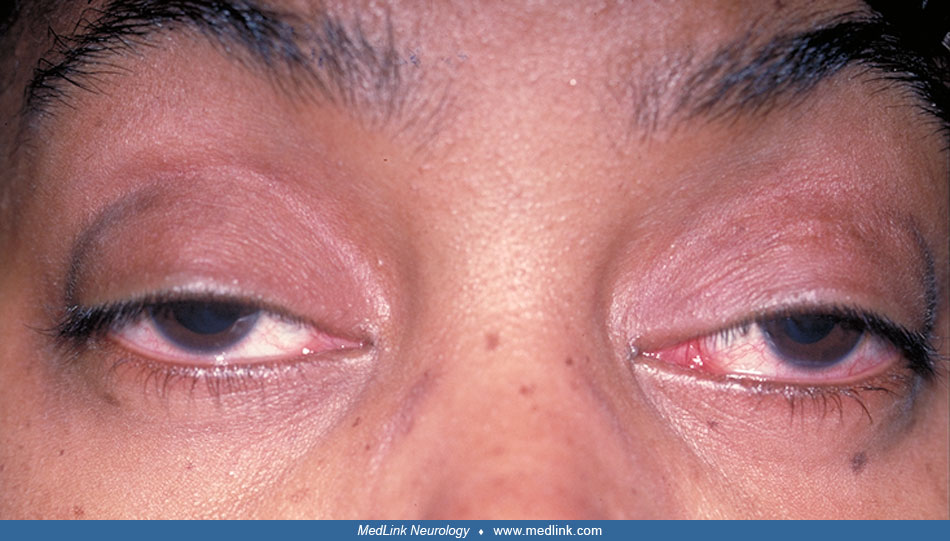
Onset is usually in childhood, with insidious ptosis, ophthalmoparesis, or both.
The ophthalmoparesis sometimes causes blurred or double vision. Weakness of the orbicularis oculi can lead to corneal ulcerations and, in one patient, caused corneal perforation (70). Corneal involvement with microcystic changes resembling Fuchs endothelial corneal dystrophy has been reported in 11 Kearns-Sayre syndrome cases (38). Vice versa, mitochondrial dysfunction has been reported in Fuchs endothelial corneal dystrophy, pointing to a role of energy metabolism in corneal diseases. Tauber and colleagues reported a patient with Kearns-Sayre syndrome and bilateral symmetrical exophthalmos, which might be secondary to ocular muscle wasting (81). Retinoschisis associated with Kearns-Sayre syndrome has been reported in a patient as a rare ophthalmic manifestation of the disease with unclear pathogenesis (12). Additionally, secondary vascular complications can threaten vision; for instance, proliferative diabetic retinopathy has been reported in a patient with Kearns-Sayre syndrome and has responded well to anti-VEGF therapy (47).
Kearns-Sayre syndrome symptoms sometimes appear at the time of, or shortly after, a febrile illness. The disease progresses over the years. If the cardiac conduction defect progresses to complete heart block, and a pacemaker is not inserted in time, the patient may die at a young age. Heart conduction block was found in 12 of 19 patients with Kearns-Sayre syndrome in a Chinese cohort (89). Di Mambro and colleagues emphasized the progressive deterioration of the cardiac conduction system in Kearns-Sayre syndrome and the need for an electrocardiographic screening and a close follow-up to determine each patient’s arrhythmic risk (16). In their cohort consisting of 15 patients with Kearns-Sayre syndrome, left anterior fascicular block preceded right bundle branch block, and bi-fascicular block rapidly degenerated into advanced or complete atrioventricular block in 40%. Timely pacemaker implantation is recommended before the development of bi-fascicular block (16). Dysphagia is not uncommon; in a study of 12 patients, nine had documented cricopharyngeal achalasia (incomplete opening of the upper esophageal sphincter during the pharyngeal phase of swallowing) (39).
