


Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Mevalonate kinase deficiency is a rare autoinflammatory disorder caused by a proximal enzymatic defect in the cholesterol and isoprenoid synthesis pathway. Specifically, mevalonate kinase deficiency results from biallelic loss-of-function or hypomorphic variants in the mevalonate kinase (MVK) gene. Although mevalonate kinase deficiency is an inborn error of metabolism, major clinical features of this disorder include inflammatory symptoms such as arthritis, malaise, arthralgias, myalgias, and recurrent fevers. The fevers and inflammatory symptoms of mevalonate kinase deficiency often appear unprovoked in the absence of a clinically evident infection. Possible triggers of the inflammatory episodes include mental or emotional stress, vaccines, temperature changes, and physical exertion. Other clinical features of mevalonate kinase deficiency often involve the skin, gastrointestinal system, or central nervous system. Dermatological findings in mevalonate kinase deficiency include oral or genital ulcers, polymorphic recurrent rashes, and porokeratosis. The gastrointestinal manifestations of mevalonate kinase deficiency include recurrent abdominal pain, vomiting, diarrhea, hepatosplenomegaly, and peritonitis. Development of inflammatory bowel disease, cholestasis, or chronic hepatitis has been noted in patients with unmanaged inflammatory symptoms. Finally, neurologic findings in mevalonate kinase deficiency are diverse and can range in severity from mild developmental delays in affected children to progressive cerebellar ataxia, aseptic meningitis, early-onset stroke, and severe intellectual disability. Neurologic manifestations in mevalonate kinase deficiency are more common in severely affected patients but should be evaluated in all patients. Due to the multisystem involvement in mevalonate kinase deficiency, patients often benefit from a multidisciplinary medical team to address the protean manifestations of the disorder.
Patients with mevalonate kinase deficiency can present initially with a wide range of clinical symptoms across a spectrum of seriousness, the severity of which often depends on the inherited pathogenic MVK variants and the resulting residual mevalonate kinase activity (104). Milder mevalonate kinase deficiency signs and symptoms include periodic fevers interspersed with prodromal periods, wherein the patient may be asymptomatic or experience mild to moderate inflammation (146). Severe mevalonate kinase deficiency symptoms and very low or absent enzyme activity are associated with persistent high-grade fevers, dysmorphic features, amyloidosis, cardiomyopathy, neurologic impairment, and liver dysfunction (45). Historically, mevalonate kinase deficiency was classified into two forms: the severe form known as mevalonic aciduria and the milder hyperimmunoglobulin D with periodic fever syndrome. However, this classification scheme has mostly gone out of favor as the condition is really a spectrum with overlap between severe and mild forms, all involving reduction or loss of function in mevalonate kinase enzyme activity.
Most patients described with mevalonate kinase deficiency are compound heterozygotes, often exhibiting the p.V377I variant, in addition to another variant, such as p.I268T or p.H20P/N (70). The p.V377I variant is the most common and appears to be associated with milder MVK enzymatic impairment. Inheritance of other variants associated with more severe disease is often associated with persistent high-grade fevers and neurologic impairments (64). However, the genotype-phenotype correlation in mevalonate kinase deficiency is imperfect; therefore, all patients should be evaluated for systemic inflammation and neurologic symptoms (17).
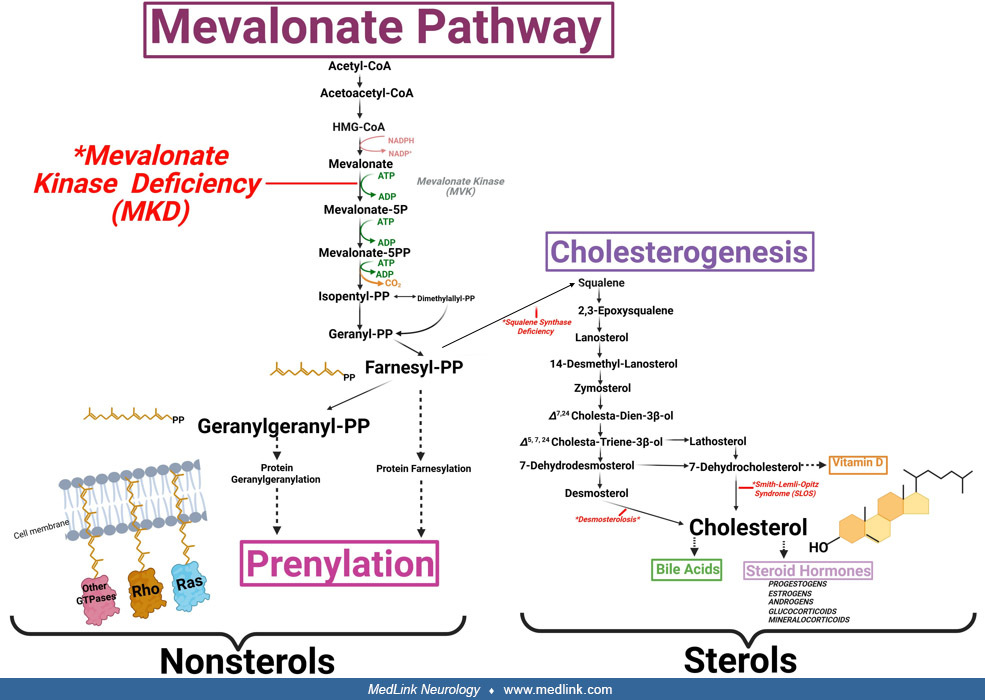
The pathophysiology of mevalonate kinase deficiency represents a complex interplay between sterol and isoprenoid metabolite deficiencies and innate immune reprogramming (03). Mevalonate kinase, the impaired enzyme in mevalonate kinase deficiency, is a proximal enzyme of the isoprenoid and cholesterol synthesis pathway. This metabolic pathway is responsible for sterol and nonsterol isoprenoid biosynthesis (44). Sterols include cholesterol, bile acids, and steroid hormones. Nonsterol isoprenoids include farnesyl and geranylgeranyl pyrophosphate. Because mevalonate kinase is a proximal enzyme in the pathway, patients with mevalonate kinase deficiency can exhibit deficiencies in both sterol and nonsterol isoprenoids (51; 64, 1997; 78). However, the autoinflammatory symptoms in mevalonate kinase deficiency appear to arise from deficiencies in nonsterol isoprenoids and protein prenylation. During prenylation, nonsterol isoprenoids function as a lipid anchor post-translational modification for protein localization to the cell membrane. Nonsterol isoprenoid deficiencies in mevalonate kinase deficiency disrupt protein prenylation, impairing protein cell membrane localization and integration into cell signaling cascades (111; 149). Prenylation defects in mevalonate kinase deficiency enhance innate immunity through inflammasome activation and enhanced NF-kB signaling (03; 112). In mevalonate kinase deficiency, innate immune system activation increases expression of multiple proinflammatory cytokines, including increased IL-1beta production due to pyrin inflammasome activation (112).
The neurologic symptoms of mevalonate kinase deficiency include both neuroinflammation and neurodevelopmental complications. Mevalonate kinase deficiency autoinflammation stemming from nonsterol isoprenoids likely contributes to neuroinflammatory symptoms, such as aseptic meningitis, uveitis, chronic fatigue, retinitis pigmentosa, and cataracts (42). The underlying pathophysiology remains unknown for the other mevalonate kinase deficiency neurologic manifestations, such as cerebellar ataxia, psychomotor impairment, and developmental delays. However, the mevalonate kinase deficiency neurodevelopmental delays retain some overlap with other cholesterol disorders caused by defects more distal in the metabolic pathway: squalene synthase deficiency, lathosterolosis, desmosterolosis, and Smith-Lemli-Opitz syndrome (79). As with mevalonate kinase deficiency, patients with these sterol disorders can have developmental delays, congenital brain malformations, and hypotonia. This overlap suggests that brain sterol deficiencies likely contribute to the neurologic phenotypes seen in mevalonate kinase deficiency. Taken together, mevalonate kinase deficiency occupies a unique position as an autoinflammatory disorder as well as an inborn error of metabolism. Consequently, the ideal management of mevalonate kinase deficiency should include suppressing the autoinflammation component of the disorder, while potentially replacing the deficient isoprenoid and cholesterol metabolites.
