









Sleep Disorders
Sudden infant death syndrome
Jul. 05, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myoclonic absences are typical absence seizures of sudden onset and offset associated with consistent, repetitive, and rhythmic myoclonic jerks, mainly of the upper body with a tonic abduction, during the ictus. The defining manifestations of typical absence seizures are impairment of consciousness and generalized 3 to 4 Hz spike-wave discharges. Clonic (rhythmic) and myoclonic (rhythmic, arrhythmic, singular) motor symptoms often feature at some stage of the absence, but these are rarely consistent, marked, and continuous. Myoclonic absences are mainly associated with the syndrome of “epilepsy with myoclonic absences” and also “facial (perioral or eyebrow) myoclonia with absences” and “eyelid myoclonia and absences” induced by eye closure. The ILAE Task Force has recognized “epilepsy with eyelid myoclonia” as a syndrome. Myoclonic absences are often misdiagnosed as focal motor seizures with adverse consequences on management. Effective antiepileptic medications are valproate, levetiracetam, ethosuximide, lamotrigine, and clonazepam, usually in combination. In this updated article, the author details developments in the clinical and EEG manifestations, etiology, prognosis, differential diagnosis, and pharmacological treatment of myoclonic absences and related epileptic syndromes.
|
• Myoclonic absences are typical absence seizures with repetitive myoclonic jerks during the ictus. | |
|
• The classical type of myoclonic absences manifests with rhythmic jerks of shoulders, arms, and legs with a concomitant tonic contraction during 3 to 4 Hz generalized spike-wave discharges. The jerks correlate with the spike, and the tonic phase with the slow wave of the spike-slow wave discharge. This is the defining symptom of epilepsy with myoclonic absences. | |
|
• Other types of myoclonic absence seizures manifest with rhythmic jerks of perioral and/or eyebrow muscles or eyelid on eye closure during 3 to 4 Hz generalized spike-wave discharges; these are the defining symptoms of facial myoclonia with absences and epilepsy with eyelid myoclonia and absence seizures induced by eye closure (previously known as Jeavons syndrome). | |
|
• Myoclonic absences start in childhood and usually continue into adult life, often combined with generalized tonic-clonic and other types of seizure, such as atonic. | |
|
• Etiology of epilepsy with myoclonic absences is varied, whereas facial myoclonia or eyelid myoclonia on eye closure with absences are probably genetically determined idiopathic generalized epilepsies. | |
|
• Myoclonic absences are often misdiagnosed as focal motor seizures, though video-EEG recordings offer unequivocal documentation of the correct diagnosis. | |
|
• Myoclonic absences are usually resistant to monotherapy with an appropriate anti-absence drug. |
The first report of absence seizures with severe clonic or myoclonic jerks appeared in 1966 (44), but it was Tassinari and his associates who described and documented myoclonic absences as a specific seizure-type and included them a few years later in the syndrome of epilepsy with myoclonic absences (88; 08; 07).
Panayiotopoulos and associates described and documented perioral myoclonia with absences as a seizure type that may also constitute an epileptic syndrome (74; 73).
Myoclonic absences are a type of typical absence seizures with significant and continuous rhythmic (2.5 to 4.5 Hz) clonic rather than myoclonic symptoms and have a tonic component.
Typical absence seizures are brief, generalized epileptic seizures of sudden onset and termination. They have two essential components: (1) clinically, the impairment of consciousness (absence) and (2) EEG generalized 3 to 4 Hz (more than 2.5 Hz) spike-and-slow wave discharges (15; 72). Impairment of consciousness may be the only clinical symptom (simple typical absence seizures), but this is often combined with other manifestations (complex typical absence seizures).
In appreciation of this diversity, the ILAE Task Force on classification recognized four types of typical absence seizures, probably of different pathophysiology and syndromic significance: (1) the classical absence seizures of childhood and juvenile absence epilepsy, (2) myoclonic absences, (3) phantom absences, and (4) eyelid myoclonia with absence (34).
Myoclonic absences (the seizures) may feature either normal or neurologically and mentally abnormal children. Epilepsy with myoclonic absences (the syndrome) was previously categorized among the “cryptogenic or symptomatic generalized epilepsies and syndromes” (16). The ILAE diagnostic scheme considers only the idiopathic form, which probably represents less than a third of the whole spectrum of epileptic disorders manifesting with myoclonic absences (33). The others are symptomatic or probably symptomatic cases. This syndrome is also recognized in the ILAE proposal and listed amongst the electroclinical syndromes with onset in childhood (03). In the last ILAE Task Force on Nosology and Definitions, epilepsy with myoclonic absences is listed, together with childhood absence epilepsy and epilepsy with eyelid myoclonia, under the heading of genetic generalized epilepsy syndromes of childhood (80). Of the three syndromes, childhood absence epilepsy is included in the idiopathic generalized epilepsies, whereas the other two do not fit but have generalized spike-waves on EEG and generalized seizure types (51).
Facial myoclonia with absences. The term “facial myoclonia” instead of “perioral myoclonia with absences” better expresses the clinical variability of the syndrome because some cases involve the perioral region or eyebrow, and few involve perioral, eyebrow, and, rarely, head-jerking together with typical absence seizures. It is an idiopathic generalized epilepsy syndrome not yet recognized by the ILAE. Facial myoclonia with absences, most commonly referred to as perioral, is a discrete seizure type that has been unequivocally documented with video-EEG recordings (74; 50; 26; 02; 19; 29; 95; 85).
A 15-month-old boy was presented with a 2-month history of frequent episodes of a sudden loss of consciousness associated with perioral and eyebrow rhythmic jerks. The discharge was associated with impairment of consciousness and concomitant eyebrow and perioral rhythmic jerky movements. The electroclinical event lasted up to 19 seconds. The absence seizures are similar to those of childhood absence epilepsy. He responded on an appropriate daily dose of sodium valproate.
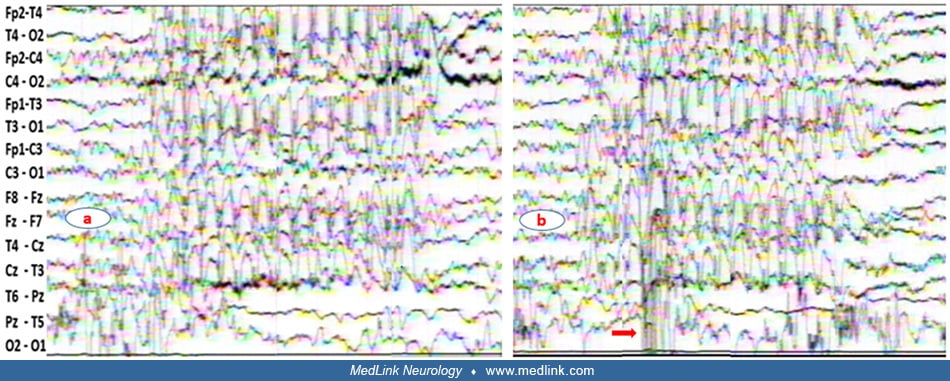
Illustrated is a generalized spike-wave discharge of a 15-month-old boy with a 2-month history of frequent episodes of sudden loss of contact associated with perioral and eyebrow rhythmic jerks. The discharge was associated wit...
A 22-month-old boy was presented with a history of daily brief episodes of staring associated with rhythmic eyebrow and head jerking. Video-EEG showed three repeated generalized spike-wave discharges, each associated with a brief typical absence seizure with concomitant rhythmic eyebrow and head jerking. During video-EEG, frequent generalized spike-wave discharges were recorded--some associated with clinical events. He was given sodium valproate and responded to it. Up to the age of 20 years old when he was last seen, a few attempts to withdraw treatment were associated with relapses, two with generalized tonic-clonic seizure during sleep. The cognitive development was assessed as borderline low. His older sister also had idiopathic generalized epilepsy.
A 5-year-old boy was presented because of frequent episodes of loss of contact concomitant with horizontal head jerky movements. EEG showed generalized spike-wave ictal discharge similar to childhood absence epilepsy, preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic horizontal head jerking. The boy’s eyes remained open with a vague look and no response. His head turned to the right side while jerking. He recovered after about 12 seconds.