Pearson syndrome (sideroblastic anemia with pancreatic exocrine insufficiency) can precede Kearns-Sayre syndrome (57). This form of anemia can be fatal in infancy and has been associated with the same mitochondrial DNA defect seen in Kearns-Sayre syndrome (65). A study investigating long-term hematopoietic dysfunction in patients with Pearson syndrome and Kearns-Sayre syndrome identified that bone marrow dysfunction is a fundamental feature of these disorders, with potential risks of clonal evolution and chromosomal aberrations, emphasizing the need for regular hematologic monitoring (27). In addition to classical clinical phenotype, unusual features such as congenital anisocoria, severe caries, liver cysts, pituitary enlargement, desquamation of hands and feet, bone chondroma, aortic ectasia, dermoid cyst, and polyposis of nasal sinuses have also been reported in an adult patient with Kearns-Sayre syndrome (22).
Cardiac conduction block is found in most patients with Kearns-Sayre syndrome. A 2012 review stresses the fact that, most frequently, syncope is the first clinical sign of the cardiac disease in Kearns-Sayre syndrome, whereas the underlying conduction defect may not always be easily identified in a 24-hour Holter ECG monitoring (82). Progression to a complete heart block can cause death; therefore, placement of a cardiac pacemaker can be a life-saving procedure (64). A literature review by Imamura and colleagues involving 112 patients with arrhythmia-associated Kearns-Sayre syndrome revealed that arrhythmia first manifested as bundle branch block, which then evolved into atrioventricular block and, in about 50% of the group, subsequently progressed to complete atrioventricular block (33). ECG should be performed every 6 months to 1 year to monitor for the development of a heart block.
The prognosis is difficult to determine without specific observation because of the variable tissue distribution of deleted mitochondrial DNA. Severe central nervous system involvement can cause debilitating ataxia, cognitive impairment, and spasticity. Retinal pigmentary degeneration may cause loss of vision, particularly loss of night (dim light) vision. Dysphagia due to pharyngeal and upper esophageal pathology is a common complication. A study of 69 patients with single mtDNA deletions suggested that neurologic complications were more likely when onset was before 9 years of age and less likely when onset was after 20 years of age (07). In addition, the presence and proportion of mtDNA deletion in blood were also associated with the development of neurologic manifestations. Brain MRI with diffusion tensor imaging may reveal early central nervous system involvement in Kearns-Sayre syndrome (19). In a cohort of 87 patients with single, large-scale mitochondrial DNA deletions, a variety of outcome measures such as COX-deficient fiber density, age-at-onset of symptoms, and progression of disease burden were significantly correlated with the size of the deletion, the deletion heteroplasmy level in skeletal muscle, and the location of the deletion within the genome (26). These authors also provide a web-based tool to model disease progression and burden in individual patients.
A young man was normal at birth and through early childhood but was noted at 8 years of age to have droopy eyelids. He was shorter and thinner than his school classmates, though he performed well academically. At 13 years of age, he complained of premature fatigue with exercise. A pediatrician noted his short, thin appearance with ptosis and ophthalmoparesis. He was referred to a pediatric neurologist who noted a pigmentary retinopathy, but no ataxia. Cerebrospinal fluid analysis revealed elevated protein of 121 mg/dL (normal level is 45 mg/dL). An electrocardiogram showed a right bundle branch and left anterior fascicular blocks with a normal PR interval. Cardiac assessment detected a prolonged His-Purkinje conduction time and incipient atrioventricular node conduction abnormalities; a cardiac pacemaker was placed. A muscle biopsy was performed that revealed numerous ragged-red and cytochrome c oxidase-negative fibers; Southern blot analysis revealed a 7.0-kilobase deletion of the mitochondrial DNA in his muscles. He was treated with L-carnitine and coenzyme Q10. At 28 years of age, evaluation for increasing dyspnea on exertion revealed a cardiomyopathy. He was five feet 6 inches in height and weighed 85 pounds. He was treated with digoxin at a dosage of 0.125 mg once a day.
Large-scale deletions of mitochondrial DNA have been found in over 90% of patients with Kearns-Sayre syndrome, with the most common deletion labeled as the “common 4977 bp deletion,” accounting for more than one third of cases (52). Most of the deletions have direct sequence repeats at the junction of the deleted segment of DNA (72; 50). Large-scale duplications of mitochondrial DNA have been found in some patients with Kearns-Sayre syndrome (58). In some patients with progressive external ophthalmoplegia, there was a point mutation at nucleotide 3243 of the mitochondrial DNA; this mutation is typically found in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome (51). In one patient who noted the onset of symptoms at 22 years of age but otherwise fulfilled clinical criteria for Kearns-Sayre syndrome, a G-to-A point mutation at nucleotide 3249 was identified (73). In a few cases, no mutation has been identified. Whole sequencing of the mitochondrial DNA genome in three patients with Kearns-Sayre syndrome revealed different classes, positions, lengths, and grades of heteroplasmy of the large-scale deletion, suggesting that size and heteroplasmy are related to the grade of this pathology (68).