|
• Mevalonate kinase deficiency is a rare, monogenic autoinflammatory disorder and inborn error of metabolism arising from biallelic variants in the mevalonate kinase gene, which encodes a key proximal enzyme of the cholesterol biosynthesis pathway. | |
|
• Patients with mevalonate kinase deficiency have reduced mevalonate kinase enzymatic activity, ranging from undetectable levels to approximately 20% normal enzyme activity. This leads to downstream deficiencies in sterol and nonsterol isoprenoids. | |
|
• Isoprenoids are a diverse class of biomolecules that include sterols (cholesterol, bile acids, and steroid hormones) and nonsterols (farnesyl and geranylgeranyl pyrophosphate). Due to the upstream defect in the mevalonate pathway, patients with mevalonate kinase deficiency can have deficiencies in both sterol and nonsterol isoprenoids. The autoinflammatory phenotype appears to arise from nonsterol isoprenoid deficiencies and resultant prenylation defects. | |
|
• Patients with mevalonate kinase deficiency can exhibit a broad range of clinical findings: periodic fevers, arthralgias, myalgias, abdominal pain, oral ulcers, diarrhea, elevated liver transaminases, and various skin rashes. Severe complications include inflammatory amyloidosis, inflammatory bowel disease, liver failure, and neurologic abnormalities ranging from neurodevelopmental delays to ataxia and stroke. | |
|
• Mevalonate kinase deficiency should be included in the differential diagnosis of patients with autoinflammatory manifestations in the absence of known triggers (eg, recurrent or periodic fevers with a negative infectious workup). Mevalonate kinase deficiency was historically classified as mevalonic aciduria or hyperimmunoglobulin D with periodic fever syndrome to distinguish the severe versus mild forms of the condition, respectively. However, this distinction has been mostly supplanted because both forms arise from pathogenic variants in MVK, neither elevated immunoglobulin D levels nor elevated levels of urinary mevalonic acid (mevalolactone) are universally present, and the condition is really characterized by a continuous spectrum of severity. Mevalonate kinase deficiency diagnosis is confirmed following clinical or laboratory suspicion by genetic testing, often through autoinflammatory gene panel screening with next-generation sequencing or by genomic testing. | |
|
• Treatment of mevalonate kinase deficiency remains challenging and unsatisfactory. Targeted therapies against different cytokines (eg, monoclonal antibodies against interleukins) remain the mainstay of mevalonate kinase deficiency management. Metabolite supplementation (eg, geranylgeranyl pyrophosphate supplementation) is an additional treatment approach, despite limited data on its efficacy. The use of statins to suppress the biochemical step immediately upstream from the enzyme defect in mevalonate kinase deficiency remains contraindicated and could exacerbate symptoms. Statin treatment in mevalonate kinase deficiency should be avoided. |
Previously, mevalonate kinase deficiency was described as two disorders, mevalonic aciduria and hyperimmunoglobulin D with periodic fever syndrome, which described severe and mild ends of the mevalonate kinase deficiency spectrum, respectively. This clinical distinction was made before routine clinical genetic testing was available. However, it is now appreciated that both exhibit pathogenic variants in the MVK gene encoding mevalonate kinase. Historically, hyperimmunoglobulin D with periodic fever syndrome was diagnosed via serum IgD elevations during fevers. Patients with mevalonic aciduria also were known to demonstrate elevated IgD, but this disorder was diagnosed with elevated urinary mevalonic acid (mevalolactone) levels (54). However, utilizing serum IgD to diagnose mevalonate kinase deficiency remained imperfect as patients with hyperimmunoglobulin D with periodic fever syndrome may present with normal IgD levels (05; 146). Moreover, patients with hyperimmunoglobulin D with periodic fever syndrome were known to display elevated urinary mevalonolactone levels during fever episodes (77). Due to this clinical and laboratory overlap and the observation that the condition is really a continuous spectrum of severity with clinical overlap between severe and mild forms, previous classification of hyperimmunoglobulin D with periodic fever syndrome and mevalonic aciduria has been largely replaced with the term “mevalonate kinase deficiency” to describe all forms of the condition. This article utilizes the updated mevalonate kinase deficiency terminology to reflect the underlying pathology of this disorder (145).
Age of onset is variable, but most patients with mevalonate kinase deficiency present initial inflammatory symptoms before 5 years of age (75). The onset of clinical features at birth is often associated with more severe MVK variants and lower residual mevalonate kinase activity than seen in patients who present later in life. Usually, most patients with mevalonate kinase deficiency present with chronic inflammation and periodic fevers by adolescence, but later onset in adulthood has been observed.
Short stature, ataxia due to progressive cerebellar atrophy, and ocular involvement with cataracts or retinal dystrophy can be manifestations in later-onset adolescent cases (64; 93; 123; 136; 130; 18). With age, the severity and the frequency of the inflammatory attacks often decline and may eventually subside in adolescence, but they mostly persist for life (18). Fifty percent of adult patients still suffer from six or more attacks per year, life-threatening infections, osteoarthritis, erosive polyarthritis, and autoimmune conditions (Sjogren syndrome or Raynaud syndrome). With age, the risk of developing organ failure increases. This is likely due to the accumulation of organ and tissue damage from years of underlying inflammation. Importantly, the risk of organ amyloidosis increases with age in mevalonate kinase deficiency. Renal amyloidosis has been observed as a long-term complication in about 4% of patients with mevalonate kinase deficiency (144).
Patients with severe mevalonate kinase deficiency tend to have more persistent inflammation that may last between flares with elevated acute phase reactants. Such severely affected patients also tend to have more neurologic manifestations, including cerebellar atrophy, microcephaly, and profound developmental delay (64). In contrast, patients with milder mevalonate kinase deficiency tend to have the classic periodic fevers and flares and may be relatively asymptomatic between flares. In a cohort of 70 patients, about 46% experienced some neurologic manifestation, with the most common being recurrent headaches seen in 36% of the patients (42). Patients with mild mevalonate kinase deficiency usually do not display developmental delays. Specifically, intellectual disability has been seen in only about 15% of patients with mild mevalonate kinase deficiency (77; 144). Although there is a spectrum of neurologic phenotypes among patients with mevalonate kinase deficiency, all of these patients should be monitored for neurologic manifestations (eg, behavioral evaluations, brain imaging, and cognitive evaluations).
Taken together, mevalonate kinase deficiency is a systemic metabolic and inflammatory disorder characterized by episodic flares, periodic fevers, and a spectrum of neurologic manifestations. The age of onset is usually in childhood, but onset into late adolescence and adulthood has been noted. Importantly, there is heterogeneity among patients with mevalonate kinase deficiency in terms of the severity of flares and neurologic manifestations, likely due to differences in residual mevalonate kinase enzymatic activity (136). Despite the clinical heterogeneity of mevalonate kinase deficiency, essentially all patients exhibit pathogenic MVK variants that should be confirmed with genetic testing.
The major clinical features of mevalonate kinase deficiency include recurrent systemic autoinflammatory episodes characterized by recurring intermittent fevers, abdominal pain, lymphadenopathy, arthralgias, or mucocutaneous lesions. These inflammatory symptoms are accompanied by elevations in serum inflammatory biomarkers such as C-reactive protein, erythrocyte sedimentation rate, ferritin, and serum amyloid A. Multiple proinflammatory cytokines can be elevated, especially during flares, namely interleukins 1beta and 6, tumor-necrosis factor-alpha, and interferon-gamma (37; 39; 03). In addition, a complete blood count with differential can show neutrophil-predominant leukocytosis. The flares occur in the absence of autoreactive lymphocytes or specific autoantibodies, as the inflammation results from constitutive innate immune activation secondary to the metabolic disturbances intrinsic to the disorder. Most mevalonate kinase deficiency flares are seemingly unprovoked, but certain non-antigen triggers have been identified, such as mental or emotional stress, minor trauma, vaccines, physical exertion, and temperature changes (68; 146; 104). Viral and bacterial respiratory infections also appear to be major inflammatory triggers that may lead to worsening flares (146; 115). Before developing a flare, some patients with mevalonate kinase deficiency experience prodromal symptoms of fatigue, chills, gastrointestinal discomfort, or arthralgias. Fever onset is usually abrupt with temperatures that may exceed 40 °C (104 °F), accompanied by cervical lymphadenopathy, abdominal pain, vomiting, or diarrhea (146).
The duration of these inflammatory episodes can last from 1 to 10 days, with a median duration of 3 to 7 days (45). The frequency of these inflammatory episodes appears to be irregular at intervals of 2 to 8 weeks. The duration and frequency of these periodic fevers show significant clinical heterogeneity and should, therefore, not be exclusionary when considering mevalonate kinase deficiency in a differential diagnosis. It is worth highlighting that the mevalonate kinase enzyme is thermolabile, which means that any febrile illness can further reduce residual enzyme activity, thereby promoting a mevalonate kinase deficiency flare (104).
Mevalonate kinase deficiency often shows considerable clinical heterogeneity, likely due to different residual mevalonate kinase activity in patients (71). Some may experience an almost asymptomatic course between flares, whereas others may have persistently elevated inflammatory markers (18). The diversity of flare symptoms and severity appears to arise from impaired mevalonate kinase activity across all cell types, causing inflammation across multiple organ systems. The following is a review of organ systems pertinent to mevalonate kinase deficiency clinical manifestations.
Gastrointestinal. Gastrointestinal involvement is the most common manifestation of mevalonate kinase deficiency, with reports describing that up to 98% of patients experience some gastrointestinal-related symptoms (144). Symptoms include abdominal pain, vomiting, diarrhea, and inflammatory bowel disease. Indeed, a significant portion of patients with mevalonate kinase deficiency first present with abdominal symptoms and may be misdiagnosed with inflammatory bowel disease or colitis (13; 09; 43). During flares, sterile peritonitis can mimic appendicitis and lead to significant discomfort.
Severe inflammatory bowel disease in patients with mevalonate kinase deficiency has been noted, leading to intestinal obstruction or perforation (08; 61). Surgical interventions have been described for patients with mevalonate kinase deficiency and severe inflammatory bowel disease, including debridement of adhesions and intestinal resections (13).
The liver appears to be particularly affected in patients with mevalonate kinase deficiency and should be continually monitored with serum transaminases or imaging. At birth, hepatosplenomegaly is frequently noted in patients with mevalonate kinase deficiency, especially those with severe mevalonate kinase activity impairment (123). Cholestasis and steatohepatitis progressing into liver failure have been noted in some patients with severe mevalonate kinase deficiency, even newborns and adolescents (59; 133; 61).
Finally, serum transaminases may be elevated in patients with mevalonate kinase deficiency during flares and should be evaluated as well (133; 142). Taken together, severe mevalonate kinase deficiency gastrointestinal complications include hepatosplenomegaly, cholestasis, hepatitis, liver failure, aseptic peritonitis, severe inflammatory bowel disease, and intra-abdominal adhesions that can result in bowel obstruction.
Musculoskeletal and mucocutaneous. Musculoskeletal involvement is another common mevalonate kinase deficiency manifestation, with approximately 79% of patients experiencing some musculoskeletal-related symptoms (144). Symptoms often include arthralgias and myalgias that can persist between flares. During flares, acute arthritis can develop, especially in the large joints. Erosive polyarthritis has also been noted in mevalonate kinase deficiency, but this manifestation is not as common (09). Skeletal manifestations have been noted, including bone erosion, flexion contractures, osteolytic lesions, and osteoporosis that may be compounded due to chronic corticosteroid use (152; 25).