EEG in 5-year-old boy shows generalized spike-wave ictal discharge similar to childhood absence epilepsy preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic ho...
An 8-year-old girl started having episodes with eyelid blinking, not responding to verbal commands, and de novo mouth movement. She was experiencing 10 to 15 episodes daily. Basic sleep-awake video-EEG did not show abnormalities. During hyperventilation, the generalized spike-wave discharge evoked was associated with the following seizure: eyelid flutter, no response to verbal commands, and concomitant de novo mouth movements. Some high-amplitude, 3 Hz slow waves heralded or followed the generalized discharges. A few other episodes recorded lasted from 11 to 14 seconds with similar electroclinical expression. Her seizures were finally controlled on ethosuximide and lamotrigine. After 4 years of good electroclinical response, lamotrigine was phased out. Eight months later, her EEG, during hyperventilation, showed some irregular spike-wave discharges, and during intermittent photic stimulation at 10 flashes per second, a generalized spike-wave discharge was evoked, but there were no concomitant clinical events. About 6 months later, she started having similar absence seizures, and lamotrigine was added to ethosuximide again. The past history was noncontributory except for 10 febrile seizures when he was younger than 5 years of age. Neurologic assessment was normal. Family history revealed her father had three febrile seizures in infancy.
Basic sleep-awake video-EEG from girl at the age of 8 years old who started having episodes with eyelid blinking, no responsiveness, and de novo mouth movement. She was having 10 to 15 episodes daily. The video-EEG did not show...
The symptoms of facial myoclonia may also occur in absence seizures of idiopathic generalized epilepsy, and, as such, facial myoclonia alone cannot be taken as sole evidence of the syndrome of facial myoclonia with absences. However, there is often a nonfortuitous electroclinical clustering of symptoms, indicating that these absences may often constitute the main symptom of a syndrome within the broad spectrum of idiopathic generalized epilepsy, for which proposed terminology is either perioral or, as preferred by this author, facial (perioral and/or eyebrow) myoclonia with absences (74; 26; 71; 19). Other manifestations of this syndrome include head clonic-myoclonic jerks with absences; generalized tonic-clonic seizures (GTCS), which often start early before or after the absences; frequent occurrence of absence status epilepticus; resistance to treatment; and persistence in adult life.
In children, the term facial myoclonia with absences is used for the reason that some cases may present with perioral (45.5%) or eyebrow (41%) myoclonia with absences or even a combination of perioral and eyebrow myoclonia with absences (13.5%). The age of onset varies from 1 to 13 years (mean 5±3). EEG is generalized spike-wave or polyspike-wave discharges 2 to 4.5 Hz, duration 0.25 to 20 seconds (26). The response to sodium valproate is 77%, increased to 91% if combined with ethosuximide. Typical absence seizures with facial or other myoclonias have variable clinical and EEG expression, and those with GTCS have less favorable outcomes (26). In the literature, the syndrome is described with the title perioral myoclonia with absences, which partially reflects the phenotype. Absence status epilepticus is very uncommon in children.
Cases with absence and myoclonic seizures, the predominant type of seizures in their phenotype, characterize the syndromes epilepsy with myoclonic absences and eyelid myoclonia and absences on eye closure-Jeavons syndrome (22; 21); genetic generalized syndrome named “epilepsy with eyelid myoclonia” has also been recognized (96). Facial (perioral and/or eyebrow) myoclonia with absences is not recognized as a syndrome by ILAE (19; 20).
See also the Medlink article on “Epilepsy with eyelid myoclonia and absences.”
In the ILAE “Epilepsy Diagnosis” manual, myoclonic absence seizures are considered as one of the four types of generalized absence seizures (typical, atypical, myoclonic, with eyelid myoclonia) and are described as follows (14):
Myoclonic absences. Myoclonic absences are rhythmic myoclonic jerks of the shoulders and arms with tonic abduction, which results in progressive lifting of the arms during the seizure. The myoclonic jerks are typically bilateral but may be unilateral or asymmetric. Perioral myoclonias and rhythmic jerks of the head and legs may occur. Seizures last 10 to 60 seconds and typically occur daily. The level of awareness varies from complete loss of awareness to retained awareness.
EEG background. Please refer to the specific syndrome in which this seizure type occurs.
Ictal EEG. Around 3 Hz generalized spike-and-wave. EMG recordings from the upper arm show a constant relationship between the bilateral myoclonic jerks and spike-and-waves.
Differential diagnosis.
|
• Typical absence (with marked clonic-myoclonic components) |
Related syndromes.
|
• Epilepsy with myoclonic absences |
According to the position paper of the ILAE Commission for Classification and Terminology:
|
A myoclonic absence seizure refers to an absence seizure with rhythmic three-per-second myoclonic movements, causing ratcheting abduction of the upper limbs leading to progressive arm elevation, and associated with three-per-second generalized spike-wave discharges. Duration is typically 10 to 60 s. Impairment of consciousness may not be obvious. Myoclonic absence seizures, classified as nonmotor (absence) by ILAE occur in a variety of genetic conditions and also without known associations (38; 37). |
However, this ILAE operational classification of epileptic seizures has been criticized by a number of experts (91; 60). A comprehensive assessment of absence seizures concludes that:
|
The classification as “generalized motor and nonmotor (absence) seizure” does not convey the complex semiology of a patient's clinical events (91). |
According to the various ILAE classifications and definitions, generalized-onset clonic seizures are mainly distinguished from generalized myoclonic seizures by their rhythmicity only and nothing else. This has created significant overlap in what to call clonic or myoclonic seizures, and often these terms are used interchangeably.
The ILAE position paper states that “clonic refers to sustained rhythmic jerking and myoclonic to regular unsustained jerking” and that “the distinction between clonic and myoclonic is somewhat arbitrary, but clonic implies sustained, regularly spaced stereotypical jerks, whereas myoclonus is less regular and in briefer runs. Myoclonus differs from clonus by being briefer and not regularly repetitive. Myoclonus as a symptom has possible epileptic and nonepileptic etiologies” (37). In the glossary of the same paper, myoclonic is defined as “sudden, brief (< 100 msec) involuntary single or multiple contraction(s) of muscles(s) or muscle groups of variable topography (axial, proximal limb, distal). Myoclonus is less regularly repetitive and less sustained than is clonus”, as opposed to clonic, which is “jerking, either symmetric or asymmetric, that is regularly repetitive and involves the same muscle groups” (37).
Myoclonic seizures in childhood are usually brief, sudden, involuntary muscle contractions that may involve the whole or part of the body (focal, segmental) and may be single or repetitive, rhythmic or arrhythmic, unilateral or bilateral (massive), symmetrical or asymmetrical. The upper part of the body, particularly the head, is usually involved, and the jerks may be subjective or objective, recorded as slight, moderate, or marked jerks. The jerks occur at the onset, in the course of, or immediately after the generalized spike-wave discharges and absence seizure; usually, brief (eyes open and stare) may precede or follow the jerk. During intermittent photic stimulation, the provoked clinical phenomena are similar to the spontaneous or may be the first manifestation. The cardinal symptoms of the syndrome epilepsy with myoclonic absences are typical absences characterized by the impairment of consciousness and myoclonias, mainly of the upper part of the body, with concomitant tonic contraction (20). The onset and termination of the seizure are sudden. The seizures may start in half to one second after the onset of the discharge; the patient seems to regain consciousness and stops jerking about one second before the discharge ends (20). The associated 3Hz spike-wave discharges are bilateral, synchronous, and symmetrical. The spikes coincide with the jerks, and the silent period corresponds with the tonic phase. The duration in the majority of cases varies from 5 to 20 seconds. The ILAE position paper on epilepsy syndromes in children, based on Zanzmera’s study, states that myoclonic absences last 10 to 60 seconds (98; 80). A large comprehensive list of various seizure durations studies provides a maximum seizure duration for myoclonic absence at 18 seconds (59).
A few typical examples of “myoclonic absence seizures” are as follows.