The human disorder has been well modeled in a transmitochondrial mouse model (mito-miceΔ) displaying a heteroplasmic state for wild-type and deleted mtDNA (34). Mice with less than 10% deleted mtDNA remain normal throughout life. Mice with an initial deletion load between 10% and 50% manifest features of Kearns-Sayre syndrome at middle age, including lactic acidosis, heart block, renal failure, deafness, and memory impairment, as well as male infertility. Interestingly, mice with more than 50% deleted mtDNA develop a Pearson syndrome-like phenotype; those who survive the first weeks show a subsequent decrease in deletion load and clinical remission, followed, however, by a new increase in deletion load and the multisystemic phenotype at middle age. As all mice in this model share the same nuclear genomic background, the level and dynamics of mtDNA deletion load in affected tissues seem to be the main determinant for onset and severity of the phenotype (36). Challenging traditional pathogenic thresholds, functional analyses have shown that single, large-scale mtDNA deletions can cause significant defects in the mitochondrial electron transport chain and drive Kearns-Sayre syndrome phenotypes even at very low heteroplasmy levels less than 10% (46).
In a Caenorhabditis elegans model that stably expresses wild-type mtDNA and mtDNA with a 3.1-kilobase deletion, it was found that cells respond to OXPHOS dysfunction by activating the mitochondrial unfolded protein response (UPR(mt)), a transcriptional response mediated by the transcription factor ATFS-1 that promotes the recovery and regeneration of defective mitochondria. It seems that UPR(mt) activation caused by OXPHOS defects propagates or maintains the deleterious mtDNA in an attempt to recover OXPHOS activity by promoting mitochondrial biogenesis and dynamics. Interestingly, downregulation of ATFS-1 reduced the percentage of deleted mtDNA from 60% to 7% (48).
Almost all reported patients with Kearns-Sayre syndrome have been sporadic, suggesting that the mitochondrial DNA rearrangements originate in the ovum or the embryo. Mitochondrial DNA is transmitted only through the mother; therefore, the mutant mitochondrial DNA cannot come from the sperm. Chen and colleagues demonstrated the presence of rearranged mitochondrial DNA molecules in human oocytes and found up to 0.1% of the so-called common deletion (11). These data support the hypothesis that the mitochondrial DNA deletions arise during germline development. Direct sequence repeats at the junctions of most deletions may contribute to the spontaneous generation of these mutations (72).
Clinical expression of the mutation depends on three factors: (1) mitochondrial DNA heteroplasmy, (2) mitochondrial DNA tissue distribution, and (3) tissue threshold. Each mitochondrion contains multiple copies of mitochondrial DNA, and each cell contains multiple mitochondria. Therefore, each cell can contain a variable number of normal and mutant mitochondrial DNA molecules, and this is termed “heteroplasmy.” The tissue distribution of mutant mitochondrial DNA is also heterogeneous (75). If a neonate has a large number of deleted mitochondrial DNA in the bone marrow, the baby will develop Pearson syndrome. If the infant survives the anemia because the bone marrow cells with a high percentage of deleted mitochondrial DNA are at a selective disadvantage and are gradually eliminated, then the child may develop Kearns-Sayre syndrome. Thus, the clinical phenotype depends on the tissue distribution of the mutation. In addition, the most metabolically active tissues are more susceptible to the deleterious effects of the mitochondrial DNA mutations (tissue threshold). Extraocular muscles seem to be particularly sensitive to the presence of mitochondrial DNA deletions. A natural history study demonstrated that Kearns-Sayre syndrome and related mtDNA deletion syndromes represent an evolving clinical spectrum characterized by recurrent MT-ND5 deletions and elevated GDF15 levels, with higher blood heteroplasmy correlating with earlier disease onset (25).