Mucocutaneous symptoms in patients with mevalonate kinase deficiency include aphthous stomatitis, pharyngitis, and skin rashes. The associated rashes do not appear to be diagnostic of mevalonate kinase deficiency. The most common dermatological manifestations include a maculopapular rash, erythema nodosum, and erythema localized to the inflamed muscle or joint (147). Some patients may also develop painful and erythematous raised skin lesions in conjunction with signs of systemic inflammation (eg, fever and arthralgias), thereby phenocopying Sweet syndrome (acute febrile neutrophilic dermatosis). Therefore, a Sweet-like syndrome has been noted in patients with mevalonate kinase deficiency (43). Histological evaluation of these lesions can reveal neutrophilic dermatitis with squamous syringometaplasia (110). Purpuric rashes have been noted in patients with mevalonate kinase deficiency, wherein some patients may have vasculitis (108). Aphthous ulcers in the oral mucosa are common in mevalonate kinase deficiency, and genital or rectal ulcers may occur as well (54).
Hematological, renal, and cardiopulmonary. Hematological manifestations of mevalonate kinase deficiency are variable and can include anemia, thrombocytopenia, and leukocytosis. The more severe hematological complications of mevalonate kinase deficiency include macrophage activation syndrome, characterized by fevers, cytopenias, hyperferritinemia, and multi-organ dysfunction (133). Bone marrow and hematologic abnormalities include normocytic hypoplastic anemia, leukocytosis, thrombocytopenia, and abnormal blood cell forms (59). These abnormalities may lead to the diagnoses of myelodysplastic syndromes or dyserythropoietic anemia (09).
Other complications of mevalonate kinase deficiency include renal involvement, such as proliferative glomerulonephritis, renal amyloidosis, and end-stage kidney failure (09; 04). Renal angiomyolipoma, a rare kidney tumor, has been described in 6% of patients with mevalonate kinase deficiency from a 50-person cohort (09). Interstitial lung disease is an uncommon but serious complication of mevalonate kinase deficiency, particularly in neonatal patients (117). Cardiac manifestations of severe mevalonate kinase deficiency, although rare, can be detrimental and include cardiac amyloidosis, pericarditis, and progressive cardiomyopathy (123; 114). Serious bacterial infections, such as otitis, cellulitis, meningitis, and pneumonia, can occur in patients with mevalonate kinase deficiency. Such bacterial infections may be related to immunosuppressive therapies, but profiling immune cell populations in mevalonate kinase deficiency suggests that patients are immunocompromised at baseline and more prone to infection (131).
Developmental delays and dysmorphia. The more severe forms of mevalonate kinase deficiency may be dysmorphic, showing distinct facial features that can include frontal bossing, microcephaly, hypertelorism, long eyelashes, and down-slanting palpebral fissures (113). Moreover, patients with severe forms of mevalonate kinase deficiency can also exhibit cerebellar ataxia, epilepsy, failure to thrive, variable degrees of global developmental delays, intellectual disability, and central nervous system manifestations, which were historically used to differentiate hyperimmunoglobulin D with periodic fever syndrome from mevalonic aciduria (136; 54). Most patients with mild mevalonate kinase deficiency experience minimal to no neurologic manifestations, with the most common symptom being recurrent headaches (18). In a cohort of 114 patients with mild mevalonate kinase deficiency, intellectual disability was reported in 3.5% of the patients, whereas persistent headaches were reported in 63% (144). In another cohort of 13 patients with mild mevalonate kinase deficiency, up to 15% displayed intellectual disability (77). On the other hand, neurologic manifestations in severe mevalonate kinase deficiency are broad and can include global hypotonia, cerebellar ataxia, strokes, aseptic meningitis, and seizures. Peripheral neuropathy has also been reported in a subset of cases (42).
Stroke. Mevalonate kinase deficiency has been recognized as a potential cause of ischemic stroke. This is a severe mevalonate kinase deficiency complication that significantly impacts patient morbidity and mortality, deserving careful consideration.
Given its underdiagnosed nature, it is plausible that a subset of cryptogenic ischemic strokes in young adults could be attributed to mevalonate kinase deficiency. However, due to the limited number of reported cases, its prevalence remains unknown. To date, only two cases of ischemic stroke associated with mevalonate kinase deficiency have been documented in the literature (136; 14). Blais and colleagues reported a case where anti-IL-1 therapy with anakinra successfully prevented recurrent inflammatory attacks and ischemic strokes, highlighting the potential role of targeted cytokine therapy in managing mevalonate kinase deficiency-related strokes (14).
Although stroke may have only recently been described in mevalonate kinase deficiency, early onset stroke and increased stroke prevalence in other autoinflammatory disorders have been well established, such as deficiency of adenosine deaminase 2 (154). The autoinflammatory nature of both mevalonate kinase deficiency and deficiency of adenosine deaminase 2 suggests that vascular inflammation contributes to the ischemic stroke pathophysiology in both disorders. The stroke prevalence in deficiency of adenosine deaminase 2 is high, with approximately 50% of patients experiencing at least one ischemic stroke (41). Behcet disease is another autoinflammatory vasculitis condition, and patients have a 2.77-fold increase in stroke risk compared to unaffected individuals (151). Although the underlying pathophysiology of mevalonate kinase deficiency is different than Behcet disease, increased IL-1, TNF-alpha, IL-6, and IFN-gamma are elevated in patients with either condition (155). Lastly, an increased risk of stroke in familial Mediterranean fever has been well established (11).
Taken together, stroke remains a common complication of many autoinflammatory disorders due to systemic inflammation and cerebral vasculitis. The estimated stroke prevalence in mevalonate kinase deficiency remains unknown; however, the prevalence will likely increase as patients with early-onset or cryptogenic stroke undergo genetic screening for autoinflammatory conditions. Taken together, early-onset stroke remains an important consideration in mevalonate kinase deficiency neurologic management.
Clinical presentation, diagnosis, and management of stroke. Patients with mevalonate kinase deficiency experiencing ischemic strokes present clinically with focal neurologic deficits of motor or sensory nature, including hemiparesis, hemianesthesia, aphasia, and visual disturbances, depending on the location of the brain ischemia (14). When ischemic strokes occur secondary to mevalonate kinase deficiency, the presentation mirrors that of other ischemic strokes. These events may coincide with an inflammatory attack, depending on the disease activity at the time. However, it is hypothesized that ischemic strokes in mevalonate kinase deficiency result from chronic neuroinflammation rather than acute inflammatory episodes. This hypothesis is supported by stroke studies in other autoinflammatory disorders, wherein chronic proinflammatory cytokine production and immune dysregulation lead to vascular inflammation (87). Because mevalonate kinase deficiency is an episodic condition, affected patients may not always display overt inflammatory symptoms at the time of stroke presentation. Some patients with mevalonate kinase deficiency have distinct prodromal periods between flares, where clinical inflammatory markers may be lower compared to flares (144). As such, evaluating acute inflammation and a history of chronic autoinflammation remains essential when diagnosing mevalonate kinase deficiency strokes.
The diagnostic approach for ischemic stroke in mevalonate kinase deficiency follows standard stroke protocols. A CT scan should always be performed as soon as possible to rule out a stroke of hemorrhagic nature. Treatment should be initiated, and further investigations should proceed, only after the completion of the initial scan. Cardiac evaluations, including echocardiography (TTE/TEE), ECG, and Holter monitoring, are necessary to rule out cardioembolic sources. Laboratory tests should include a lipid profile, fasting glucose, and HbA1c. Inflammatory markers such as C-reactive protein and erythrocyte sedimentation rate, as well as coagulation studies including prothrombin time and activated partial thromboplastin time, can be ordered according to the clinical picture. In cryptogenic stroke, especially in younger patients, screening for autoinflammatory disorders and mevalonate kinase deficiency may be warranted. A positive patient medical history of periodic fevers of unknown origin should prompt genetic testing for known autoinflammatory diseases, such as familial Mediterranean fever, deficiency of adenosine deaminase 2, Behcet disease, etc.
The treatment of ischemic stroke in mevalonate kinase deficiency requires a comprehensive approach that addresses both vascular and inflammatory components. As for ischemic stroke of any origin, immediate management includes thrombolysis when appropriate, provided the patient meets eligibility criteria and there are no contraindications, such as a heightened risk of bleeding due to systemic inflammation. Antiplatelet therapy with aspirin, clopidogrel, or both is recommended for secondary stroke prevention. Supportive care in an intensive care unit or stroke unit is crucial for monitoring and managing complications. Additional management of ischemic stroke secondary to mevalonate kinase deficiency lies in the treatment of the underlying disease. The documented cases suggest that IL-1 blockade with anakinra or canakinumab significantly reduces neuroinflammation and prevents recurrent ischemic events (136; 14).
Suspected pathophysiology of stroke. The pathophysiology of ischemic stroke as a complication of mevalonate kinase deficiency likely arises in the uncontrolled neuroinflammation provoked by this autoinflammatory disorder. Mevalonate kinase deficiency leads to a shortage of isoprenoids, which are essential for protein prenylation. This defect leads to IL-1beta hypersecretion through inflammasome activation, as well as pyroptosis and mitochondrial defects in the central nervous system (03; 42). Isoprenoid deficiency and increased stroke risk has been described in stroke-prone animal models (100). Brain tissues in stroke-prone rats had significantly lower expression of mevalonate pyrophosphate decarboxylase, an enzyme two biochemical steps following mevalonate kinase. Reduced isoprenoid biosynthesis was noted in these stroke-prone hypertensive rat models, suggesting that impaired mevalonate pathway activity may increase stroke risk (101). However, the mechanism of isoprenoid-deficiency and stroke has not been elucidated, and inflammation was not evaluated in these models.
It is unclear whether the increased risk of ischemic stroke in mevalonate kinase deficiency is due to chronic neuroinflammation or the acute inflammatory episode during flares, the latter of which could trigger a transient prothrombotic state. At baseline, patients with mevalonate kinase deficiency have elevated serum cytokines and persistent, chronic inflammation. However, during acute flare episodes, immune activation contributes to a hyperinflammatory state characterized by systemic release of cytokines, reactive oxygen species, and endothelial dysfunction (45; 125). Blood-brain barrier impairment, upregulation of adhesion molecules, and tissue factor expression facilitate a hypercoagulable environment, promoting thrombus formation and ischemic stroke (87). The activation of von Willebrand factor and the coagulation cascade further exacerbates the thrombotic risk.