Myoclonic absence seizures, example 1. An 11-year-old boy suffered from epilepsy with myoclonic absences of idiopathic (unknown) cause since the age of 12 months, when he was noticed to have jerks of the upper part of his body (head, upper limbs) mostly induced by noise. These episodes, observed a few times per week, were described as earthquake-like. At the age of 6-years-old, he was reported to have about 10 episodes daily of vague looks concomitant with rhythmic jerking of the head and upper limbs. The episodes had not responded to high doses of sodium valproate, lamotrigine, clobazam, or levetiracetam. When ethosuximide was introduced to his treatment instead of lamotrigine, absence episodes responded, but he had, for the first time, three generalized tonic-clonic seizures during which his head and eyes turned left and mouth twisted; clonic-myoclonic movements of upper limbs occurred for a few minutes, and he did not respond for about 45 minutes.
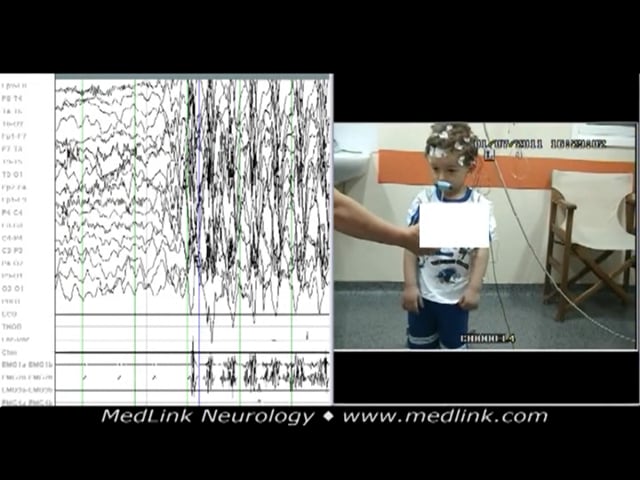
Myoclonic absence seizures, example 2. A 5-year-old boy presented with a month’s history of sudden episodes of abnormal movement of the upper part of his body and poor response; some movements, mainly of the right upper limb, were reported. The episodes were diagnosed as focal impaired awareness seizures, and he was given a sodium channel-blocking antiseizure drug. As a consequence, seizures increased in frequency to many episodes daily.
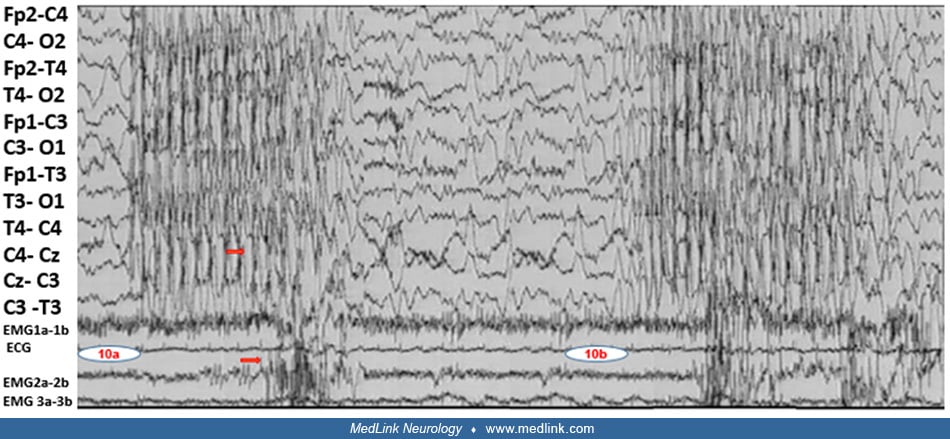
The polygraphic video-EEG spike-wave generalized discharge was recorded in a 5-year-old boy during hyperventilation while standing. The recorded generalized spike-wave discharge was initially associated with abrupt onset of rhy...
Myoclonic absence seizures, example 3. A boy presented with a variety of typical myoclonic absence seizures while standing. Synchronous with the generalized spike-wave discharge, the upper part of his body, upper limbs, and right leg rhythmically jerk with concomitant unsteadiness, but he didn’t fall.
Myoclonic absence seizures are characterized by typical absences associated with rhythmic bilateral clonic jerking, mainly of the upper body, with a tonic component. The upper limbs fling open (tonic phase) and, as they jerk, elevate progressively. The body may also deviate slightly and, with each rhythmic jerk, bend toward the floor. The ictal EEG shows bilateral generalized spike-wave discharges at 3 Hz (20).
Clonic (rhythmic) and myoclonic (arrhythmic, singular) symptoms are particularly frequent at the onset of typical absence seizures but may also occur at any other stage of the seizure randomly, continuously, or repetitively (09; 82; 72; 73; 20). The most common symptoms are clonic or myoclonic jerking of the eyelids, eyebrows, and eyeballs, including random or repetitive eye closures and horizontal or vertical nystagmus-like ocular movements.
A few other episodes recorded lasted from 11 to 14 seconds, with similar electroclinical expression. Her seizures were finally controlled on ethosuximide and lamotrigine. After 4 years of good electroclinical response, lamotrigine was phased out. Eight months later, her EEG during hyperventilation showed some irregular spike-wave discharges, and during intermittent photic stimulation at 10 flashes/second, a generalized spike-wave discharge was evoked, but no concomitant clinical events were noted. About 6 months later, she started having similar absence seizures, and lamotrigine was added to ethosuximide again. The past history was noncontributory except for 10 febrile seizures before she was 5 years old. Neurologic assessment was normal. Family history revealed that her father had three febrile seizures in infancy.
Fast eyelid flickering is probably the most common ictal clinical manifestation and may occur during brief generalized discharges without discernible impairment of consciousness. This should not be confused with eyelid myoclonia and absences on eye closure, the cardinal symptom of what was previously called Jeavons syndrome, where most cases are photosensitive (18; 22; 21; 25). Perioral myoclonias at the corner of the mouth and jerking of the jaw are less common. Clonic or myoclonic jerks of the head, body, and limbs are less frequent than those of the face and may be mild or violent, singular, continuous, or repetitive. In some patients with absence seizures, single myoclonic jerks of the head and, less often, the limbs may occur during the progression of ictus.
A 5-year-old boy was presented because of frequent episodes of loss of contact concomitant with horizontal head jerky movements. EEG showed generalized spike-wave ictal discharge similar to childhood absence epilepsy, preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic horizontal head jerking. The boy’s eyes remained open with a vague look and no response. His head turned to the right side while jerking. He recovered after about 12 seconds.
EEG in 5-year-old boy shows generalized spike-wave ictal discharge similar to childhood absence epilepsy preceded by high amplitude delta-theta waves in centroposterior regions. The discharge immediately starts with rhythmic ho...
However, not any typical absence seizures with the above myoclonic and clonic motor manifestations are classified as myoclonic absence seizures, which are mainly of two types according to the muscles involved: (a) the classical type manifests with rhythmic jerks of shoulders, arms, and legs with a concomitant tonic contraction during absence seizures and constitutes the defining symptoms of epilepsy with myoclonic absences; and, (b) myoclonic absences manifest with rhythmic jerks of perioral or eyebrow muscles during absence seizures, and they are the defining symptom of facial myoclonia with absences.
Classical type of myoclonic absences. Classical myoclonic absences manifest with rhythmic jerks of the shoulders, arms, and legs with a concomitant tonic contraction during absence seizures and constitute the defining symptoms of epilepsy with myoclonic absences (88; 92; 08; 07; 42; 82; 20). The myoclonic absence seizures consist of impairment of consciousness, which is usually severe.
An 11-year-old boy suffered from epilepsy with myoclonic absences of idiopathic (unknown) cause since the age of 12 months, when he was noticed to have jerks of the upper part of his body (head, upper limbs) mostly induced by noise. These episodes, observed a few times per week, were described as earthquake-like. At the age of 6 years old, he was reported to have about 10 episodes daily of vague looks concomitant with rhythmic jerking of the head and upper limbs. The episodes had not responded to high doses of sodium valproate, lamotrigine, clobazam, or levetiracetam. When ethosuximide was introduced to his treatment instead of lamotrigine, absence episodes responded, but he had, for the first time, three generalized tonic-clonic seizures during which his head and eyes turned left and mouth twisted; clonic-myoclonic movements of upper limbs occurred for few minutes, and he did not respond for about 45 minutes.
A 5-year-old boy presented with a month’s history of sudden episodes of abnormal movement of the upper part of his body and poor response; some movements, mainly of the right upper limb, were reported. The episodes were diagnosed as focal impaired awareness seizures, and he was given a sodium channel-blocking antiseizure drug. As a consequence, seizures increased in frequency to many episodes daily.
