Epilepsy & Seizures
Febrile seizures
Jun. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Myoclonic epilepsy in infancy is a rare self-limited idiopathic generalized epilepsy that typically appears between 6 months and 2 years of age. It is characterized by the occurrence of myoclonic seizures as the unique type of seizure (expect simple febrile seizure) occurring in normal infants, either spontaneously or induced by unexpected acoustic or tactile stimuli (reflex variant). The long-term outcome is usually favorable, with disappearance of seizures and normal cognitive functioning. However, reports of protracted evolution show that about 20% of patients have other seizure types, mainly at adolescence, and that about 39% have mainly mild to severe cognitive impairment. The reflex variant of this syndrome has a better prognosis, with nearly all patients becoming seizure-free within weeks or months from onset even without treatment. The syndrome should be differentiated from other epileptic and nonepileptic conditions presented with myoclonic jerks. The author presents clinical and EEG manifestations, epidemiology, pathophysiology, differential diagnosis, and management of this rather benign early onset idiopathic myoclonic epilepsy.
• Myoclonic epilepsy in infancy (formerly named “benign myoclonic epilepsy in infancy”) encompasses two forms: one with predominant spontaneous seizures and one with predominant reflex seizures elicited by unexpected acoustic and tactile stimulation. Spontaneous and reflex seizures may occur in the same infant. | |
• Myoclonic epilepsy in infancy is an early form of possibly genetic idiopathic generalized epilepsy. | |
• Seizures are usually self-limiting, pharmacoresponsive, and may remit without treatment. | |
• In spite of complete remission of seizures, long-term cognitive outcome is abnormal in around 30% of patients, usually in the range of mild impairment and rarely severe. The factors of this unfavorable outcome remain unknown. | |
• The reflex variant is usually benign, with earlier age at onset, higher remission rate, better response to medications, and normal neurocognitive development. |
Dravet and Bureau in 1981 described “benign myoclonic epilepsy in infancy” in seven normal children with onset of myoclonic seizures in the first 3 years of life (14). The syndrome was defined as including myoclonic seizures only, except rare simple febrile seizures, with good prognosis regarding response to therapy and cognitive functions. Prior to this, early benign myoclonic epilepsy was reported in three infants in a study of early-onset epilepsies (12), but without known follow-up. In 1977 Jeavons gave the name ‘‘myoclonic epilepsy of childhood’’ to a similar type of syndrome, beginning at 3 years of age (23). The benignity of the syndrome has been questioned, leading to a change in its name to “myoclonic epilepsy in infancy” (17; 44). “Idiopathic myoclonic epilepsy in infancy” has been proposed (15). The ILAE Commission on Classification and Terminology initially proposed replacing “idiopathic” with “genetic” (06) but more recently recommended that either idiopathic or genetic can be used (35). In the more recent proposal for ILAE classification and definition of syndromes proposed by the Task Force on Nosology and Definitions, the myoclonic epilepsy of infancy is classified under the “self-limited epilepsies” of infancy (45).
Ricci and associates described six normal infants with reflex myoclonic jerks provoked by unexpected (startle) auditory or tactile stimuli (32). Seizure onset was before the age of 2 years with rapid remission after 3 to 12 months. They proposed that this constitutes a new “reflex myoclonic epilepsy of infancy”, which is an age-dependent idiopathic generalized epilepsy syndrome, with an apparently good prognosis (32). Until 2013, about 80 cases had been described by other authors (41). It is debated whether this reflex form is a variant of the myoclonic epilepsy in infancy or an independent stimulus-sensitive syndrome.
In the 2010, 2014, and 2021 ILAE proposals, myoclonic epilepsy in infancy, including the reflex variant is classified among neonatal/infantile epileptic syndromes (06; 11; 45). The following is a complete description from the ILAE epilepsy manual (11) and the ILAE position paper on classification of epilepsy syndromes with onset in neonates and infants (45).