In the reported cases, ischemic stroke seems to coincide with mevalonate kinase deficiency flares. However, the stroke itself may provide a mechanism for exacerbating neuroinflammation. With low perfusion and ischemia, reactive oxygen species production increases. Reactive oxygen species production is a potent activator of the NLRP3 inflammasome, which could further upregulate inflammation in a positive feedback mechanism (01; 138; 125). This interplay between immune dysregulation and cerebrovascular ischemia underscores the necessity of targeted immunomodulatory therapies to reduce stroke risk in patients with mevalonate kinase deficiency (14).
Ocular symptoms. Ocular manifestations are common in patients with mevalonate kinase deficiency, with about 10% to 15% developing some form of eye manifestations that include the following: blue sclera, choroideremia, early development of cataracts, recurrent conjunctivitis, or uveitis (54; 144; 02). Moreover, several cases of retinitis pigmentosa with previously unidentified underlying genetic changes have been found to carry biallelic MVK variants (81). Retinal dystrophy may become an important long-term manifestation of mevalonate kinase deficiency, along with cataracts and optic atrophy (42). Therefore, diagnostic evaluation and follow-up care should, where possible, include ocular electrophysiology tests. The pathophysiology of these ocular manifestations appears to be due to both inflammatory and ocular patterning defects. The inflammatory ocular phenotype (eg, uveitis or conjunctivitis) likely arises due to deficient prenylation, upregulating inflammasome activation, and proinflammatory cytokine expression. Choroideremia has been seen in mevalonate kinase deficiency, likely due to deficient Rab protein prenylation that normally maintains retina patterning and photoreceptor localization (84). A detailed review of prenylation disorders and ocular phenotypes can be found in a paper by Ashok and Ramachandra Rao (07).
The overall prognosis for patients with mevalonate kinase deficiency generally depends on the severity of the condition as well as early intervention and inflammatory management. Poorly controlled systemic autoinflammation and metabolic dysfunction can lead to severe complications, such as organ failure or death (140). Patients with more severe MVK variants and lower residual mevalonate kinase activity are often at a higher risk of developing severe complications: p.N205D, p.I268T, p.A334T, p.H20P, p.H20N, R388X, R215X, and Y114fs (64; 123; 130; 152; 18). Failure to thrive has been noted in some severely affected patients (64; 140; 144). Defining a genotype-phenotype correlation in mevalonate kinase deficiency remains difficult due to the genetic and phenoptypic heterogeneity of the disorder. In general, however, variants p.L264F, p.H20N, p.H20P, or p.A334T carry a higher risk of neurologic and ophthalmic complications (17).
Early developmental milestones can be delayed in patients with mevalonate kinase deficiency, including rolling over, crawling, walking, and babbling. Borderline intellectual impairment may be present in some young, mildly affected patients. However, the clinical course appears to improve with age, likely due to metabolic compensation (18). Beyond adolescence, the clinical course appears to be relatively stable, but short stature, cerebellar ataxia, muscular hypotonia, and fatigue may be present. In contrast, patients with severe mevalonate kinase deficiency (known previously as mevalonic aciduria) often have shortened lifespans (64). Absent or extremely low mevalonate kinase activity is often fatal in infancy or early childhood, with most patients dying of sepsis or multisystem organ failure, such as liver failure (64; 126).
Severe complications can arise due to amyloidosis from chronic inflammation (106; 128). Unmanaged inflammatory attacks and chronic subclinical inflammation may lead to amyloidosis, decreased kidney eGFR, and increased serum creatinine (23). End-stage kidney failure and kidney transplant have been noted as a rare but significant complication of mevalonate kinase deficiency (128). Furthermore, liver amyloidosis may occur in patients with mevalonate kinase deficiency, but cholestasis and steatohepatitis leading to end-stage cirrhosis appear to be more common liver complications. Retinal dystrophy may become an important long-term manifestation of the disease, along with cataracts and optic atrophy (17). Therefore, diagnostic evaluation and follow-up care should include, where possible, ocular electrophysiology tests. Lastly, the chronic inflammation and fatigue associated with mevalonate kinase deficiency can also adversely impact growth in children, quality of life, educational achievements, and employment status (146; 48). Due to the rarity of mevalonate kinase deficiency, some patients may feel isolated or have difficulty explaining their disorder to others. Evaluating mental health and social support is critical for ensuring the well-being of these patients.
The most severe, acute mevalonate kinase deficiency neurologic complication is ischemic stroke. Long-term severe complications are organ failure due to uncontrolled inflammation and atlantoaxial joint instability due to arthritis..
The underlying cause of mevalonate kinase deficiency is a metabolic defect in nonsterol and sterol isoprenoid synthesis. The genetics follows an autosomal recessive inheritance pattern, wherein most patients are compound heterozygous for the p.V377I variant in addition to a more severe variant. Although mevalonate kinase deficiency may be metabolic in etiology, the hallmark of this disorder is innate immune activation that manifests as periodic fevers, arthralgias, and myalgias. Deficiencies in nonsterol isoprenoids, such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate, cause downstream prenylation defects, disrupting protein post-translational localization to the plasma membrane. Prenylation defects interact with signal transduction pathways to upregulate proinflammatory cytokine transcription and processing, resulting in a predisposed inflammatory state. The severity of mevalonate kinase deficiency appears to be inversely proportional to the residual activity of mevalonate kinase. However, the mevalonate kinase enzyme is thermolabile, meaning that increased body temperature during fevers can further reduce residual enzyme activity, thereby promoting mevalonate kinase deficiency flares. This positive feedback mechanism likely results in the hyperinflammatory states seen in mevalonate kinase deficiency. The neurologic manifestations in mevalonate kinase deficiency remain less understood but likely arise both from cholesterol deficiency during neurodevelopment and neuroinflammation due to prenylation defects.
Mevalonate kinase deficiency arises from biallelic loss-of-function or hypomorphic variants in the MVK gene that encodes mevalonate kinase (69). Mevalonate kinase is an essential enzyme of the mevalonate pathway that phosphorylates mevalonate into mevalonate-5-phosphate and ultimately feeds into the cholesterol synthesis pathway (44). The downstream products of mevalonate kinase are isoprenoids, lipid molecules that are ubiquitously expressed throughout human tissues. There are about 300 unique MVK variants that have been reported on Infevers, an online database for autoinflammatory variants.
Mevalonate kinase deficiency exists on a spectrum, as some variants correspond to more deleterious reductions in residual mevalonate kinase activity. The most common variant is a conservative p.V377I substitution, with a residual mevalonate kinase activity of about 10% to 28% wild type activity (27; 135; 70). Other common variants include p.I268T, p.H20P, p.H20N, and p.A334T. These variants tend to be more severe, ranging from undetectable to 10% mevalonate kinase activity (60; 27). Highly penetrant nonsense and frameshift variants have also been noted, such as R388X, R215X, and Y114fs (94).
Most patients with mevalonate kinase deficiency are compound heterozygous, usually possessing the common, hypomorphic p.V377I variant in addition to another more penetrant loss-of-function variant, such as p.I268T or p.H20P/N. 71.5% of all mevalonate kinase deficiency variants are accounted for by the p.V377I, p.I268T, and p.H20P/N variants (70). However, V377I is the most prevalent with a 42% allele frequency in patients with mevalonate kinase deficiency. Although uncommon, individuals with p.V377I homozygous recessive mevalonate kinase deficiency have been reported, but such patients are asymptomatic or retain milder symptoms (99; 19). Taken together, mevalonate kinase deficiency often follows a pattern where inheritance of variants with lower mevalonate kinase activity leads to more severe phenotypes. The mevalonate kinase deficiency phenotypic diversity arises from inheriting different combinations of pathogenic variants that interact to create a spectrum of mevalonate kinase activity and resulting clinical severity.
Patients with mevalonate kinase deficiency retain a defect in the mevalonate pathway, an essential metabolic pathway responsible for isoprenoid biosynthesis. Although isoprenoids are a diverse lipid group, all contain isoprene units: five-carbon, unsaturated branched hydrocarbons. Isoprenoids are some of the most diverse and ubiquitous biomolecules across life, with virtually all organisms retaining mevalonate pathway enzymes (86). Two different isoprenoid classes are synthesized from the mevalonate pathway: sterols and nonsterols. Sterol isoprenoids are derived from isoprene units arranged in a fused ring structure, with the classic example being cholesterol. Additional cholesterol hydroxylation modifications create sterol derivatives with vastly different physiological properties, such as bile acids and steroid hormones (mineralocorticoids, glucocorticoids, androgens, etc.) (118). In contrast, nonsterol isoprenoids have isoprene units arranged linearly, with examples including farnesyl pyrophosphate and geranylgeranyl pyrophosphate (44).
(Created in BioRender. Drda, J. 2025. https://BioRender.com/icnk9pu.)
The first step of the mevalonate pathway involves the condensation of three acetyl-CoA molecules into 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA). Reduction of HMG-CoA into mevalonate occurs at HMG-CoA reductase, the rate-limiting step of the pathway. Mevalonate is then phosphorylated into mevalonate 5-phosphate by mevalonate kinase, the impaired enzyme in mevalonate kinase deficiency (122). Under normal conditions, mevalonate 5-phosphate undergoes additional phosphorylation and decarboxylation steps to produce isopentenyl pyrophosphate and its isomer, dimethylallyl pyrophosphate. Together, isopentenyl pyrophosphate and dimethylallyl pyrophosphate are the five-carbon units that combine to form all endogenous isoprenoids.
The condensation of two isopentenyl pyrophosphates and one dimethylallyl pyrophosphate produces farnesyl pyrophosphate, a fifteen-carbon lipid molecule. Following farnesyl pyrophosphate synthesis, the mevalonate pathway bifurcates into two distinct pathways: the sterol and the nonsterol pathways. During sterol generation, condensation of two farnesyl pyrophosphate molecules generates squalene, which is subsequently converted into cholesterol through a complex ring-forming mechanism (35). In contrast, nonsterol generation involves condensation of farnesyl pyrophosphate and isopentenyl pyrophosphate to generate geranylgeranyl pyrophosphate, a linear, twenty-carbon lipid molecule (103). Importantly, patients with mevalonate kinase deficiency retain synthesis impairments in both sterol and nonsterol isoprenoids because mevalonate kinase is upstream of the mevalonate pathway bifurcation (64).