The polygraphic video-EEG spike-wave generalized discharge was recorded in a 5-year-old boy during hyperventilation while standing. The recorded generalized spike-wave discharge was initially associated with abrupt onset of rhy...
A boy presented with a variety of typical myoclonic absence seizures while standing. Synchronous with the generalized spike-wave discharge, the upper part of his body, upper limbs, and right leg rhythmically jerk with concomitant unsteadiness, but he didn’t fall.
The duration of the absences varies from a few seconds to 60 seconds, with a high daily frequency of occurrence. They are precipitated by hyperventilation and occur mainly on awakening. Photosensitivity is uncommon (14%). Myoclonic absences precipitated by eye-closure or eye-opening in non-photosensitive patients have been described (75; 88). Other types of seizures also occur in two thirds of the patients (75; 88; 31; 73). These are infrequent generalized tonic-clonic seizures, “absences” or “falling attacks,” and atonic seizures that may precede or follow the onset of myoclonic absences. Absence status is rare. Absences with persistent localized neck myoclonia have been described (55). Complex gestural automatisms can rarely occur during myoclonic absence seizures (67).
Myoclonic absences are a rare seizure type (probably 0.5% in selected cases of epilepsy) with a male preponderance (70%) and a mean age of onset at 7 years (07), starting as early as 4 years (31) and 6 months (92). Neurologic examination is usually normal, but half (45%) have impaired cognitive functioning before the onset of absences. A family history of epilepsy is found in a fifth of the cases. Etiological factors, mainly chromosomal abnormalities, are found in one third of patients (31; 32), prematurity, and perinatal damage (07).
See also MedLink Neurology article: Epilepsy with myoclonic absences.
Second type of myoclonic absences. The second type of myoclonic absences manifests with facial rhythmic jerks of perioral or eyebrow muscles during absence seizures, and they are the defining symptom of facial (perioral) myoclonia with absences (74; 26; 18; 22; 50; 04; 02; 19; 20; 29; 95). The characteristic feature is perioral myoclonia, which, as studied on video-EEG, consists of rhythmic contractions of the orbicularis oris muscle (causing protrusion of the lips), contractions of the depressor anguli oris (resulting in twitching of the corners of the mouth), or, rarely, more widespread involvement to include the muscles of mastication (producing jaw jerking). Another motor manifestation during the absence is eyebrow or eyelid myoclonus, which is synchronous but less prominent than the perioral myoclonus (74; 70; 29). Because not only perioral muscles are involved, the term facial instead of perioral myoclonia with absence should be preferred (26; 19; 20). Impairment of consciousness varies from severe to mild, and most patients are usually aware of the perioral myoclonia. Duration is usually brief, lasting a mean of 4 seconds (range 2 to 20 seconds). Absences of facial myoclonia may occur many times per day, one to two times per week, or they may be rare. Patients with facial myoclonia with absences may present from early childhood to early adolescence, usually following a GTCS, or less commonly with absences or absence status epilepticus. All patients suffer generalized tonic-clonic seizures, which often start before or soon after the onset of clinically apparent absences. Exceptionally, GTCS may start many years after the onset of absences. GTCS are usually infrequent (ranging from once in a lifetime to 12 per year) and are often heralded by clusters of absences or absence status. Absence status epilepticus is very common (57%) and frequently ends with GTCS.
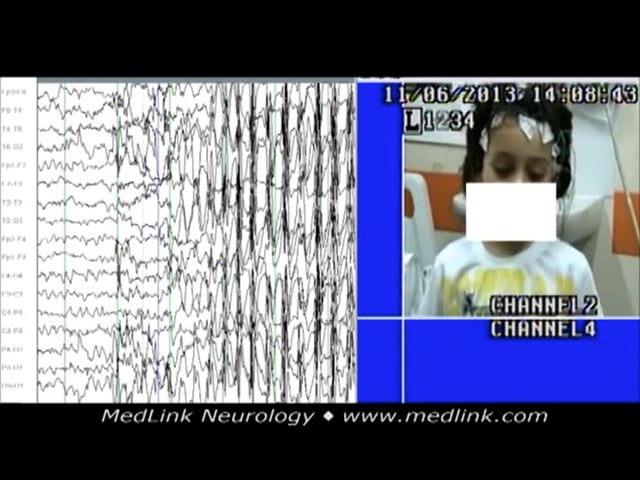
A 15-month-old boy was presented with a 2-month history of frequent episodes of sudden loss of consciousness associated with perioral and eyebrow rhythmic jerks. The discharge was associated with impairment of consciousness and concomitant eyebrow and perioral rhythmic jerky movements. The electroclinical event lasted up to 19 seconds. The absence seizures are similar to those of childhood absence epilepsy. He responded to an appropriate daily dose of sodium valproate.
Illustrated is a generalized spike-wave discharge of a 15-month-old boy with a 2-month history of frequent episodes of sudden loss of contact associated with perioral and eyebrow rhythmic jerks. The discharge was associated wit...
A 22-month-old boy was presented with a history of daily brief episodes of staring associated with rhythmic eyebrow and head jerking. Video-EEG showed three repeated generalized spike-wave discharges, each associated with a brief typical absence seizure with concomitant rhythmic eyebrow and head jerking. During video-EEG, frequent generalized spike-wave discharges were recorded--some associated with clinical events. He was given sodium valproate and responded to it. Up to the age of 20 years old, when he was last seen, a few attempts to withdraw treatment were associated with relapses, two with generalized tonic-clonic seizure during sleep. The cognitive development was assessed as borderline low. His older sister also had idiopathic generalized epilepsy.
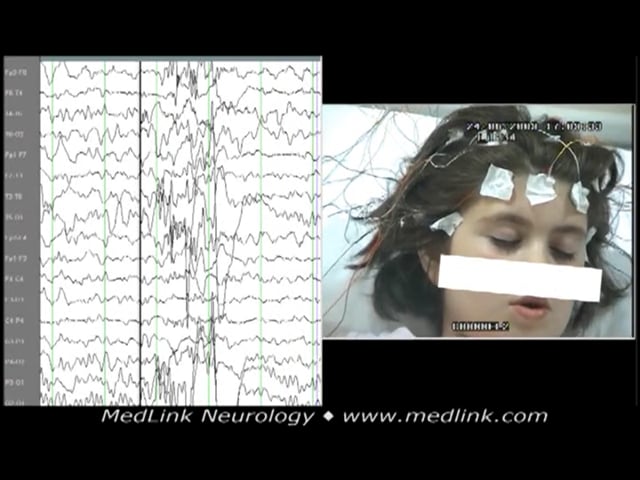
An 8-year-old girl started having episodes with eyelid blinking, not responding to verbal commands, and de novo mouth movement. She was experiencing 10 to 15 episodes daily. Basic sleep-awake video-EEG did not show abnormalities. During hyperventilation, the generalized spike-wave discharge evoked was associated with the following seizure: eyelid flutter, no response to verbal commands, and concomitant de novo mouth movements. Some high-amplitude, 3 Hz slow waves heralded or followed the generalized discharges. A few other episodes recorded lasted from 11 to 14 seconds with similar electroclinical expression. Her seizures were finally controlled on ethosuximide and lamotrigine. After 4 years of good electroclinical response, lamotrigine was phased out. Eight months later, her EEG during hyperventilation showed some irregular spike-wave discharges, and during intermittent photic stimulation at 10 flashes per second, a generalized spike-wave discharge was evoked, but there were no concomitant clinical events. About 6 months later, she started having similar absence seizures, and lamotrigine was added to ethosuximide again. The history was noncontributory except for 10 febrile seizures when he was younger than 5 years of age. Neurologic assessment was normal. Family history revealed her father had three febrile seizures in infancy.
Basic sleep-awake video-EEG from girl at the age of 8 years old who started having episodes with eyelid blinking, no responsiveness, and de novo mouth movement. She was having 10 to 15 episodes daily. The video-EEG did not show...
There are three characteristic idiopathic generalized syndromes wherein absence and myoclonic seizures predominate in the phenotype, namely: epilepsy with myoclonic absences, eyelid myoclonia and absences precipitated on voluntary or on-command eye closure in the presence of light (22; 21; 25), and facial myoclonia with absences (19; 20).