Patients in whom mutant mitochondrial DNA segregates predominantly in muscles can have progressive external ophthalmoplegia as the only clinical manifestation or progressive external ophthalmoplegia plus myopathy affecting limbs, the oropharynx, or both. Some individuals have progressive external ophthalmoplegia and other manifestations of Kearns-Sayre syndrome but do not fulfill all the clinical criteria for the diagnosis of Kearns-Sayre syndrome (67). These cases have been called "progressive external ophthalmoplegia plus" or "Kearns-Sayre syndrome minus." Patients with Kearns-Sayre syndrome generally harbor higher proportions of mtDNA deletions in muscle than individuals with progressive external ophthalmoplegia (85; 49). In a study involving 155 Chinese patients with mitochondrial progressive ophthalmoplegia with large-scale mtDNA deletions, the size and locations of deletions were shown to be useful in differentiating chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome (92). Longer deletions and a higher number of deleted genes encoding respiratory chain complex subunits and tRNA genes were observed in the Kearns-Sayre syndrome group. In a Chinese pediatric cohort of 28 patients with single large-scale mtDNA deletions, Kearns-Sayre syndrome accounted for 64% of cases and showed a distinct genetic profile characterized by larger mtDNA deletions affecting a greater number of tRNAs and respiratory chain complexes, correlating with later onset and broader multisystem involvement compared to non-Kearns-Sayre syndrome phenotypes (84).
Evidence from sequential muscle biopsies of patients with Kearns-Sayre syndrome shows that the percentage of deleted mitochondrial DNA can increase in skeletal muscle (41), perhaps because the mutant mitochondrial DNA, being smaller, replicates more rapidly. This increase in deleted mitochondrial DNA may account for the delayed onset of symptoms and signs.
Analyses of respiratory chain enzymes in muscle have demonstrated variably severe combined defects of complexes containing mitochondrial DNA-encoded subunits. Cytochrome c oxidase (also called complex IV) seems to be particularly affected.
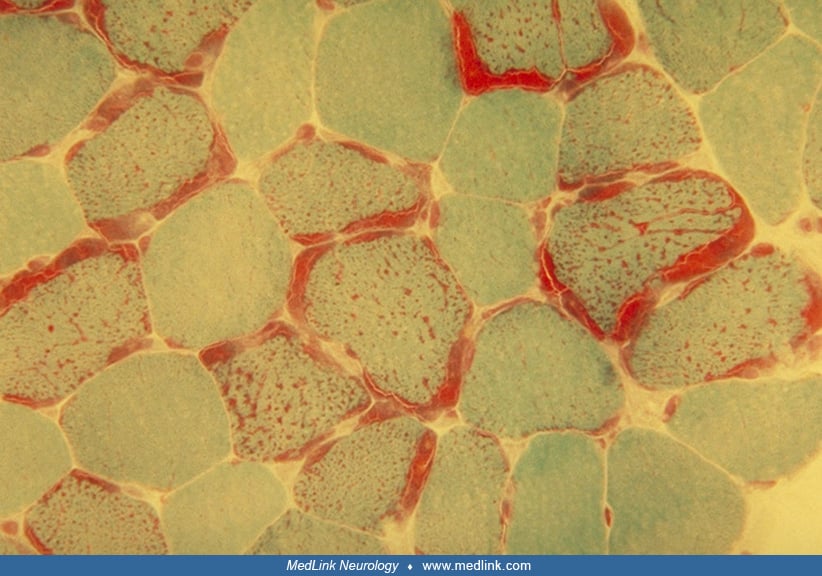
Muscle biopsies demonstrate mitochondrial abnormalities. Modified Gomori trichrome stain shows ragged-red fibers; the red stain in the subsarcolemmal regions of muscle fibers is due to accumulations of mitochondria.
Similarly, the succinate dehydrogenase histochemical stain reveals fibers equivalent to ragged-red fibers, with darker than normal reaction. Ultrastructural studies have confirmed the increased number of mitochondria, as well as morphologically abnormal mitochondria that sometimes contain paracrystalline inclusions. Cytochrome c oxidase histochemical stain shows individual muscle fibers with absent or reduced cytochrome c oxidase activity.
Muscle biopsies revealed a high prevalence of morphological neuromuscular junction defects, such as remodeled, neoformed, dispersed, and dilated endplates, in mitochondrial disorders, including Kearns-Sayre syndrome (44).