Overview. This epilepsy syndrome is uncommon. Myoclonic seizures are the only seizure type seen at onset. Infrequent febrile seizures may also occur. Myoclonic seizures may be activated by photic stimulation in one fifth of the patients, whereas others may have myoclonic seizures that are induced by sudden noise or touch. Cognitive, behavioral, and motor difficulties may exist. Seizures are self-limiting, ceasing within 6 months to 5 years from onset. Generalized convulsive seizures may be seen infrequently during the teen years.
Note. Self-limiting refers to seizures having a high likelihood of spontaneously remitting at a predictable age. |
Clinical context. This syndrome is characterized by the onset of myoclonic seizures between the ages of 6 months and 2 years, and in some cases, earlier (4 months) or later (2 to 4 years) onset has been reported. Myoclonic seizures may be induced by photic stimulation in some patients or by sudden noise or touch in others. The term “reflex myoclonic epilepsy in infancy” has been used to refer to this scenario. Infrequent febrile seizures may be seen in approximately 10% of patients. Seizures remit within 6 months to 5 years from onset, but generalized convulsions may be seen in teenage years in 10% to 20% of patients. Patients with photosensitivity may have seizures that are more difficult to control. Males are twice as likely to be affected as females. Antecedent and birth history is unremarkable. Head size and neurologic examination are normal. Cognitive, motor, and behavioral difficulties are reported, especially if seizures are not controlled. Developmental outcome is normal in 63% to 85% of cases.
Mandatory seizures. Myoclonic seizures are seen. These are mainly of the head (causing nodding), eyeballs (which roll upwards), upper extremities (causing the arms to fling up and out), and the diaphragm (resulting in vocalization). Rarely, the lower limbs are affected, causing falls. Jerks can be singular or can occur in a series and may vary in severity. Responsiveness is preserved; however, it may be reduced if there are clusters of jerks in a series. Jerks may occur in all states (alert, drowsiness, slow sleep, and on wakening).
Exclusion criteria. The presence of absence seizures, atonic seizures, epileptic spasms, focal impaired awareness seizures, hemiclonic seizures, myoclonic-absence seizures, and tonic seizures is incompatible with the diagnosis of myoclonic epilepsy of infancy.
Patients may have:
• Simple febrile convulsions (seen in 10%, infrequent) | |
• Generalized convulsive seizures (seen in 10% to 20% of patients in teens, infrequent) | |
• Early-onset absence seizures have been reported in up to 20% of patients with myoclonic epilepsy in infancy. | |
• Evolution to juvenile myoclonic epilepsy may occur. | |
• Evolution to drug-resistant epilepsy has been reported in case reports. | |
• Evolution to myoclonic-astatic epilepsy has been reported in case reports. |
Exclusionary. Other seizure types are exclusionary.
EEG Background. The background EEG is normal.
Interictal EEG. The interictal EEG is normal.
Activation. Sleep may activate the EEG, and generalized spike-and-wave and polyspike-and-wave may occur, with or without accompanying myoclonic jerks clinically. One fifth of patients have photosensitivity, with myoclonic jerks precipitated by photic stimulation. In 10% of patients, myoclonic jerks may be activated by sudden noise or touch in either awake or sleep states.
Ictal EEG. Myoclonic jerks are associated with generalized spike-and-wave or polyspike-and-wave discharges.
Imaging. Neuroimaging is normal.
Genetics. The pattern of inheritance is unknown but is likely genetic.
Family history of seizures/epilepsy. A family history of febrile seizures or epilepsy is seen in one third of patients.