Although cholesterol is given particular emphasis in the medical literature, nonsterol isoprenoids exert equally profound biological functions. Nonsterols comprise the following: (1) dolichols, which act as oligosaccharide carriers during protein glycosylation; (2) ubiquinone, which functions as an electron carrier in the electron transport chain during mitochondrial oxidative phosphorylation; (3) isopentenylated transfer RNAs, which are important for protein synthesis; and (4) protein prenylation, which is a protein post-translational modification where nonsterol isoprenoids are attached near the C-terminal region (52). Depending on the nonsterol metabolite used for attachment, farnesyl pyrophosphate or geranylgeranyl pyrophosphate, prenylation consists of either protein farnesylation or geranylgeranylation, respectively. The nonsterol isoprenoid moiety is added to a conserved C-terminal prenyl localization signal that determines farnesyl pyrophosphate or geranylgeranyl pyrophosphate attachment depending on the amino acid sequence (153). Prenylation serves as a stable, hydrolysis-resistant lipid anchor that allows for membrane-directed protein localization (111). Such membrane localization appears to be critical for proper integration of prenylated proteins into signal transduction cascades.
Protein prenylation is a diverse process, with over 300 proteins in the human proteome undergoing prenylation (149). The largest group of prenylated proteins is the Ras GTPase superfamily, comprising Ras, Rho, and Rab proteins. These proteins exert immense biological regulation of signal transduction, such as growth control and cell cycle control; cytoskeletal rearrangements and cell mobility; and vesicle trafficking through the endomembrane system, respectively (24).
Autoinflammation and period fevers define mevalonate kinase deficiency symptoms and clinical management. Mevalonate kinase deficiency appears to be a multi-cytokine disorder with increases in classic proinflammatory cytokines, such as IL-1beta, IL-6, and TNF-alpha (37; 39; 03). Evidence suggests that IL-12 and IFN-gamma also contribute to mevalonate kinase deficiency inflammation (47). The primary immunological driver in mevalonate kinase deficiency appears to be inflammasome activation and proinflammatory expression due to prenylation errors from nonsterol isoprenoid metabolism. A more detailed explanation arises from three key events that serve as a feedback mechanism to systemically activate inflammation: (1) nonsterol isoprenoid metabolite deficiencies, (2) reduced protein prenylation, and (3) further mevalonate kinase activity impairments during fevers (95; 03; 78; 112; 104).
In some metabolic disorders, symptoms arise due to the accumulation of toxic intermediates. Because patients with mevalonate kinase deficiency accumulate mevalonic acid and its metabolites, often presenting with increased serum and urinary mevalonic acid, it was originally hypothesized that the inflammation stemmed from mevalonate toxicity. In part to test this hypothesis, intervention with lovastatin, an HMG-CoA reductase inhibitor, was administered in two patients with mevalonate kinase deficiency to decrease mevalonate concentrations. Doses of 0.3 to 0.6 mg/kg per day for more than 2 weeks resulted in severe critical clinical decompensation manifesting as elevated body temperature, acute myopathy, highly elevated creatine kinase, diarrhea, vomiting, and worsening ataxia (64). After cessation of statin therapy, clinical and biochemical changes reversed. Because decreasing mevalonic acid exacerbated symptoms, it was determined that mevalonate kinase deficiency was caused by a deficiency of mevalonate pathway end products, not mevalonic acid toxicity.
The mevalonate kinase deficiency cell culture model with peripheral blood mononuclear cells demonstrated that exogenous cholesterol supplementation was not sufficient to attenuate inflammation (95). Instead, only exogenous nonsterol geranylgeranyl pyrophosphate supplementation decreased the expression of proinflammatory cytokines such as IL-1beta (97). Taken together, nonsterol isoprenoid deficiencies drive the mevalonate kinase deficiency inflammatory phenotype in immune cells, as opposed to sterol deficiencies. However, the role of other metabolites in mevalonate kinase deficiency symptoms has been proposed. Cholesterol and other sterol deficiencies have been suggested as major contributors to the neurologic symptoms seen in mevalonate kinase deficiency (42). Sterol turnover in mevalonate kinase deficiency nervous tissue has not been evaluated, but the neurologic phenotypic overlap between mevalonate kinase deficiency and cholesterol synthesis disorders such as Smith-Lemli-Opitz syndrome suggests cholesterol deficiency may contribute to mevalonate kinase deficiency pathophysiology (56). Interestingly, serum cholesterol levels in patients with mevalonate kinase deficiency may be normal or moderately decreased (49; 64). However, there is likely a difference between serum and extracellular and intracellular cholesterol in patients with mevalonate kinase deficiency. These patients likely compensate for decreased intracellular cholesterol by upregulating exogenous cholesterol absorption from the diet, hence the moderately decreased or normal serum cholesterol (49). Finally, deficient ubiquinone has been proposed to contribute to mevalonate kinase deficiency inflammation due to oxidative stress and mitochondrial dysfunction arising from inappropriate electron transport during oxidative phosphorylation (64; 72). Oxidative stress has been widely implicated in inflammation through NLRP3 activation and IL-1beta production, which has been linked to mevalonate kinase deficiency (01; 138; 125). Nevertheless, although other isoprenoids may contribute to the mevalonate kinase deficiency symptoms, nonsterol isoprenoid deficits appear to be the most salient factor in systemic autoinflammation.
Deficient protein prenylation appears to be the link between nonsterol isoprenoids and mevalonate kinase deficiency inflammation. When treated with prenyltransferases to inhibit nonsterol isoprenoid attachment, peripheral blood mononuclear cells from healthy human donors increased TNF-alpha, IL-1beta, and IL-6 proinflammatory cytokine expression (95; 46). Mice models deficient in prenyltransferases also showed similar increases in proinflammatory cytokine expression, similar to mevalonate kinase deficiency (82; 03). Unfortunately, these experiments were not specific to a given prenylated protein. Instead, prenylation was systemically downregulated in these models. As such, which protein prenylation deficiencies contribute to inflammation remains an active area of mevalonate kinase deficiency research.
Evidence suggests that both RhoA and Kras prenylation deficiencies appear to upregulate mevalonate kinase deficiency inflammation through the inflammasome. Park and colleagues demonstrated that geranylgeranylated RhoA normally inhibits pyrin inflammasome through phospho-inactivation (112). Without an immune challenge, RhoA-mediated inhibition of the pyrin inflammasome serves as a molecular brake on inflammation, preventing cleavage of inactive pro-IL-1beta into active IL-1beta. In the presence of an immune challenge, bacterial products inhibit RhoA, dampening the molecular brake on pyrin and, therefore, activating IL-1beta processing. However, in mevalonate kinase deficiency, loss of prenylation removes RhoA from the cell membrane, constitutively abolishing the molecular brake that RhoA asserts over the pyrin inflammasome (112). RhoA prenylation losses in mevalonate kinase deficiency, therefore, promote pyrin inflammasome auto-activation, constitutively IL-1beta processing in the absence of immune challenges. Akula and colleagues further expanded these findings by demonstrating that deficient Kras also contributes to mevalonate kinase deficiency inflammation (03). Mice deficient in prenyltransferases had marked increases in IL-1beta expression that coincided with AKT inactivation. Further experiments demonstrated that prenylated Kras normally associates with PI(3)K to activate AKT. However, in prenyltransferase-deficient mice, loss of Kras prenylation promoted dissociation from PI(3)K, inactivating AKT. The resulting AKT suppression from Kras prenylation loss promoted GSK3beta/NF-κB activation and downstream proinflammatory cytokine transcription of TNF-alpha, IL-6, IL-12, and IL-1beta (03). Further analysis of peripheral blood mononuclear cells from patients with mevalonate kinase deficiency confirmed a similar mechanism of NF-κB activation and proinflammatory cytokine expression.
It must be noted that RhoA and Kras represent only two of over 300 known prenylated proteins (149). In mevalonate kinase deficiency, systemic nonsterol isoprenoid deficiencies means that all known prenylated proteins within the proteome could be impacted. Determining which prenylated proteins activate inflammatory signaling pathways remains a significant knowledge gap, but there are a few key proteins that may potentiate mevalonate kinase deficiency inflammation. Deficient Rab protein prenylation may impact CD14 trafficking and TLR4 signaling, serving to promote LPS-mediated proinflammatory cytokine release (46). Deficient CD42 prenylation may dysregulate pyrin assembly to promote increased IL-1beta processing (139). This CDC42 prenylation mechanism appears to be supported clinically, as autoinflammatory patients with genetic defects proximal to the CDC42 prenylation site have been discovered (85; 150). Taken together, prenylation exerts a profound regulatory function over the innate immune system due to the diverse protein classes that undergo this post-translational modification. Due to the systemic metabolic defect in mevalonate kinase deficiency, there is potential for all prenylated proteins to be impacted. Determining which prenylated proteins contribute to autoinflammation remains a significant challenge in mevalonate kinase deficiency research.
Finally, there appears to be another mechanism promoting mevalonate kinase deficiency autoinflammation: elevated body temperatures during fevers further impairing residual mevalonate kinase activity. This mechanism was proposed by Houten and colleagues when they showed that cultured mevalonate kinase deficiency fibroblasts displayed remarkable temperature sensitivity (68). Specifically, fibroblasts from patients with mevalonate kinase deficiency with the p.V377I variant had a significant reduction in mevalonate kinase activity when cultured at 39°C compared to 37°C (68). Testing this hypothesis in vivo proved to be difficult because original mevalonate kinase deficiency mouse models were highly penetrant. However, Munoz and colleagues generated a milder mevalonate kinase deficiency mouse model that harbored the common p.V377I variant (104). Similar to human patients with mevalonate kinase deficiency, these mevalonate kinase deficiency mouse models demonstrated prenylation defects that coincided with proinflammatory cytokine expression. Importantly, when mevalonate kinase deficiency mice were heat shocked to elevate body temperatures by 2°C to 3°C, there was a significant increase in plasma mevalonic acid (104). During heating, prenylation defects were also potentiated, suggesting that increased temperatures exacerbate the intrinsic mevalonate kinase defect. Prenylation was restored to the baseline defect following an extended cooling period (104). Thus, temperature increases amplify the intrinsic protein instability in variant mevalonate kinase, and a return to the baseline defect only occurs following de novo enzyme expression.