Epilepsy with myoclonic absences is often resistant to treatment; 50% of patients (probably with structural causes) continue having seizures in adult life, with a high incidence of generalized tonic-clonic and atonic types. Rarely, patients may develop features of other types of epilepsy such as Lennox-Gastaut syndrome or juvenile myoclonic epilepsy. Of those who were normal before the onset of absences, 50% develop cognitive and behavioral impairment. Mental retardation is present only in patients with poor seizure control (92). Frequent GTCS is a predictor of bad prognosis (07). The fact that 45% have impaired cognitive functioning before the onset of absences may indicate the negative effect seizures and EEG discharges have on cognition and evolution.
See also MedLink Neurology article: Epilepsy with myoclonic absences.
Death in the rare occasion of generalized tonic-clonic status epilepticus may occur (98). Absence status epilepticus continues in adult life for half of the patients.
Clinical vignette 1. A 7.5-year-old boy suffered from idiopathic (unknown) episodes of myoclonic absences. The absences started about 2 months prior to being seen and consisted of vague looks and occasional dropping of items from his hands. In the previous week, the frequency had increased to five to seven episodes daily.
During hyperventilation in another episode, a single eyelid jerk and a single upper limb myoclonic jerk were part of the described myoclonic absence seizure. Perinatal and past history were noncontributory. Neurologic examination, neuroimaging, and relevant investigations were normal. The electroclinical events responded to sodium valproate, and he had remained free of seizures and EEG discharges for 3 years when last seen.
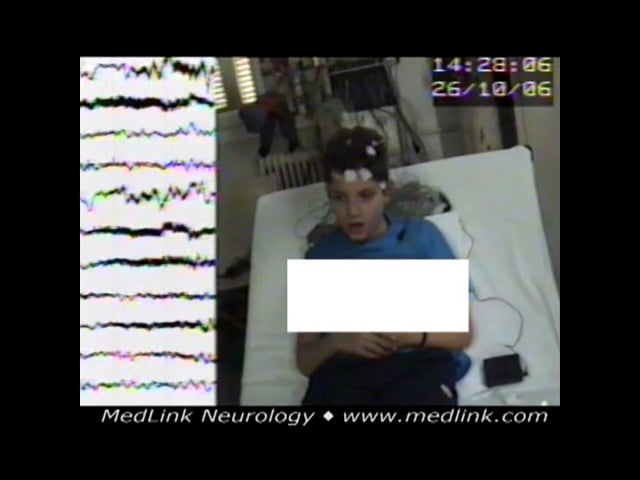
Clinical vignette 2. An 8-year-old boy was brought for examination because of some jerking of his upper limbs close to his chest. The jerks had first appeared 4 months before examination at a frequency of three to four per week but had progressed to one to two per day. The parents also noticed some “vague looks.”
Neurologic assessment revealed hyperkinesis, stuttering, poor attention span, immature behavior, and clumsiness. There were no lateralized focal findings. MRI was normal. Cognitive assessment determined the patient performed at the level of a 4- to 5-year-old, and he was reported to have severe educational problems. Past and family history were noncontributory. He was given sodium valproate up to 30mg/kg and remained free of seizures for 2.5 years when his treatment was withdrawn. He relapsed with up to 10 myoclonic absence seizures per day. Valproate was reintroduced at a daily dose of 24mg/kg. Four years later, he was free of seizures, and the daily dose was reduced to 11mg/kg; but 18 months later (age 16 years old), he did not take treatment for 5 days, and he relapsed again. Subsequently, he decided to comply with treatment and manage his weight gain, due to sodium valproate, with alternative measures.
Clinical vignette 3. An 11-year-old boy suffered from epilepsy with myoclonic absences of idiopathic (unknown) cause since the age of 12 months, when he was noticed to have jerks of the upper part of his body (head, upper limbs) mostly induced by noise. These episodes, observed a few times per week, were described as earthquake-like. At the age of 6 years old, he was reported to have about 10 episodes daily of vague looks concomitant with rhythmic jerking of the head and upper limbs. The episodes had not responded to high doses of sodium valproate, lamotrigine, clobazam, or levetiracetam. When ethosuximide was introduced to his treatment instead of lamotrigine, absence episodes responded, but he had, for the first time, three generalized tonic-clonic seizures during which his head and eyes turned left and mouth twisted; clonic-myoclonic movements of upper limbs occurred for a few minutes, and he did not respond for about 45 minutes.
Clinical vignette 4. A 6-year-old girl suffered from myoclonic absences of idiopathic (unknown) cause since the age of 5.9 years. Her head would bend forward, her eyes were open with a vague look concomitant with upper limb and right leg jerking, and unsteadiness; occasionally, she would fall. If an episode happened while she was doing homework, she would be surprised to find all her work disorganized when she recovered. If she held things, she dropped them. She had daily episodes.
She responded to sodium valproate 30mg/kg. Five years later, valproate was reduced to 17 mg/kg, and treatment was discontinued a year later. From the start, she was discovered to have moderate to severe educational difficulties that made her discontinue secondary level education, and she was receiving special care. She had remained free of seizures with normal follow-up EEG for 9 years off treatment when she was last seen.
Myoclonic absences are generalized epileptic seizures with mainly 3 to 6 Hz generalized multiple spike-and-slow wave discharges of higher amplitude in the anterior regions (14).
The etiology of myoclonic absences is unknown. A seemingly homogenous electroclinical phenotype may reflect a variable degree of genetic complex/polygenic heterogeneity with a family history positive for epilepsy in approximately 25% of the cases. Additional etiological factors have been found, such as prematurity, perinatal hypoxia, congenital hemiplegia, partial trisomy of the long arm of chromosome 14 (08), trisomy 12p syndrome (32), mutation in the SLC2A1 gene (46), and de novo variants in SETD1B gene-related disorders (49).
The underlying pathophysiology of myoclonic absences is unknown but may have similarities with the pathophysiology of typical absence seizures. It appears that the basic pathophysiological mechanisms involved in the generation of spike and wave discharges in the thalamic relay neurons depend on inhibitory inputs from neurons of the nucleus reticularis thalami that engage different hyperexcitable cortical areas. The degree of cortical involvement may be related proportionally to the instability of the T-type Ca²+ channel in the thalamus, and the ionized, not protein-bound calcium affects the excitability of muscle cells and neurons. Neither the cortex nor the thalamus alone can sustain these discharges, indicating that both structures are involved in their generation. Meeren and colleagues described a consistent focus within the perioral region of the somatosensory cortex in epileptic rats with absence seizures (64). Assessment of their electroclinical characteristics supports the cortical focus theory of absence seizures. During the first cycles of the seizure, the cortex drives the thalamus, whereas thereafter, the cortex and thalamus drive each other, thus amplifying and maintaining the rhythmic discharge (64). Simultaneous EEG and fMRI measurements document cortical deactivation and thalamic activation. Cortical deactivation is related to slow waves and disturbances of consciousness of varying degrees. Motor symptoms correspond to the spike component of the 3 Hz spike-and-wave discharges. Thalamic activation can be interpreted as a response to overcome cortical deactivation and to restore normal cortical functions (91).
Ikeda and colleagues studied myoclonic absences with ictal single-photon-emission computed tomography in two patients (53). They performed ictal and interictal (99 m)Tc-ethyl cysteinate dimer SPECT. They then generated images of subtraction ictal SPECT coregistered to MRI from the interictal and ictal data to evaluate topographic changes in cerebral blood flow during myoclonic absences as compared to the interictal state. In one patient, the cerebral blood flow increased in the perirolandic areas, thalamus, caudate nucleus, and precuneus, and decreased in the middle frontal gyrus and bilateral orbitofrontal regions. In the other patients, cerebral blood flow increased in the thalamus, putamen, and globus pallidus but was decreased in the precuneus. These results indicate that in addition to the thalamus and basal ganglia, the perirolandic cortical motor area is involved in myoclonic absences. The authors hypothesized that in myoclonic absences, the blood perfusion in the perirolandic cortical motor area might have changed under the influence of the cortico-thalamic network oscillation features (53).