The central nervous system shows spongiform changes predominantly in the white matter, including neuronal degeneration, gliosis, and demyelination (54; 77). Mineral deposits of calcium and iron have been noted in and around blood vessels in the basal ganglia and thalamus. Vascular proliferation has also been described but without a consistent geographic pattern. Histological studies have revealed mitochondrial abnormalities in the cerebellar dentate nucleus, loss of Purkinje cells, spongiform degeneration of the cerebellar white matter, and disconnection of Purkinje cells at the dentate nucleus; these abnormalities may contribute to the cerebellar ataxia of Kearns-Sayre syndrome (79; 78). Analysis of brain tissue from three patients showed loss of myelin-associated glycoprotein and oligodendrocytes, whereas inflammation, neuronal loss, and axonal injury were minimal (43). This indicates that energetic failure can lead directly to CNS demyelination. Cerebral folate deficiency has been detected in patients with Kearns-Sayre syndrome and may contribute to cerebral white matter changes (03; 74). It has been suggested that the accumulation of mtDNA deletions in the choroid plexus of patients with Kearns-Sayre syndrome leads to an energetic defect of metabolite transport via the blood-brain barrier. The resulting deficiency of 5-methyltetrahydrofolate (5-MTHF) may directly compromise the methylation and stability of myelin. In a study, untargeted metabolomics analysis of two patients with Kearns-Sayre syndrome revealed increased levels of free sialic acid, sphingomyelin, and tau protein with low 5-methyltetrahydrofolate (5-MTHF) levels on CSF samples (69).
The high frequency of cardiac conduction block in Kearns-Sayre syndrome may be due to preferential accumulation of deleted mitochondrial DNA in the conduction system (53).
The pathophysiology whereby defects in mitochondrial DNA cause the histological and clinical features is not known. Alterations in respiratory chain complexes and energy metabolism are the main consequences of mtDNA deletions in Kearns-Sayre syndrome. In the last two decades, researches have been made to shed light on other pathological pathways. Cells containing mtDNA deletions showed signs of increased protein damage, inhibition of the ubiquitin-proteosome system, and induction of autophagy, which may contribute to the pathogenesis (01). Potential associated mechanisms include reactive oxygen species overproduction, protein synthesis inhibition, myelin vacuolation, demyelination, apoptosis, autophagy, and lipid raft involvement. In addition, the contributions of oligodendrocytes and astrocytes have been demonstrated (86).
The majority of Kearns-Sayre syndrome cases have been sporadic. A few reports of familial Kearns-Sayre syndrome may represent families with the autosomal dominant progressive external ophthalmoplegia with multiple mitochondrial DNA deletions (71; 15; 91). Patients have been described from numerous countries; no ethnic predisposition to Kearns-Sayre syndrome has been noted. An epidemiological study of an adult population in northern Finland estimated the prevalence of large-scale mtDNA deletions (that cause Kearns-Sayre syndrome) to be 1.6/100,000 based on analysis of buccal epithelial samples (63).
There are no reliable means to prevent this genetic disorder.
Kearns-Sayre syndrome must be differentiated from other disorders that cause ophthalmoplegia, such as myasthenia gravis, oculopharyngeal muscular dystrophy, myotonic dystrophy, spinocerebellar ataxia type 7 (28), and other mitochondrial myopathies with progressive external ophthalmoplegia (these include autosomal dominantly transmitted multiple deletions and mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes).
The differential diagnosis of chronic progressive external ophthalmoplegia includes oculopharyngeal muscular dystrophy, myotonic dystrophy, oculopharyngodistal myopathy, congenital fibrosis of the extraocular muscles, thyroid eye disease, myasthenia gravis, and isolated CPEO. Kearns-Sayre syndrome should be differentiated from other possible mitochondrial diseases with multisystemic involvement, such as MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes), MERRF syndrome (myoclonus epilepsy associated with ragged red fibers), Leigh syndrome, polymerase gamma (POLG) disease, thymidine kinase 2 deficiency, Pearson syndrome, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), and sensory ataxic neuropathy dysarthria ophthalmoplegia (SANDO) (30).