• Dravet syndrome: myoclonic seizures are frequent, however, typically occur in the second year of life and are preceded by a period of susceptibility to febrile convulsions. | |
• Hypnogogic jerks | |
• Hyperekplexia | |
• Benign myoclonus of early infancy or benign spasms of infancy |
|
• Myoclonic epilepsy in infancy manifests with myoclonic seizures typically between 6 months and 2 years. | |
|
• Febrile seizures or early-onset absence seizures may also occur in a minority of patients with myoclonic epilepsy in infancy. | |
|
• In reflex myoclonic epilepsy in infancy, myoclonic seizures may be triggered by tactile or auditory and, less frequently, photic or proprioceptive or thermal stimuli. | |
|
• Generalized convulsions may occur in 10% to 20% of patients in their teen years. |
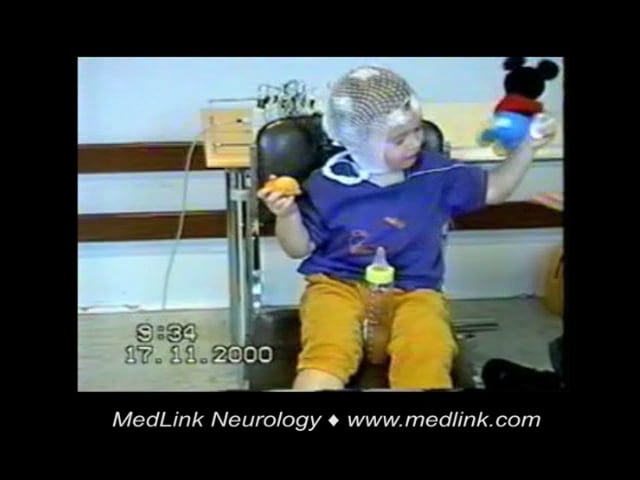
Myoclonic epilepsy in infancy is characterized by brief myoclonic attacks in normal infants between the ages of 6 months and 2 years without any other type of seizure (except simple febrile seizure). An earlier onset is exceptional, although later onset (up to 4 to 6 years) has been reported. Boys are twice as likely to be affected as girls (103/60) (19). The jerks are brief and singular or clusters that vary in frequency and intensity. They mainly affect the head, eyeballs, upper extremities, and the diaphragm. They manifest clinically with head nodding and, more rarely, flexion or extension of the body. The upper limbs usually fling upwards and outwards, whereas the eyeballs may roll upwards. A brief yell, probably resulting from the contraction of the diaphragm, sometimes accompanies the jerks. Falls may occur in the rare event that the lower limbs are affected.
The mild forms of myoclonic jerks consist only of brief forward movement of the head, or even a simple closure of the eyes, which are usually inconspicuous or dismissed as nonpathological. The most severe forms consist of clusters of two or three pseudo-rhythmic repeated jerks lasting up to 5 to 10 seconds. These are usually massive, involving the axis of the body and the limbs.
See also video presentation of a case with myoclonic epilepsy in infancy: YouTube.com.
Consciousness during the attacks is difficult to assess but it does not appear to be disturbed except during severe clusters of jerks.
Clinical examination is normal.
Circadian distribution. Myoclonic seizures are usually spontaneous, occurring randomly in alert stages and exaggerated by drowsiness and NREM sleep. In some patients they tend to cluster on awakening or during the first hours of sleep. There is a report of a group of five children with excessive myoclonic jerks, only during sleep, with only one third of these events being associated with EEG epileptiform discharges (31).
Precipitating factors. One fifth of patients have clinical and EEG photosensitivity (08; 19). In 10% the myoclonic jerks are predominantly or exclusively elicited by unexpected sudden acoustic or tactile or visual or proprioceptive stimuli (reflex variant) (09).
Types of seizure other than myoclonic.
Febrile seizures. Febrile seizures have been reported in 10% to 20% of patients with myoclonic epilepsy in infancy (03; 09). They are rare simple febrile convulsions usually preceding the onset of myoclonias.
Generalized convulsions. These occur in 10% to 20% of patients in their teens (11). Rarely, generalized tonic-clonic seizures can also occur rarely before the onset of myoclonic seizures and these mostly disappear within several months when they are replaced by myoclonic seizures (22; 42). The age of onset and ictal video-polygraphic features of myoclonic seizures, as well as the long-term seizure and developmental outcome in these children, were similar to those of children with typical myoclonic epilepsy in infancy (22; 42).
Brief absence seizures. These are captured with video-EEG and are found in around 19% of patients with myoclonic epilepsy in infancy (05). These absence seizures occur mainly during brief clusters of rhythmic myoclonic jerks and ictal EEG are similar to the ictal EEG patterns associated with only myoclonias. No differences in relation to gender, family history, or ictal EEG discharge are found between patients with myoclonic seizures with absence seizures and myoclonias without absence seizures. Seizures respond well to antiseizure medications and prognosis is excellent (05).