De Saint Martin and colleagues reported a young girl with rolandic seizures that started at the age of 3 years 6 months (28). In addition, between the ages of 5 and 6 years, the girl had (1) increased seizure frequency; (2) brief perioral and palpebral myoclonic jerks, concomitant with the spike component of interictal spike-waves, and (3) persistent but fluctuating oromotor deficits (drooling, dysarthria, dysphagia). The EEG showed a marked increase in abundance and amplitude of wake and sleep interictal abnormalities, which became bilateral. Awake FDG-PET revealed a bilateral increase of glucose metabolism in opercular regions. A complete and definitive EEG and clinical remission occurred at age 5 years 11 months and persisted through the last interview at age 7 years 9 months. The authors concluded that epileptiform dysfunctions within rolandic areas may induce "interictal" positive or negative oromotor symptoms, independent of classic seizures.
The classical type of myoclonic absences is a rare seizure type (probably 0.5% in selected cases of epilepsy) with a male preponderance (70%) and a mean age of onset at 7 years (07), starting as early as 4 years (31) and 6 months (92) or even earlier from the age of 1 year (54).
Perioral myoclonia with absences is also very rare, probably less than 1% of typical absence seizures in children, but because it fails to remit, it is higher in adults with absences (9.3%). Girls are affected far more frequently than boys (73). In children, based on 13 cases with facial myoclonia, 46.5% had perioral, 38.5% eyebrow, and 15% perioral combined with eyebrow myoclonia with absences. The male-to-female ratio was 1.6:1 (26).
The prevention is based on early correct diagnosis, using appropriate antiseizure medication at an optimum daily dose, considering the risk-to-benefit ratio. Reducing underlying causes, preventing triggering risk factors, assuring compliance, and avoiding sodium channel blocker antiepileptic drugs will help prevent seizures. Early diagnosis and treatment of comorbidities will also improve quality of life.
Myoclonic absences are usually typical, and the diagnosis is mainly based on a detailed history, careful clinical observations, and video-EEG studies with polygraphic recording (EMG of various shoulder and arm muscles) of the electroclinical episodes (75; 09; 72; 20; 07). Clinically, the rhythmic myoclonic movements can be overlooked due to tonic contraction of the arm, or the intensity of the myoclonias can be reduced due to treatment. When the motor manifestations are asymmetric, focal motor seizures are erroneously diagnosed.
Myoclonic absences differ from typical absence seizures of childhood absence epilepsy because myoclonic jerks are generally not seen in childhood absence epilepsy. If they are present, they are subtle. The lack of atonic component also distinguishes myoclonic absences from myoclonic atonic seizures.
The differential diagnosis of epilepsy with myoclonic absences from other syndromes with absences is easy because of the characteristic type of myoclonic absences. The difficulty is between idiopathic and symptomatic and probably symptomatic (structural, unknown) cases that manifest with the same seizure type (myoclonic absences). Symptomatic cases often have an abnormal neurologic state, abnormal background EEG, and abnormal brain MRI, and some may necessitate meticulous biochemical assessment. A minority may continue to be unknown. Chromosomal abnormalities are common. Additionally, absences with rhythmic myoclonic jerking but less than 2.5 Hz generalized spike-wave discharges and other characteristics of atypical absences may occur in epileptic encephalopathies (36; 07; 23; 24), and these may account for some of the cases with chromosomal abnormalities (31).
GLUT1 deficiency is a metabolic disorder due to defective glucose transport across the blood-brain barrier. It should be suspected when absence seizures are associated with at least one among the following: irregular ictal EEG discharges, mild intellectual disability, migraine, microcephaly, drug resistance, and worsening during fasting (78); also, if it coexists with complex movement disorders characterized by ataxia, dystonia, and chorea, which could be paroxysmal or continuous, and may be influenced by fasting, fever, or infection. However, many patients with Glut1-deficiency syndrome remain undiagnosed or misdiagnosed on account of the disorder’s varied phenotype (97).
Facial (perioral or eyebrow) myoclonia with absences is frequently misdiagnosed as focal motor seizures. Also, patients with focal motor seizures are unlikely to suffer from nonconvulsive status epilepticus, which is observed in facial myoclonia with absences. The main differential diagnosis of facial myoclonia with absences is from other syndromes of idiopathic generalized epilepsy with absences. Video-EEG invariably reveals the electroclinical characteristics of facial (perioral or eyebrow) myoclonia that sometimes may be subtle, particularly in treated patients. Useful clinical indicators in favor of facial myoclonia with absences and against the syndrome of childhood absence epilepsy are: (1) onset of generalized tonic-clonic seizures before or at the same age as typical absences, (2) relatively brief duration of absences with facial myoclonia, and (3) frequent occurrence of absence status epilepticus. Perioral myoclonia may rarely occur in absence seizures of other idiopathic generalized epilepsies (50).
Long-lasting atypical status epilepticus associated with an impairment of attention and continuous polymorphous jerks mixed with other complex abnormal movements in infants suffering from a nonprogressive encephalopathy was described by Caraballo and colleagues (10). One subset of the clinical manifestations includes absences, subcontinuous jerks (at times rhythmic or arrhythmic), and mainly positive brief myoclonic absences. A second subset is characterized by absence status and mainly negative, continuous, rhythmic myoclonus and dyskinetic movements. And a third subset shows continuous spike activity in rolandic regions as well as bilateral rhythmic myoclonias, followed by inhibitory phenomenon. These cases with nonprogressive encephalopathies have to be differentiated from other forms, including neuronal ceroid-lipofuscinosis.
Myoclonic absences are seen in the syndromes of epilepsy with myoclonic absences (recognized by the ILAE commission) as well as facial (perioral or eyebrow) myoclonia with absences and eyelid myoclonia and absences induced immediately after voluntary or on-command eye closure in the presence of light.
Epilepsy with myoclonic absences. Briefly, epilepsy with myoclonic absences is a rare syndrome of idiopathic generalized epilepsy that demands scrupulous exclusion of other forms of structural cases manifesting with the same seizure (myoclonic absences). Myoclonic absences are the defining symptom with rhythmic myoclonic jerks mainly of the shoulders, arms, and legs. GTCS or atonic fits occur in two thirds of patients, predicting an unfavorable prognosis. These are cases of structural epilepsy or cases with other types of seizures, or may indicate a wider genetic expression (20). Absence status epilepticus is rare (73; 07; 98).
The ILAE diagnostic scheme considers only the idiopathic form, which probably represents less than a third of the whole spectrum of epileptic disorders manifesting with myoclonic absences (34). Prevalence is very low (less than 0.5% to 1%) among selected patients with epileptic disorders.
Age at onset is between 5 months and 13 years (median 7 years), and males (70%) predominate.
Neurologic and mental state. One third of patients are normal (idiopathic form). The others are of a structural cause, mainly with learning difficulties before seizures.
Etiology. The etiology of epilepsy with myoclonic absences is unknown but presumed to be genetic. In a seemingly homogenous electroclinical phenotype, there appears to be the expression of a variable genetic heterogeneity (complex or polygenic) (20). One quarter of patients have a family history of epilepsy. Two siblings with epilepsy with myoclonic absences have been reported (13).
Myoclonic absences (the seizures, not the syndrome) are due to idiopathic, genetic, structural, or cryptogenic causes, including chromosomal abnormalities. Elia and associates investigated etiology in 14 patients with epilepsy with myoclonic absences (31; 32). Five were cryptogenic. Two had myoclonic absences associated with other seizure types due to neuronal migration abnormality or a metabolic disorder. Seven, with or without other seizure types, had chromosomopathies (two trisomy 12p, four Angelman syndrome, and one with inv dup (15)). One of these cases with trisomy 12p syndrome was also detailed.
Of interest are the few cases of myoclonic absences occurring in glucose transporter type 1 (GLUT1) deficiency syndrome with mutations in the SLC2A1 gene (46). This syndrome often manifests with a variety of absence seizures that are usually of early onset (84; 65; 76; 83; 78; 68).