The screening of patients for Kearns-Sayre syndrome should begin with routine blood tests, including complete blood count, serum electrolytes, liver function tests, blood urea nitrogen, creatinine, lactate, and pyruvate. These tests may reveal sideroblastic anemia, or kidney or liver dysfunction. Elevated lactate and pyruvate at rest are common in patients with Kearns-Sayre syndrome, and these values may increase dramatically after moderate exercise. The blood leukocyte DNA may be screened for mitochondrial DNA rearrangements, but mutations will be detected only in a minority of patients with Kearns-Sayre syndrome and are almost never found in patients with pure progressive external ophthalmoplegia.
ECG must be performed to screen for heart block. Lumbar puncture typically shows elevated cerebrospinal fluid protein (usually over 100 mg/dL), whereas pleocytosis is usually absent. Oligoclonal bands in the cerebrospinal fluid may be a nonspecific abnormality. Electromyography and nerve conduction studies are usually consistent with a myogenic process, although a neuropathy may also be present. A report characterizing the imaging patterns of mitochondrial leukodystrophies showed that patients with Kearns-Sayre syndrome predominantly have abnormalities in the subcortical white matter, globus pallidus, and substantia nigra (66). Early involvement of the subcortical white matter is highly characteristic of Kearns-Sayre syndrome, which may help in the differential diagnosis of the lysosomal and peroxisomal disorders. Proton MRI-spectroscopy features include low levels of N-acetyl aspartate in the CNS and increased CSF lactate (04). Pasquini and colleagues reported different patterns of spinal cord involvement in six of 11 patients with Kearns-Sayre syndrome, including gray and white matter lesions (56). In a multicenter longitudinal study, early selective upper brainstem tegmental involvement on MRI predicted a characteristic progression to additional brainstem and white matter regions, forming a distinct and consistent neuroimaging phenotype regardless of the initial clinical presentation (05).
Finally, a muscle biopsy should be performed to confirm the diagnosis. Ragged-red fibers on modified Gomori trichrome stain are the hallmark histological feature. In addition, other features characteristic of abnormal mitochondria may be found. Mitochondrial enzyme activities can be measured in whole muscle homogenate or in isolated mitochondria. Muscle mitochondrial DNA should be screened by long-range polymerase chain reaction and by Southern blot analysis for the presence of deleted or duplicated mitochondrial DNA.
No treatment for the genetic defect is available. Management is supportive and symptomatic; it includes treatment of complications: eyelid slings and surgical repair for ptosis, cochlear implants and hearing aids can be used for sensorineural hearing loss, and hormone replacement therapy for endocrinopathies.
Cardiac involvement in Kearns-Sayre syndrome requires comprehensive management, and cardiac conduction block is a preventable cause of mortality. The College of Cardiology, American Heart Association, Heart Rhythm Society, and European Society of Cardiology guidelines recommend prophylactic pacemaker implantation for advanced atrioventricular block in patients with Kearns-Sayre syndrome. Although pacemaker implantation alone may not be effective enough to prevent sudden death, an implantable cardioverter-defibrillator may also be required for selective cases. Imamura and colleagues reported a patient with Kearns-Sayre syndrome who developed polymorphic ventricular tachycardia, ventricular fibrillation, and QT prolongation who was treated with mexiletine and successfully managed with an implantable cardioverter-defibrillator (33). Heart transplantation due to dilated cardiomyopathy has been reported only in a few patients with Kearns-Sayre syndrome (17).
Coenzyme Q10 (50 to 100 mg three times a day) and L-carnitine (330 mg three times a day) have been used to improve mitochondrial function and have had anecdotal success. EPI-743 is a synthetic structurally modified analog of CoQ 10 in which the bis(methoxy) groups of the quinone have been replaced with bis-methyl groups, and the tail has three rather than 10 isoprenyl units. Double-blind, placebo-controlled, randomized clinical trials of EPI-743 are currently in progress for Kearns-Sayre syndrome https://clinicaltrials.gov/ct2/show/NCT01370447.
Promising results of the first mitochondrial augmentation therapy (MAT; using autologous CD34+ hematopoietic stem cells of the recipient enriched with normal mitochondria from his mother) on a 14-year-old patient with Kearns-Sayre syndrome have been reported. Marked improvement in both neurologic functions and metabolic status 7 months after treatment suggests that MAT may be a potential therapy for Kearns-Sayre syndrome (88). Data on the effect of mitochondrial augmentation therapy in six pediatric patients with single large-scale mtDNA deletion syndromes (including four patients with Pearson syndrome and two patients with Kearns-Sayre syndrome) showed some clinical improvement in aerobic functions and quality of life assessments (35).