Major mevalonate kinase deficiency clinical manifestations include episodic fevers, followed by a prodromal period where patients may appear asymptomatic or mildly inflamed. From these temperature experiments, emerging evidence suggests that mevalonate kinase deficiency’s episodic nature may parallel the residual activity of mevalonate kinase (68; 104). Within this framework, at baseline, patients with mevalonate kinase deficiency experience constitutively active inflammation due to intrinsic prenylation defects (112). However, immune challenges such as bacterial or viral infections initiate an immune response and fevers. As the fever progresses and core body temperature rises, mevalonate kinase activity is further impaired. Further enzymatic impairment reduces nonsterol isoprenoid biosynthesis beyond the baseline defect, exacerbating prenylation losses and inflammasome activation. This positive feedback loop likely generates the periods of hyperinflammation seen in patients with mevalonate kinase deficiency, where C-reactive protein may exceed 200 mg/dL (104). The mechanism that breaks this vicious cycle of autoinflammation remains unknown, but pathogen clearance, increased dietary isoprenoid absorption, or metabolic compensation are possible explanations (50; 67; 71).
Taken together, the pathophysiology of mevalonate kinase deficiency arises due to a complex interplay between metabolism and inflammation. The link between these two seemingly distinct processes occurs through prenylation of critical signaling proteins. Due to a proximal defect in mevalonate kinase, patients with mevalonate kinase deficiency retain defects in both nonsterol and sterol isoprenoids, but nonsterol deficiencies appear to be the key factor in autoinflammation. As such, clinical mevalonate kinase deficiency management should manage the inflammatory symptoms while targeting the underlying metabolite deficiencies.
Mevalonate kinase deficiency incidence appears to be highest in Northern European populations, particularly the Netherlands, Belgium, France, Italy, and Germany (146). In the Netherlands, mevalonate kinase deficiency incidence remains high due to a 1:65 p.V377I heterozygote frequency (70). This high heterozygote frequency in Northern Europe suggests that some patients with mevalonate kinase deficiency may escape clinical diagnosis, perhaps due to milder symptoms or a lack of knowledge surrounding mevalonate kinase deficiency within the medical community. Interestingly, there appears to be a sizeable mevalonate kinase deficiency cohort in Turkey, Japan, India, and China (107; 142; 26; 53; 83; 91; 08; 89). The common p.V377I variant has been noted in these patients, in addition to other region-specific variants.
The high p.V377I heterozygote frequency suggests that this variant may be retained through selection or the founder effect. The Netherlands population retaining a high p.V377I heterozygote frequency appears to partly arise from the founder effect (137). Other explanations include evolutionary selection of certain MVK variants that reduce mevalonate kinase activity. These variants may be beneficial in regions with high fat, high cholesterol diets (148). Lastly, the high p.V377I heterozygote frequency also suggests that homozygous recessive individuals for this variant may be subclinical. Patients with mevalonate kinase deficiency homozygous for the p.V377I variant have been noted, but these patients remain uncommon and may not experience autoinflammation (99; 19).
Mevalonate kinase deficiency is an inherited genetic disorder, but de novo mutations have very rarely been noted. The most common inherited variant is the p.V377I seen in Northern European populations, such as the Netherlands, France, and Italy (70). Genetic testing for pathogenic MVK variants is advised if there is a family history of mevalonate kinase deficiency, fevers of unknown origin, or Northern European ancestry. Genetic counseling for a family with a previously documented child is strongly recommended. In locations where the p.V377I is more prevalent, such as Northern Europe and the Netherlands, biallelic homozygous patients appear to be more common (70; 99). In contrast, for populations in which heterozygote frequencies of MVK variants may not be as high, consanguineous matings may significantly increase the risk of mevalonate kinase deficiency (09).
Prenatal diagnosis is possible by using biochemical or genetic testing. Genetic testing is now more widely available but is aided by knowledge of the genotype of the previously affected child in the family. Mevalonic acid may be measured prenatally in amniotic fluid by use of an isotope dilution gas chromatography/mass spectrometry assay method employing deuterium-labeled mevalonic acid as an internal standard (66). Mevalonate kinase activity can be measured in cultured amniocytes and biopsied chorionic villus tissue (65; 63), and molecular genetic diagnosis (eg, MVK gene sequencing) can be performed.
Due to the metabolic and inflammatory nature of mevalonate kinase deficiency, the differential diagnosis remains broad and should include both metabolic and inflammatory disorders. For more severe forms of mevalonate kinase deficiency, the differential diagnosis is more likely to include inborn errors of metabolism. Organic acid disorders and mevalonate kinase deficiency retain common symptoms, including neonatal-onset hepatosplenomegaly, developmental delays, craniofacial abnormalities, and hypotonia. If mevalonate kinase deficiency is suspected, other inborn errors of cholesterol biosynthesis should be evaluated, especially Smith-Lemli-Opitz syndrome, the most common inborn error of cholesterol metabolism (121). The phenotypic overlap of mevalonate kinase deficiency and Smith-Lemli-Opitz syndrome includes hypocholesterolemia, skin rashes, craniofacial abnormalities, hypotonia, and hepatic involvement (eg, cholestasis) (141). However, some of the physical malformations seen in Smith-Lemli-Opitz syndrome are absent in mevalonate kinase deficiency, such as cleft palate and digit malformations, whereas the autoinflammatory phenotype is unique to mevalonate kinase deficiency (120). Other cholesterol metabolic disorders should be evaluated, such as desmosterolosis, lathosterolosis, X-linked dominant chondrodysplasia punctata, congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome, and SC4MOL deficiency (58; 56).
For mild mevalonate kinase deficiency, the differential diagnosis is most likely to focus within the group of systemic autoinflammatory disorders (80). This group consists of other recurrent fever syndromes: familial Mediterranean fever; tumor necrosis factor receptor-associated periodic syndrome; cryopyrin-associated periodic syndromes; and periodic fever, aphthous ulcers, pharyngitis, and adenitis. A detailed list of autoinflammatory disorders and the associated clinical manifestations can be reviewed in a paper by An and colleagues (06). Other inflammatory disorders to keep within the differential diagnosis include hereditary angioedema, systemic juvenile idiopathic arthritis, and macrophage activation syndrome, which exhibit clinical findings that can overlap with those seen in mevalonate kinase deficiency (33).
Mevalonate kinase deficiency could be misdiagnosed as familial Mediterranean fever due to the similar pathophysiology of these autoinflammatory disorders (38; 34). Patients with familial Mediterranean fever carry pathogenic variants in the MEFV gene, encoding for pyrin (21). Due to constitutive activation of the pyrin inflammasome in familial Mediterranean fever, affected patients produce more IL-1beta. This mirrors the cellular and biochemical findings seen in mevalonate kinase deficiency with inflammasome activation, albeit through a mechanistically distinct process without defective prenylation. Patients with familial Mediterranean fever or mevalonate kinase deficiency experience recurrent, systemic inflammatory bouts characterized by periodic fevers and elevated IL-1beta levels (92). Patients with familial Mediterranean fever usually respond well to colchicine treatment, whereas patients with mevalonate kinase deficiency are generally colchicine nonresponsive (34). This difference is due to colchicine’s mechanism of action, where it binds to tubulin and depolymerizes microtubules. This cytoskeletal restructuring releases RhoA activators, leading to RhoA inhibiting the pyrin inflammasome. With mevalonate kinase deficiency, this inhibitory mechanism cannot be activated as RhoA membrane localization is lost due to geranylgeranylation deficits (112). Another clinical cues to differentiate familial Mediterranean fever from mevalonate kinase deficiency is that the attacks are usually shorter in familial Mediterranean fever, lasting about 6 hours to 4 days. Skin rashes are also more common during flares in mevalonate kinase deficiency compared to familial Mediterranean fever (Bhatt and Cascella 2023). Mevalonate kinase deficiency could also be misdiagnosed as periodic fever, aphthous ulcers, pharyngitis, and adenitis syndrome because both disorders can have elevated serum IgD (10). Periodic fever, aphthous ulcers, pharyngitis, and adenitis syndrome is the most common recurrent fever in children, and patients are responsive to single-dose steroids, similar to mevalonate kinase deficiency. As such, differentiating periodic fever, aphthous ulcers, pharyngitis, and adenitis syndrome from mevalonate kinase deficiency may be difficult from patient history and physical examination alone (10). Taken together, mevalonate kinase deficiency may retain phenotypic overlap with other autoinflammatory disorders. However, clinical genetic testing should always be pursued to confirm mevalonate kinase deficiency diagnosis instead of relying on clinical history or IgD levels (88).
Although patients with mevalonate kinase deficiency, have a recognizable phenotype of serious clinical manifestations, there often remains a significant delay between symptom onset and diagnosis (88). Timely diagnosis of mevalonate kinase deficiency is essential to initiate treatment and prevent the more serious complications of this disorder (88). More severely affected patients may have early-onset failure to thrive, congenital malformations, developmental delays, hepatosplenomegaly, and persistent inflammation. If developmental delay and neurologic symptoms are not prominent, the differential diagnosis is likely to focus within the group of systemic autoinflammatory disorders or milder mevalonate kinase deficiency.
Mevalonate kinase deficiency diagnosis can be delayed due to the lack of awareness of this disorder and the variable clinical phenotypes (88). This difficulty in diagnosis arises in part due to the lack of definitive clinical biomarkers. Historically, elevated IgD has been utilized for diagnosis, but this marker may only be elevated during fever episodes and not at all in some patients with mevalonate kinase deficiency, especially in pediatric patients (88). The role of IgD in mevalonate kinase deficiency remains uncertain because there is no correlation between IgD and inflammation severity (27; 31; 05). Taken together, elevated IgD is neither necessary nor sufficient for mevalonate kinase deficiency diagnosis and must, therefore, not drive clinical diagnosis. Because IgD elevation is not specific and may not be present in all patients, IgD testing is no longer recommended for those with mevalonate kinase deficiency, and classification of hyperimmunoglobulin D syndrome has been replaced by mevalonate kinase deficiency (88).