Reflex variant of myoclonic epilepsy in infancy. Reflex myoclonic epilepsy in infancy manifests with myoclonic jerks elicited by sudden, unexpected auditory or tactile stimuli and, less frequently, by thermal (eg, cold water) or visual (eg, flash of light) or proprioceptive stimuli (32; 13; 09; 40; 41; 37). Startle is fundamental. Repetition of stimulus at short intervals may attenuate the ictal response. The age at onset of reflex myoclonic jerks is usually between 3 and 24 months in otherwise normal children (41). Reflex myoclonic jerks are triggered both in wakefulness and during sleep, with a lower threshold in stage 1 and a gradual increase during slower stages of sleep (32). Intermittent photic stimulation can provoke myoclonic jerks in a minority of patients. Photosensitive benign myoclonic epilepsy in infancy has also been described when myoclonic seizures were always triggered by photic stimulation (08).
See associated video in the cited paper by Turco and associates (38) at: http://www.jpeds.com/cms/attachment/2056515187/2061413337/mmc1.mp4.
One third (32.5%) of patients also have spontaneous attacks that usually appear months after the reflex attacks and are facilitated by drowsiness and sleep.
Auvin and colleagues did not find significant differences in terms of clinical and EEG parameters in the reflex group compared to the non-reflex group (03), though prognosis is much better in the reflex variant where seizures often disappear without treatment and neuropsychological development is normal in nearly all patients.
Seizures. Prognosis is good in regard to myoclonic seizures, which are easily treated with appropriate antiseizure medication and remit within 1 to 2 years from onset. Only 20% of patients required a combination of two antiseizure medications to be seizure-free. However, 16% of patients (26 out of 162) presented with other types of seizure after the remission of myoclonic jerks (19). These are mainly generalized tonic clonic seizures (17 patients) either during a premature drug withdrawal or later on at adolescence. In addition, and exceptionally, a few patients developed symptoms of juvenile myoclonic epilepsy, childhood absence epilepsy, eyelid myoclonia with absences, or myoclonic astatic epilepsy long after the remission of the initial manifestations of myoclonic epilepsy in infancy (03; 02; 26; 19; 33).
Reflex seizures provoked by noise or contact are more easily controlled than the spontaneous ones (40; 41).
Neuropsychological development. The neuropsychological outcome is not as favorable as initially thought. Although most patients are reported to have normal cognitive skills, the follow-up duration was variable and many studies did not report neuropsychological assessment (19).
Of 23 patients followed up to at least 15 years, 14 patients (61%) had a normal level, confirmed by psychometric evaluation in five, sometimes with learning difficulties at school, and nine (39%) had a low level, from mild to severe cognitive impairment.
The prognosis is excellent in regard to seizures and neurocognitive development of patients featuring the association of absences and myoclonic epilepsy of infancy (05) as well as those of the reflex variant of this syndrome (40; 41; 38; 37).
Case 1. The patient was the second of two brothers. The oldest brother was in good health. There were febrile convulsions in one of the father’s cousins and a hormonal deficit in the first months of pregnancy. The birth delivery was normal and there was normal psychomotor development. The first myoclonic attacks were between 8 and 9 months of age, with a slight forward movement of the head, upward movement of the upper limbs, and rolling of the eyeballs several times a day. At 14 months the first EEG showed generalized spike and polyspike wave. Low doses of valproate and clonazepam did not completely control seizures that were sometimes provoked by sudden contacts or noises. First referred at 16 months, several spontaneous or reflex myoclonic jerks associated with brief generalized spike wave in polygraphic video EEG were recorded. There was no photosensitivity. Clinical examination was normal. No neuroimaging was performed. The development quotient measured by the Brunet-Lézine scale was 96, and the child attended normal kindergarten. Plasma level of valproate was low (62 mg/mL). Clonazepam was not well tolerated. Freedom of seizures was achieved with 30 mg/kg/day valproate monotherapy. At 5 years 6 months, valproate was stopped. The last EEG control showed a normal awake trace and the persistence of rare generalized spike wave during drowsiness and slow sleep. Intermittent photic stimulation and videotape watching did not provoke discharges. At 6 years 3 months, he attended his first elementary class without difficulties.