Short stature in these patients has been attributed to a lack of growth hormone, but the response to growth hormone treatment is heterogeneous (62). In eight patients with Kearns-Sayre syndrome, the mean improvement in height was from -3.9 to -2.9 standard deviation score, but two patients did not show growth improvement at all. The additional index patient in this report achieved a final adult height at -0.8 standard deviation score (62).
In patients with severe dysphagia due to cricopharyngeal achalasia, myotomy can be helpful (39).
Kidney transplantation has been shown to be a feasible therapeutic option for patients with Kearns-Sayre syndrome who develop end-stage kidney disease, as transplant surgery and standard immunosuppressive regimens are generally well-tolerated without triggering acute metabolic decompensation (40).
Endurance exercise training may improve exercise intolerance and quality of life. Resistance exercise training may increase muscle strength and may lower mutational burden in patients with mitochondrial DNA deletions. Both modes of exercise appear to be well tolerated. Patients with Kearns-Sayre syndrome should start their training at low intensity and duration. They should "listen to their body" and not exercise on days they have a fever or superimposed illness (80).
Preliminary data support the notion that folinic acid therapy may be useful in treating patients with Kearns-Sayre syndrome. In six patients treated with oral folinic acid at doses of 1 to 3 mg/kg/day for up to 8 years, decreased 5-methyltetrahydrofolate levels in CSF recovered to normal values (three of three patients), and two of six patients improved neurologically (61). Absence of clinical and radiological response to folinic acid therapy in a patient with Kearns-Sayre syndrome is also reported (55). Early treatment with high-dose folinic acid therapy may be an option for the treatment of Kearns-Sayre syndrome, and a randomized trial seems warranted.
Because hypomagnesemia in Kearns-Sayre syndrome may contribute to neuromuscular irritability, seizures, and cardiac arrhythmias, magnesium should be supplemented in case of deficiency (83).
A more causal therapy approach is site-specific elimination of pathogenic human mtDNA mitochondrially targeted zinc finger nucleases. In a hybrid cell model harboring the common deletion, expression of these nucleases led to a reduction in deletion load, and subsequent repopulation of wild-type mtDNA restored mitochondrial respiratory function (24). This study constitutes proof-of-principle that, through heteroplasmy manipulation, delivery of site-specific nuclease activity to mitochondria can alleviate a severe biochemical phenotype in primary mitochondrial disease arising from deleted mtDNA species. Generation and evaluation of isogenic-induced pluripotent stem cells (iPSC) as a source of cell replacement therapies in patients with Kearns-Sayre syndrome has been reported (45).
A retrospective study of 40 women with disorders due to a mitochondrial DNA deletion revealed that three of 73 offspring had clinical evidence of a mitochondrial disease, giving a recurrence risk of 4% to 11% (13). The three mothers of the affected children had progressive external ophthalmoplegia with or without limb myopathy. Two of the children had Pearson syndrome, and the other developed chronic progressive external ophthalmoplegia.
Lauwers and colleagues describe sudden third-degree atrioventricular block as the major anesthetic complication (42).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Didem Ardicli MD
Dr. Ardicli of Ankara Bilkent City Hospital has no relevant financial relationships to disclose.
See Profile
Haluk Topaloglu MD
Dr. Topaloglu of Hacettepe Children's Hospital in Ankara, Turkey, has no relevant financial relationships to disclose.
See Profile
Nicholas E Johnson MD MSCI FAAN
Dr. Johnson of Virginia Commonwealth University received consulting fees and/or research grants from AMO Pharma, Avidity, Dyne, Novartis, Pepgen, Sanofi Genzyme, Sarepta Therapeutics, Takeda, and Vertex, consulting fees and stock options from Juvena, and honorariums from Biogen Idec and Fulcrum Therapeutics as a drug safety monitoring board member.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neurogenetic Disorders
Jun. 01, 2026
Neuromuscular Disorders
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
General Child Neurology
May. 12, 2026
Developmental Malformations
May. 08, 2026
Neurogenetic Disorders
May. 08, 2026