Clinical biomarkers that strongly suggest mevalonate kinase deficiency include elevated urinary and serum mevalonic acid, which is often in its cyclical lactone structure, mevalolactone. Patients with severe mevalonate kinase deficiency excrete about 500 to 56,200 mmol mevalonic acid/mol creatinine in urine compared to 0.09 to 0.43 mmol/mol creatinine in controls (64). In more mildly affected patients, significant elevations may only be found during febrile crises with only slight elevations between attacks, or they may even remain normal (144). Accurate quantification of mevalonic acid/mevalonolactone is achieved using liquid chromatography-mass spectrometry assay, employing deuterium-labeled mevalonic acid as an internal standard (66). Elevated acute phase reactants such as C-reactive protein, erythrocyte sedimentation rate, serum amyloid A, ferritin, and fecal calprotectin are nonspecific markers of inflammation that support mevalonate kinase deficiency diagnosis. Mevalonic aciduria observed with elevated acute phase reactants directly suggests mevalonate kinase deficiency diagnosis and should prompt genetic testing. Taken together, elevated inflammatory markers, IgD, or mevalonic acid are not sufficient for a definitive mevalonate kinase deficiency diagnosis, but should instead immediately prompt genetic testing (88).
Definitive mevalonate kinase deficiency diagnosis is only achieved through genetic testing or evaluation of mevalonate kinase activity (05; 45). Genetic testing should be the first definitive diagnostic test if mevalonate kinase deficiency is suspected. If known pathogenic MVK variants (such as p.V377I, p.I268T, p.H20P, p.H20N, p.A334T) are identified through genetic testing, then mevalonate kinase deficiency diagnosis can be confirmed because enzymatic activity assays are already known for these variants. However, if novel MVK variants or no MVK variants are identified, then mevalonate kinase enzymatic activity assays should be performed in patient-derived white blood cells or fibroblasts (63). Less than 20% mevalonate kinase activity in patient-derived cells is often considered sufficient to confirm mevalonate kinase deficiency diagnosis. Unfortunately, access to this testing is becoming less available, and most clinical settings do not offer this test.
If mevalonate kinase activity assays cannot be performed, critical analysis of mevalonate kinase structure can be performed in silico using computational modeling and protein folding software. Variants clustered near the activated site residues are more likely to be deleterious: K13, H20, N55, S135, S146, Y149, E193, D204, and A334 (102). Variants within a conserved hydrophobic core of mevalonate kinase from P375-L396 also carry a high suspicion of being deleterious. However, direct enzymatic activity assays are considered the gold standard for mevalonate kinase deficiency diagnosis if genetic testing remains inconclusive, as is the case, for example, when variants of uncertain significance in MVK are identified. Due to the inaccessibility of direct mevalonate kinase activity assays, in silico predictions of MVK variants with protein modeling are becoming more accepted. If mevalonate kinase enzyme activity assays are needed to evaluate variants of uncertain significance or novel variants, the readers are encouraged to contact the authors of this article.
The primary mevalonate kinase deficiency symptoms are episodic fevers and hyperinflammation that may last approximately 3 to 7 days. Most of the mevalonate kinase deficiency therapies only address these inflammatory symptoms. As such, this section describes only the management of the inflammatory complications of mevalonate kinase deficiency. These treatments, by and large, do not impact the other systemic manifestations.
Before biological therapies, nonsteroidal anti-inflammatory drugs were used to manage mevalonate kinase deficiency inflammation (116; 75). NSAIDs are often used on-demand during mevalonate kinase deficiency flares, but NSAIDs as the sole treatment for mevalonate kinase deficiency remain ineffective (30). Corticosteroids do appear to be somewhat therapeutic for patients with mevalonate kinase deficiency at least in symptom management, but long-term corticosteroid usage remains contraindicated due to its multiple adverse effects, including but not limited to osteopenia, weight gain, metabolic sequala, cataracts, hypertension, adrenal suppression, gastric ulcers, and growth restriction in children (127). Therefore, corticosteroids are only recommended if biologicals are not available, tolerated, or effective. Providing corticosteroids as needed during breakthrough flares may help prevent hospitalization.
For the current international guidelines of mevalonate kinase deficiency management and treatment, the 2024 SHARE (88) and the 2021 EULAR/American College of Rheumatology (129) guidelines provide the basis for the recommendations provided in this section on biological therapies.
Anti-IL-1 targeted treatments remain the first-line treatment due to the contributions of inflammasome activation and IL-1beta production to mevalonate kinase deficiency pathogenesis. Canakinumab remains the only mevalonate kinase deficiency treatment approved by the U.S. FDA and European Medicines Agency. However, the efficacy of canakinumab appears to be variable. Some patients report minimal flares, and others experience routine breakthrough autoinflammation. A longitudinal, open-label canakinumab study with 74 patients with mevalonate kinase deficiency reported that 63% experienced no flares, whereas 20% experienced breakthrough flares (76). However, a randomized, double-blind, placebo-controlled canakinumab study with 37 patients with mevalonate kinase deficiency only showed initial therapeutic effects. After the initial 16 weeks, only 23% experienced disease control (28).
Although canakinumab may be the recommended treatment, breakthrough flares remain common. Unfortunately, large cohort mevalonate kinase deficiency studies on the effectiveness of canakinumab remain lacking. In a cohort of eight patients with mevalonate kinase deficiency, inflammatory markers were normalized in only 50% of the patients on canakinumab (109). Further prospective studies are needed to evaluate the efficacy of canakinumab regarding inflammation management and its long-term efficacy on other mevalonate kinase deficiency symptoms, such as the gastrointestinal, dermatological, and neurologic phenotypes (48).
If canakinumab alone does not adequately manage flares, combination therapy with anakinra, an anti-IL-1 receptor antibody, has been shown to be effective. In these patients, canakinumab with on-demand use of anakinra can significantly reduce relapses (15). Anakinra is rapidly eliminated with a serum half-life of 4 to 6 hours. In contrast, the half-life of canakinumab is 22 to 25 days, meaning that it only needs to be administered once every 4 to 8 weeks (16; 146; 48; 143; 55). As such, canakinumab provides longer inflammatory management, but anakinra can provide shorter-term, rapid management when there are breakthrough fevers. In patients for whom canakinumab is unsuccessful, one might consider targeting TNF-alpha with etanercept or adalimumab or IL-6 (eg, tocilizumab) (124; 89; 98).
Mevalonate kinase deficiency treatment involves biological therapies that target increased proinflammatory cytokines. As a first-line treatment, canakinumab should be initiated due to its long half-life. However, if fever flares persist, changing the targeted cytokine or initiating combination therapies could be considered. Decisions on combination therapies with canakinumab or changing the targeted cytokine should be based on clinical features and serum cytokine testing, if available. If IL-1 levels are highly elevated, canakinumab with add-on on-demand anakinra could be considered. If TNF-alpha levels remain high, switching to anti-TNF drugs may be warranted.
Mevalonate kinase deficiency pathophysiology encompasses multiple proinflammatory cytokines, in particular IL-1beta, TNF-alpha, and IL-6 (36; 45). As such, evaluating expression of these cytokines will lead to a more personalized strategy for mevalonate kinase deficiency management, optimized based on the patient’s underlying disease course, financial situation, and age.
Interleukin-1 antagonists are the first-line treatment for mevalonate kinase deficiency, especially canakinumab and anakinra, which target IL-1beta and the IL-1 receptor, respectively. Both canakinumab and anakinra are administered as subcutaneous injections. However, the pharmacokinetics differ dramatically; canakinumab is administered every 4 to 8 weeks, whereas anakinra is administered every day.
Canakinumab often remains the preferred mevalonate kinase deficiency treatment because it provides a more extended period of coverage (129). The dosage of canakinumab should be informed based on inflammatory markers, such as C-reactive protein and serum amyloid A. The standard therapy for mevalonate kinase deficiency remains 150 to 300 mg canakinumab every 4 to 8 weeks (76; 129). If symptoms persist, titration up to 300 mg every 4 weeks or adding on-demand anakinra in a combination therapy should be considered (15). In pediatric patients less than 40 kg, the dosage of canakinumab is 2 mg/kg every 4 weeks, or titrating up to 4 mg/kg every 4 weeks if needed (129). The suggested dosage of canakinumab, anakinra, and other common biologicals can be reviewed in the paper by Du and colleagues (40).
Finally, cost or supply issues may be a significant barrier in mevalonate kinase deficiency management. Although canakinumab may provide more sustained treatment, it generally costs about 7 to 10 times more than anakinra. Patients who initiate on-demand anakinra treatment starting at the prodromal stage of the attack can have decreased fever duration and severity. Inherent in this regimen is the patient recognizing their prodromal symptoms and initiating treatment before a flare. Employing this fever-based anakinra regimen may be an alternative to canakinumab if needed (15; 48).
The guidelines for choosing the most effective biological therapy for mevalonate kinase deficiency can be found in the 2020 international PRO-KIND guidelines (55). In general, anti-IL-1 therapies with canakinumab or anakinra are recommended as first-line treatments. Choosing between canakinumab and anakinra often remains a clinical judgment based on the availability of the therapy and the cost. However, canakinumab appears to be more effective in treating mevalonate kinase deficiency inflammatory symptoms and flare frequency compared to anakinra (48). If the patient does not respond to anti-IL-1 therapies, anti-TNF therapies remain the next therapeutic option (55; 129). If neither anti-IL-1 nor anti-TNF therapies are effective, switching to anti-IL-6 therapies may be warranted.
Long-term inflammation follow-up should include general physical examination and basic laboratory work-up with a complete blood count, and acute phase reactants such as erythrocyte sedimentation rate, C-reactive protein, or serum amyloid A. Monitoring for skin lesions and rashes should be performed, including oral and genital ulcers. Importantly, culturing these lesions may be sterile, as autoinflammation may be driving these manifestations as opposed to infection. As such, a strong clinical indication of a primary infection must be noted before antibiotics or debridement are initiated.