Case 2. An otherwise normal girl, developed at age 3 years frequent and violent myoclonic jerks mainly in the head and shoulders with grunting noises. She was referred for a routine EEG 6 weeks from the onset of the jerks. On the basis of the history, the EEG technologist applied video-EEG recording and deliberately extended the recording for clinical events. Two electroclinical seizures were captured. Clinically, the first symptom was a sudden jerk of the head and shoulders backwards. This coincided with a giant (approximately 1 mV) spike or multispike-slow-wave followed by rhythmic slow activity at around 3 to 5 Hz, together with some random spikes and slow waves. The whole discharge lasted for approximately 5 to 7 seconds.
The seizures stopped only when small doses of clonazepam were added to valproate (valproate was probably not needed). Subsequent serial EEGs were normal. At age 6 years she was no longer on medication, had developed well, was free of seizures, and had a normal EEG.
|
• Myoclonic epilepsy in infancy is considered as idiopathic generalized epilepsy with usually favorable outcomes. | |
|
• Sporadic associations with SLCA2C1 or TBC1D24 gene variants or chromosomal locus 16p13 have been reported. |
Genetics. This is an early-onset self-limited idiopathic generalized epilepsy (10) but genetics are unclear. Possible relationships with other types of idiopathic generalized epilepsies have been speculated (19).
There is a report of a large Italian pedigree segregating an autosomal recessive idiopathic myoclonic epilepsy that starts in early infancy with myoclonic seizures, febrile convulsions, and tonic-clonic seizures. The locus has been mapped on chromosome 16p13 by linkage analysis (43).
Two brothers with myoclonic epilepsy in infancy were found to have p.Arg550Cys (c.1648C> T) heterozygous mutation on HCN4 (07). For more information, see the MedLink Neurology article on myoclonus and an external review of the pathophysiology of myoclonic seizures (36).
Case reports of myoclonic epilepsy in infancy with glucose transporter 1 (GLUT1) deficiency (GLUT1DS) have been described. A boy with de novo heterozygous missense mutation of SLCA2C1 gene (c.1199G> T; p.R400L) presented with spontaneous myoclonic jerks associated with generalized spike-and-wave discharges that initially resolved with valproate (18). However, subsequent emergence of myoclonic seizures, generalized tonic-clonic, and absence seizures occurred at 18 months of age as well as developmental delay. Antiseizure medications (valproate, lamotrigine, topiramate) and ketogenic diet failed to control seizures. GLUT1DS has also been reported in a girl who presented with very frequent epileptic myoclonus events at the age of 8 months, which resolved within a few days of starting levetiracetam and was, therefore, diagnosed with myoclonic epilepsy in infancy (01). At the age of 12 months, a pathogenic frameshift mutation in the SLC2A1 gene (del.c.505-507) was identified; ketogenic diet was initiated with good response.
Association with a homozygous missense mutation in TBC1D24 gene (TBC/LysM-Associated Domain Containing 6) (c.457G> A/p.Glu153Lys) has been reported in two brothers, one of whom had reflex myoclonic seizures (30). TBC1D24 is involved in synaptic vesicle trafficking.
A case report of a 5-year-old girl with a milder phenotype of YWHAG-associated epilepsy, presenting as myoclonic epilepsy of infancy at 2 years of age, was published by Iodice and colleagues (21). However, subsequent evolution entailed focal unaware seizures, which are exclusionary for this syndrome, and global developmental delay. The YWHAG gene codes for tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein gamma (14-3-3γ) (Chr 7q11.23), which regulates neuronal migration. The existing rare associations of YWHAG variants with epilepsy usually present as developmental and epileptic encephalopathies.
|
• Myoclonic epilepsy in infancy is a rare syndrome occurring in less than 1 of 1,000,000 population. | |
|
• The reflex variant of myoclonic epilepsy in infancy is extremely rare. |
Myoclonic epilepsy in infancy is a rare syndrome probably occurring in less than 1 of 1,000,000 population (ORPHA86909). According to the few available epidemiological data, it seems to represent less than 1% of all the epilepsies, 1.3% to 1.7% of epilepsies with onset in the first year of life (34), around 2% of epilepsies beginning in the first 3 years of life, and 0.39% when onset is in the first 6 years of life (28). In a tertiary center, myoclonic epilepsy in infancy occurred in 0.8% of children between 1 month and 15 years of age, whereas Lennox-Gastaut syndrome accounted for 0.5%, Dravet syndrome for 1.4%, and West syndrome for 4.1% (16).