Bahi-Buisson and colleagues described a family with a dominantly inherited mutation in glutamate dehydrogenase (GDH), with the mother, brother, and both sisters having myoclonic absence seizures, but only the mother and one sister had the complete hyperinsulinism/hyperammonemia pattern (01). For the two sisters with myoclonic absences, epilepsy started during the second year of life, whereas in the brother, it started at 6 years. All three children showed the same EEG pattern characterized by photosensitive, generalized, and irregular spike-wave discharges and runs of multiple spikes. The mother's EEG recordings were normal without photosensitivity. Magnetic resonance imaging and spectroscopy were normal (01).
Klitten and colleagues described a patient with epilepsy with myoclonic absences and learning disability who was found to have a de novo balanced translocation: t(6; 22)(p21.32; q11.21) (57). The breakpoint at 6p21.32 was found to truncate the N-methyl-d-aspartate (NMDA) receptor-associated gene SYNGAP1. The breakpoint at 22q11.21 was within a highly variable region without known protein-coding genes. Mutations of SYNGAP1 are associated with nonsyndromal intellectual disability, and two thirds of the patients have generalized epilepsy (57).
Talarico and colleagues reported that a spectrum of myoclonic epilepsy syndromes, including myoclonic absences, was associated with RORA-related neurodevelopmental disorder, with the RORA gene being significantly involved in cerebellar maturation (86).
A 2025 case report identified a pathogenic CREBBP variant in a patient with epilepsy with myoclonic absences, highlighting a potential link to Rubenstein-Taybi syndrome (63).
Myoclonic absences can rarely occur in SCN8A infantile developmental and epileptic encephalopathy (41). Generalized epilepsy seizures with an important subset of myoclonic absences are also reported in SYNGAP1 and SETD1B-related intellectual disability and autism spectrum disorder (49; 52). Seizures are frequent and drug-resistant in 50% of cases. Adding cannabidiol in three cases with SYNGAP1 drug-resistant cases showed reported improvement from 80% to 95% (58). Larger studies are needed for confirmation and the obvious impact on quality of life.
Rarely, myoclonic absences may occur in children with Dravet syndrome. In 2017, Myers and Scheffer reported a 20-year-old man with Dravet syndrome for whom, by 5 years of age, photosensitive myoclonic absence seizures had become his dominant seizure type, implicating the role of SCN1A mutations (66). This rare seizure type may be underreported in Dravet syndrome, as the myoclonic features may be subtle and can be missed if thorough history taking and video recordings are not available (66). An infant diagnosed with Dravet syndrome with focal cortical myoclonus on conventional EEG when he was 6 months old has been reported, which may broaden the types of seizures of Dravet syndrome and may provide some diagnostic clues for Dravet syndrome (61).
See the article on Epilepsy with myoclonic absences.
Facial myoclonia with absences. Facial myoclonia with absences, as such, has not been recognized either as a seizure type or as a syndrome by the ILAE; however, absences with facial (perioral or eyebrow) myoclonia is a discrete seizure type unequivocally documented with video-EEG recordings. Furthermore, there is often a nonfortuitous clustering of other clinical and EEG features, indicating that these absences may constitute the main symptom of an interesting syndrome of “facial myoclonia with absences” within the idiopathic generalized epilepsies.
Prevalence is small, occurring in less than 1% of children (9.3% in adults) with typical absences. Age at onset is at 2 to 13 years (median 10 years). Females predominate (about 80%).
Neurologic and mental state. Normal.
Etiology. Facial myoclonia with absences is probably genetically determined. Half of the patients have first-degree relatives, mainly siblings, with idiopathic generalized epilepsy and absences.
Clinical manifestations. Typical absence seizures with perioral or eyebrow myoclonia are the defining symptoms. Facial myoclonia consists of rhythmic protrusion of the lips, twitching of the corners of the mouth, or jaw or eyebrow jerking. Impairment of consciousness varies from severe to mild. The absences last 2 to 9 seconds (median 4 seconds), with varying frequency from many times per day to one to two per week or less frequently.
Most patients suffer from GTCS, starting before, soon after, or, exceptionally, many years after the absences. GTCS are usually infrequent and are often heralded by clusters of absences or absence status epilepticus.
Absence status epilepticus is very common (57%) and frequently ends with GTCS.
In children, the term facial instead of perioral myoclonia with absences is preferably used because some cases present with perioral, others with eyebrow, or perioral and eyebrow. Based on a population of 13 children, 46% presented with perioral, 38% with eyebrow, and 15% with perioral/eyebrow myoclonias with absences. The age of onset varied from 1 to 13 years (mean 5±3 years). All children were assessed with sleep-awake video EEG recording following sleep deprivation. The duration of the recorded generalized spike-wave discharges varied from 0.25 to 20 seconds, and 15% showed a positive response to intermittent photic or pattern stimulation. The response to sodium valproate was 85%. No absence status epilepticus was recorded in this population of children. Those cases with GTCS in their phenotype had a less favorable outcome (26). Absence and myoclonic seizures, the predominant type of seizures in the phenotype, characterize the syndrome epilepsy with myoclonic absences (formerly known as Tassinari syndrome) and also two syndromes in development, eyelid myoclonia and absences (formerly known as Jeavons syndrome) and facial myoclonia with absences (22; 20; 21).
Early-onset absence epilepsy. Nasser and colleagues described the electroclinical features of nine children with normal development and typical absence seizures starting before the age of 4 years (68). Eight patients had rhythmic myoclonic jerks involving the muscles of the upper face (eyebrows and eyelids) or neck, present from the onset to the end of the typical absence discharge. The myoclonia were synchronous with spike-wave complexes. One patient with GLUT-1 deficiency was refractory to antiepileptic polytherapy. The other eight became seizure-free; five with one antiepileptic drug and three with a combination of two drugs. The authors suggested that the motor symptoms during the absence seizures may represent a distinctive age-related feature of early-onset absence epilepsy (68).
In the author’s experience, based on a study of 19 children (F: M =1.7:1) with early-onset absence seizures (< 3 years, mean 2.7±0.7 years), two groups were identified: nonmyoclonic (37%) and myoclonic type (63%). In the nonmyoclonic group, there are cases with spanioleptic and pyknoleptic absence seizures. The spanioleptic are reminiscent of juvenile absence epilepsy, whereas the pyknoleptic are either childhood absence-like or an early expression of what later evolves to juvenile myoclonic epilepsy with or without positive response to intermittent photic stimulation. Early predictors are marked by clonic-myoclonic component and brief generalized spike/poly-spike wave discharges. In the nonmyoclonic group, the cases that are mostly associated with cognitive and educational problems are the CAE-like, followed by the JAE-like. Those who evolved to juvenile myoclonic epilepsy do well.
The absence seizures in the myoclonic group are mostly pyknoleptic. Spanioleptic absence seizures in this category may be a false impression, as many absence seizures are inconspicuous and not easily recognized if not recorded by video-EEG, and the transient events during brief generalized discharges are not meticulously studied. Some cases can be identified as myoclonic epilepsy of infancy or childhood. Others are reminiscent of epilepsy with myoclonic absences or facial (perioral) myoclonia with absences or myoclonic atonic epilepsy. One category, which can be early identified, is eyelid myoclonia and absences evoked on eye closure (formerly known as Jeavons syndrome).
Almost 75% of the cases respond to monotherapy with sodium valproate in nonresponders. Ethosuximide, levetiracetam, and lamotrigine can be combined with valproate. Relapses are frequent in the myoclonic group (70%) as compared to the nonmyoclonic (20%), even after a long duration of successful treatment, with a mean 4.3 ± 0.8 years and 6.6±3 years, respectively. Brief generalized spike-wave discharges, photosensitivity, and GTCS are the most unfavorable relapse risk factors.
Early-onset absence epilepsy includes a variety of epileptic syndromes. All children are developmentally normal at onset, but most develop cognitive and educational problems with advancing age despite an early and successful treatment. Their phenotypic heterogeneity reflects genetic differences that most likely predetermine evolution. It is also probable that environmental variables, duration of symptoms, and response to treatment contribute to the outcome (12; 17).
See the article on Typical absences.