At each primary care visit, urinalysis should be performed to assess kidney function and proteinuria, especially amyloidosis (129). Moreover, urine analysis for injury markers should be routinely performed for drug toxicity. Liver synthetic function and injury markers tests should be performed at each visit (55).
Finally, periodic ophthalmic examination should be performed to evaluate visual acuity, cataracts, or other inflammatory eye conditions (129). Periodic neuropsychological examination should be performed as well in all patients with mevalonate kinase deficiency. In severely affected patients, periodic brain imaging with MRI or CT scans could be used to assess neurodevelopment (55).
Expanding the targetable cytokines in mevalonate kinase deficiency management holds significant potential. Out of the available biologicals, anti-IFN-gamma therapies likely hold the most promise as this cytokine is routinely elevated in patients with mevalonate kinase deficiency and in vitro models (37; 47). Emapalumab, an anti-IFN-gamma antibody, has shown exceptional promise in reducing the inflammation in primary hemophagocytic lymphohistiocytosis and systemic juvenile idiopathic arthritis (90; 29). Exploring emapalumab as a therapeutic for mevalonate kinase deficiency remains a viable, evidence-based treatment strategy, especially if the disease course is complicated with macrophage activation syndrome (133).
Employing JAK inhibitors to treat mevalonate kinase deficiency has not been explored, but the efficacy of targeting the JAK-STAT pathway in other autoinflammatory diseases has been widely documented (73). Blocking the JAK-STAT pathway remains another enticing option to reduce IFN-gamma and proinflammatory cytokine signaling (62).
Hematopoietic stem cell transplantation has been shown to treat severe mevalonate kinase deficiency. The first such stem cell transplant for mevalonate kinase deficiency was reported in the literature in a 3-year-old male with severe mevalonate kinase deficiency (mevalonic aciduria) (105). Allogenic bone marrow stem cell transplantation from the patient’s HLA-matched sister was performed. With transplantation, the autoinflammatory phenotype was reversed, but the neurologic manifestations, such as ataxia, developmental delays, and brain structural abnormalities, were unchanged. The patient still had mevalonic aciduria, likely due to deficient mevalonate kinase activity in nonhematopoietic tissues, such as the liver (105). However, subsequent bone marrow stem cell transplantations for mevalonate kinase deficiency have been less successful. A study of nine patients with mevalonate kinase deficiency who received an allogenic stem cell transplant showed severe adverse effects. Four patients developed acute graft-versus-host disease, and two died following further complications (74). Allogenic stem cell transplantation holds promise, but this treatment should only be reserved for the patients most severely affected by mevalonate kinase deficiency due to the significant risks. Moreover, allogenic stem cell transplantation does not address the other systemic symptoms of mevalonate kinase deficiency, such as the neurologic manifestations and liver complications. A combined allogenic stem cell and liver transplant was well tolerated in a patient with severe mevalonate kinase deficiency with persistent fever episodes and cholestatic liver disease (22). A combined stem cell and liver transplant likely would remain more effective in combating the autoinflammatory and underlying metabolic defect in mevalonate kinase deficiency. However, this combined approach may not be accessible and carries a significant risk of post-transplant complications.
Isoprenoid metabolite supplementation holds promise because it can mitigate the lack of nonsterol isoprenoids in patients with mevalonate kinase deficiency. Oral geranylgeraniol supplementation led to a moderate improvemens in C-reactive protein and serum amyloid A in three patients with mevalonate kinase deficiency, but this sample size necessitates further expansion (134). Oral isoprenoid supplementation holds therapeutic potential, but intestinal isoprenoid absorption remains low, and concentrations may need to be evaluated on a personalized basis as patients with mevalonate kinase deficiency retain a range of mevalonate kinase activity (27). Once absorbed, geranylgeraniol needs to be phosphorylated twice into the active geranylgeranyl pyrophosphate metabolite (57). For mevalonate kinase deficiency isoprenoid supplementation to be effective, the mechanisms of this phosphorylation pathway and gut absorption should be evaluated in humans.
Supplementation of ubiquinone (CoQ10, Co-Enzyme Q10) could also be considered in patients with mevalonate kinase deficiency, as this lipid molecule requires a nonsterol isoprenoid moiety for function. Patients with mevalonate kinase deficiency appear to have a decrease in serum ubiquinone concentrations, providing evidence that this metabolite may be deficient intracellularly (64; 72; 132). Ubiquinone is an essential electron-transfer molecule within the electron transport chain during oxidative phosphorylation. Loss of ubiquinone may promote mitochondrial dysfunction and reactive oxygen species, both of which have been implicated in inflammasome activation and neurologic complications. Specifically, oxidative stress has been widely implicated in inflammation through NLRP3 activation and IL-1beta production, which has been linked to mevalonate kinase deficiency (01; 138; 125). Providing ubiquinone to patients with mevalonate kinase deficiency remains another evidence-based therapy to restore deficient isoprenoid metabolites.
Before the prenylation mechanism of mevalonate kinase deficiency was elucidated, it was hypothesized that mevalonate toxicity led to the autoinflammatory phenotype. Moreover, in vitro studies suggested that statins can upregulate transcription of mevalonate kinase (119). Statins pharmacologically block HMG-CoA reductase, the enzyme that catalyzes the conversion of HMG-CoA reductase into mevalonate. Statin administration was tested in a few patients with mevalonate kinase deficiency to reduce mevalonate accumulation and increase mevalonate kinase activity. Statin treatment had no effect in eight patients with mild mevalonate kinase deficiency (09). In one severely affected patient, statin treatments lowered urine mevalonic acid concentrations, but fevers and inflammatory symptoms were exacerbated (64). This adverse effect was likely due to a reduction in nonsterol isoprenoids and exacerbation of prenylation defects. As such, statins are contraindicated in mevalonate kinase deficiency treatment and should be avoided. Importantly, prevention of ischemic stroke can include the initiation of cholesterol-lowering measures such as statins. Because statins in patients with mevalonate kinase deficiency can exacerbate the frequency or intensity of flares, statins remain contraindicated, even during ischemic stroke.
Likewise, bisphosphonates, such as alendronate, could further suppress the mevalonate pathway by blocking farnesyl-PP synthase. High concentration bisphosphonate treatment of healthy control peripheral blood mononuclear cells can stimulate IL-1beta release and stimulate autoinflammation similar to mevalonate kinase deficiency (95). However, bisphosphonates may be beneficial in treating osteolytic manifestations of mevalonate kinase deficiency by reducing bone reabsorption. Bisphosphonate use in patients with mevalonate kinase deficiency should be carefully monitored for tolerability and weighed against the risks (20). If flares are exacerbated with bisphosphonate treatment, bisphosphonate should be discontinued.
Finally, farnesyl transferase inhibitors such as Manumycin A have been evaluated in mevalonate kinase deficiency. Interestingly, farnesyl transferase inhibitors may shift mevalonate pathway flux from protein farnesylation to geranylgeranylation (32). This may reduce geranylgeranylation defects and reduce inflammatory cytokine release. Prenylated proteins often get preferentially geranylgeranylated, and many of the prenylated proteins implicated in mevalonate kinase deficiency receive a geranylgeranyl anchor. As such, using farnesyl transferase inhibitors to upregulate geranylgeranylation may be beneficial (96). However, these farnesyl transferase inhibitors have not been evaluated in mevalonate kinase deficiency cell lines or patients with mevalonate kinase deficiency. Lonafarnib is an approved farnesyl transferase inhibitor, but this has not been tested in patients with mevalonate kinase deficiency. Because farnesyl transferase inhibitors may exacerbate the prenylation defect, use should be carefully evaluated as a final option if all other therapies fail. If flares are exacerbated with lonafarnib treatment, farnesyl transferase inhibitors should be discontinued.
With routine genetic testing, more variants of the MVK gene have been described, many of which are initially classified as variants of unknown significance in the absence of confirmatory biochemical assays measuring mevalonate kinase enzymatic activity. Evaluating mevalonate kinase activity often requires experimental lab analysis and mass spectroscopy experiments. If analysis cannot be performed and a patient is suspected of having mevalonate kinase deficiency or a novel MVK variant of unknown significance, providers are encouraged to contact labs that specialize in studying mevalonate kinase deficiency or cholesterol biosynthesis.
Pregnancy in patients with mevalonate kinase deficiency has not been widely described in the medical literature. However, patients heterozygous for MVK variants may present with fevers during pregnancy (61). Fetal tachycardia, intestinal obstruction, hydrops fetalis, and microcephaly may be noted during pregnancy (61).
Prenatal diagnosis of mevalonate kinase deficiency is possible via molecular genetic investigations in informative families. In at-risk families, mevalonic acid can be measured accurately in amniotic fluid by an isotope-dilution gas chromatography and mass spectrometry method, but this assay is not widely available. Determination of mevalonate kinase activity in cultured amniocytes and biopsied chorionic villus is also possible (65; 66; 63; 93). In affected pregnancies, elevated levels of mevalonic acid have been detected in maternal urine (65; 66; 93). Significant elevations of mevalonic acid were detected in autopsied tissues from an affected fetus, including lymph nodes, adrenals, ovaries, spleen, liver, and brain (66).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jack Drda
Mr. Drda of the University of Pittsburgh School of Medicine has no relevant financial relationships to disclose.
See ProfileRami Ballout MD
Dr. Ballout of UT Southwestern Medical Center has no relevant financial relationships to disclose.
See ProfileLyna-Nour Hamidi MD MSc
Dr. Hamidi of Hôpital du Sacré-Coeur de Montréal has no relevant financial relationships to disclose.
See Profile
Sylvain Lanthier MD
Dr. Lanthier of University of Montreal has no relevant financial relationships to disclose.
See Profile
Ezgi D Batu MD MSc
Dr. Batu of Hacettepe University has no relevant financial relationships to disclose.
See Profile
Robert D Steiner MD FAAP FACMG
Dr. Steiner of the University of Wisconsin School of Medicine and Public Health received consulting fees from Amgen, Astrellas, BridgeBio, Mirum, and research support from Regeneron.
See Profile
Deepa S Rajan MD
Dr. Rajan of UPMC Children's Hospital of Pittsburgh has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026