The reflex variant of myoclonic epilepsy in infancy is extremely rare, with only 89 cases found in the literature (37).
Myoclonic epilepsy in infancy should be differentiated from nonepileptic conditions such as hypnagogic jerks and Fejerman syndrome (benign nonepileptic myoclonus). Hypnagogic jerks do not occur in waking states and the EEG is normal. The attacks of Fejerman syndrome resemble epileptic spasms rather than the myoclonic jerks (see MedLink Neurology article: Fejerman syndrome). The reflex variant of myoclonic epilepsy in infancy should be differentiated from hyperekplexia (see MedLink Neurology article: Hyperekplexia).
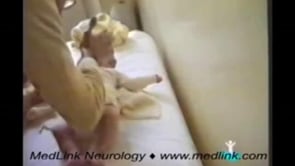
Fifteen-day-old neonate with typical startle response to tactile stimuli in the face and mainly the nose. Please note that marked generalized startle response (typical of hyperekplexia) is triggered each time the face and, main...
It should not be difficult to differentiate myoclonic epilepsy in infancy from epileptic encephalopathies such as West syndrome, Dravet syndrome, or Lennox-Gastaut syndrome with multiform seizures, significant EEG abnormalities, and often severe neurodevelopmental deficits.
Hirano and associates conducted a video-polygraphic study of myoclonic seizures in different epileptic syndromes to clarify semiologic and electroencephalography differences among them (20). The subjects were 26 children with myoclonic seizures, including myoclonic epilepsy in infancy in 10, Dravet syndrome in six, epilepsy with myoclonic-atonic seizures in four, and juvenile myoclonic epilepsy in six. Myoclonic seizures manifested with proximal predominance/forward flexion/single occurrence in myoclonic epilepsy in infancy, proximal predominance/forward astatic flexion/single occurrence in epilepsy with myoclonic-atonic seizures, proximal predominance/extension/succession in Dravet syndrome, and distal predominance/extension/succession in juvenile myoclonic epilepsy. The median frequency of generalized spike-wave discharges was 1.5, 1.3, 3.2, and 3.1 Hz, respectively, and the median duration of the myoclonic EMG activity was 387, 587, 81, and 65 ms, respectively. The authors concluded that myoclonic seizures in the four different epileptic syndromes show significant semiologic and EEG differences, as well as similarities (20).
Progressive myoclonus epilepsy usually manifest with rapid developmental decline, visual abnormalities, ataxia, fragmentary myoclonus, and grossly abnormal EEG.
In rare cases, a picture resembling myoclonic epilepsy in infancy was described in patients with symptomatic epilepsies related to chromosomopathies (Down syndrome and trisomy 12p) and glucose transporter-1 deficiency (18). Also, a paper described three cases of inherited glycosylphosphatidylinositol anchor deficiency with early onset myoclonic seizures (between 1 week and 3 months of life), which were easily evoked by sudden unexpected acoustic or tactile stimuli (27). All three patients had dysmorphic facial features.
The diagnostic workup is simple. It needs a good clinical description and EEG confirmation.
EEG. The interictal EEG is normal for the child’s age. Spontaneous spike-wave discharges are rare; some slow wave may be found over the central areas. During drowsiness, there is an enhancement of the myoclonias that usually, but not always, disappears during sleep. In one quarter of patients, intermittent photic stimulation elicits spike-and-wave discharges with or without myoclonic jerks. Photosensitivity usually appears after the myoclonic seizures have remitted and can persist for many years, usually without clinical manifestation. Focal EEG abnormalities may be recorded in childhood or in adolescence, during waking or sleep. These are usually located in the frontocentral and the vertex areas.