The EEG, preferably sleep-awake video-EEG after sleep deprivation, particularly in children, is the single most important diagnostic procedure in diagnosing myoclonic absence seizures. These are often induced by hyperventilation, though not as easily as other typical absence seizures of childhood and juvenile absence epilepsy. Ideally, these patients should have video polygraphic-EEG recordings in an untreated state, as this may reveal features favoring a specific epileptic syndrome and may, therefore, determine long-term prognosis and management. If this is not possible, the clinical manifestations of the seizures should be documented with camcorders by the parents or the treating physicians. Breath-counting during hyperventilation (wherein the patient is asked to count his or her deep breaths) is an important, often neglected, method to detect impairment of consciousness during the discharge (43; 73). In infants, without the aid of a paper windmill to blow, it is impossible to encourage continuous and effective hyperventilation. Also, counting during hyperventilation is less successful in young children because the pattern of breathing is interrupted, and effective hyperventilation is rarely achieved (19). In infancy and early childhood, video-EEG recordings need meticulous assessment in order to identify inconspicuous and brief clinical events.
In epilepsy with myoclonic absences, the interictal EEG shows normal background activity with generalized irregular spike-wave activity (in one third of the cases) that is sometimes intermingled with polyspikes; rarely, focal or multifocal spike-waves or spikes occur (07). The ictal EEG comprises rhythmic generalized and bilateral synchronous and symmetric 3 Hz spike-wave activity, as can be seen in typical absences. Occasionally, these classic discharges may be intermingled with polyspikes. The polygraphic recordings reveal bilateral and rhythmic myoclonias with a strict and constant relation with the spike wave of the discharge; the latency between EEG spikes and EMG myoclonic activity varies between 15 and 40 milliseconds in proximal muscles. Later into the seizure, the myoclonias are associated with progressive tonic contraction of the affected muscles (87; 07).
In perioral myoclonia with absences, the interictal EEG also shows normal background activity with frequent, less than 1-second generalized discharges of 3 to 7 Hz spike or multiple spike waves that are usually asymmetrical. Focal abnormalities are common, including single spikes, spike-wave complexes, or slow waves. Ictal EEG shows brief (mean 4 seconds; range 2 to 9 seconds) generalized discharges of 3 to 4 Hz spikes or multiple spike waves with frequent irregularities in terms of the number of spikes, fluctuations in spike amplitude, and fragmentations. Perioral myoclonia may occur during generalized EEG discharges of polyspikes and waves without evidence of absence (27).
In children with myoclonic absences, genetic studies should be considered if no etiology is found after a detailed clinical examination and MRI, particularly in patients with associated neurodevelopmental disorders or a structural or functional brain disorder that is known to have a genetic basis. Genetic testing should also be a primary investigation for cases presenting in infancy and early childhood.
The myoclonic absence phenotype might be a common clinical expression of several etiologies rather than a discrete entity.
First-line antiepileptic drugs in typical absences are sodium valproate or ethosuximide, which are of equal efficacy, controlling absences in approximately 75% of patients; lamotrigine is less effective, with nearly half of the patients becoming seizure-free (69; 77; 45; 94). Clonazepam, levetiracetam, sulthiame, and zonisamide are also used as add-on treatments (81). Sodium channel blocker antiseizure drugs should be avoided.
Myoclonic absences are usually resistant to monotherapy. Therefore, a combination of high doses of valproate, often combined with lamotrigine or ethosuximide, levetiracetam (35), or clonazepam is recommended (07). A favorable response to add-on treatment with rufinamide in three cases with drug-resistant myoclonic absences has been reported (48). Perampanel has also demonstrated efficacy and safety in generalized tonic-clonic seizures (06) and in myoclonic seizures (30) among patients with idiopathic generalized epilepsy and does not seem to exacerbate absence seizures.
Choosing the correct antiepileptic medications at an appropriate daily dose, taking into consideration the risk-benefit ratio and recognizing comorbidities for early intervention, contributes to better management. Sodium valproate has been proven to be most successful for this type of seizure, either as monotherapy or combined with other appropriate antiepileptic drugs. In this respect, taking into consideration the dose-related teratogenicity, it should be avoided as a first choice in adolescent females. Special care should be given to those females on valproate who intend to become pregnant or are pregnant, where the daily dose of valproate should be based on a careful risk-benefit decision (90). Levetiracetam is increasingly prescribed for these patient populations despite a scarcity of evidence of clinical effectiveness or cost-effectiveness. In the SANAD II study, levetiracetam compared to valproate was found to be neither clinically effective nor cost-effective (62). For girls and women of childbearing potential, these results inform discussions about the benefit and harm of avoiding valproate. There is an urgent need to develop up-to-date, globally applicable recommendations (89).
In a single-center retrospective study of 131 cases with typical absence seizures, eighteen cases (13.7%) were classified as difficult to treat. Absence seizures were more often difficult to treat in patients with myoclonic absence seizures and absence seizures with eyelid myoclonia (40.0%), compared with patients with typical absence seizures (11.4%) (47). Additional factors contributing to difficult-to-treat absence seizures were positive family history of epilepsy, higher seizure frequency before treatment, and longer time between seizure onset and treatment onset (47).
Carter and colleagues reviewed medical records between 2017 and 2022 to identify electroclinical variability and response to treatment of patients with myoclonic absences (11). Of the 10 patients identified, four had an atonic component, and two out of three of those who were drug-resistant underwent corpus callosotomy. One was seizure-free 8 months after surgery, and the other had greater than 50% seizure reduction over a 5-month period. A larger study is needed to evaluate the efficacy of this treatment.
Flamini and colleagues investigate the effectiveness of add-on CBD therapy for individual convulsive seizure types (clonic, tonic, tonic-clonic, atonic, focal to bilateral tonic-clonic), nonconvulsive seizure types (focal with and without impaired consciousness, absence [typical and atypical], myoclonic, myoclonic absence), and epileptic spasms (39). Among patients with treatment-resistant epilepsy, add-on CBD was associated with a reduction in the frequency of convulsive seizure types, nonconvulsive seizure types, and epileptic spasms. At least 50% reduction was reported in greater than or equal to 49% of patients across all evaluated seizure types and at nearly all intervals for up to 144 weeks of treatment.
Absence status epilepticus of perioral myoclonia with absences, for which most patients are aware, should be terminated with self-administered benzodiazepines and mainly buccal or nasal spray midazolam.
Contraindicated drugs. The treatment of myoclonic absences is demanding because many antiepileptic drugs are either ineffective or exaggerate absences and myoclonic jerks (79; 73; 95; 13). An antiepileptic drug is contraindicated not only when it exaggerates seizures but also when it is ineffective in controlling the seizures that it is supposed to treat. It may cause unnecessary adverse reactions and deprive the patient of the therapeutic effect that could be provided by another antiepileptic drug. Carbamazepine, phenytoin, oxcarbazepine, phenobarbital, gabapentin, pregabalin, vigabatrin, and tiagabine are contraindicated in the treatment of myoclonic absences, irrespective of cause and severity. This is based on clinical and experimental evidence. In particular, the GABA agonists vigabatrin and tiagabine are used to induce, not to treat, absence seizures and absence status epilepticus (73). Lacosamide, a novel antiseizure medication, is believed to exert its anticonvulsant action through selective enhancement of slow inactivation of voltage-gated sodium channels. Lacosamide has been demonstrated to be efficacious as an adjunctive therapy for primary generalized tonic-clonic seizures (93) but may have the potential to worsen or to induce fresh myoclonic seizures (05). In this respect, lacosamide should be used with caution in idiopathic generalized epilepsies.
Recognizing GLUT1 deficiency syndrome as a rare cause of myoclonic absences is important because a ketogenic diet is effective in improving epileptic seizures and may also reduce some neurologic symptoms (40; 56). Most antiepileptic drugs are contraindicated either because they are ineffective or because they exacerbate seizures. Phenobarbital, but also chloral hydrate and diazepam, may further impair GLUT1 and should not be used in these patients.
The evolution of epilepsy with myoclonic absences, the prototype of myoclonic absences, is variable. Almost 45% have intellectual disability from the start, 75% final, and half persist beyond adolescence. This variability of clinical phenotype may be an expression of the genetic heterogeneity, in addition to the effect it may have on cognition, the seizures, the interictal-ictal EEG activity, and even more, in some cases, the underlying pathology. Early diagnosis and early appropriate treatment, taking into consideration the risk-benefit ratio and treating early coexisting comorbidities, significantly contribute to better outcomes and quality of life.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Neil R Patel MD
Dr. Patel of Montefiore Einstein has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026