Ictal EEG during the myoclonias is associated with generalized spike waves and polyspike waves at approximately 3 Hz. These are more or less regular, lasting 1 to 3 seconds.
Repeated polygraphic video-EEG recordings demonstrate the presence of myoclonic attacks with generalized spike wave discharges, either spontaneous or facilitated by drowsiness, acoustic, tactile, or intermittent photic stimulation.
Neuroimaging. Neuroimaging can be useful to affirm the absence of brain lesions but is not mandatory. Neuropsychological assessment is useful in order to follow the psychomotor development. Biological investigation can be necessary in case of associated symptoms evoking another disease.
If there is any doubt regarding the presence of neurologic symptoms at the onset of the disease or during follow-up, in particular neurologic regression, all investigations to rule out progressive myoclonic epilepsy should be done.
Genetic testing. Genetic testing may be helpful in cases with family history. Although there is no consistent gene variant associated with myoclonic epilepsy in infancy, case reports of genetic variants in SLCA2C1, suggesting GLUT1DS, or TBC1D24 genes have been published.
|
• Antiseizure medications like valproate, clonazepam, or levetiracetam can be effective in seizure control. | |
|
• In reflex myoclonic epilepsy in infancy, antiseizure medications may not be always necessary. | |
|
• Ketogenic diet has been tried in myoclonic epilepsy in infancy associated with GLUT1DS. |
The first choice antiseizure medication treatment is either valproate or clonazepam, which must be applied as soon as possible, though some authors propose to postpone treatment with antiseizure medications until seizures persist for more than 6 months or if they become more frequent, longer, or spontaneous (41; 38; 37).
Valproate in a daily dose of 30 mg/kg is usually sufficient, but higher doses may be necessary (25). The treatment should be maintained for 2 years after the onset if it is well tolerated. Clonazepam, the most effective antimyoclonic drug, should be considered either as monotherapy or when valproate is ineffective or contraindicated (see clinical vignette Case 2) (29). Levetiracetam has also been useful in the treatment of myoclonic epilepsy in infancy with or without reflex myoclonic seizures (41).
In the cases of purely reflex seizures, some authors suggest that pharmacotherapy may not be needed (38; 37).
The occurrence of new seizure types in late childhood or adolescence requires a new brief period of prophylactic treatment.
Monitoring cognitive and behavioral development is necessary. Moreover, psychological support must be offered to the family (39).
Ketogenic diet has been tried in cases with GLUT1DS following trials with other antiseizure medications. Ketogenic diet had either beneficial effect on seizures (01) or no effect (18).
The outcomes are usually favorable in regards to seizure control and neurodevelopmental course. Myoclonic epilepsy in infancy usually remits 6 months to 5 years from onset (04). Response to antiseizure medications, such as valproate, occurs within 3 to 22 months in patients who have myoclonic seizures as the first seizure manifestation and antiseizure medications can be safely withdrawn (24). Two thirds usually have normal development and one third only mild deficits (04). The photosensitivity may persist years after clinical response (04). Reflex myoclonic epilepsy in infancy has been reported to have a better prognosis (02; 41). Occasional evolution to myoclonic astatic epilepsy, juvenile myoclonic epilepsy, childhood absence epilepsy, or eyelid myoclonia with absences has been reported (03; 26; 02; 19; 33).
No case of pregnancy has been reported in the literature. Given the usual favorable prognosis, affected girls are likely not to have seizures and treatment when they are of childbearing age. However, in case of pregnancy before the arrest of treatment, the recommended guidelines for the drug that is used should be applied. Valproate is highly teratogenic and should be avoided. The risk for epilepsy in the offspring has not been documented.
Caution is required with regards to antiseizure medications and their interactions with the anesthetic drugs.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Aristea S Galanopoulou MD PhD
Dr. Galanopoulou of Albert Einstein College of Medicine received consultant fees from Synergy Medical Solutions.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jun. 02, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 17, 2026
Epilepsy & Seizures
Apr. 13, 2026
Epilepsy & Seizures
Mar. 24, 2026