







Neuroimmunology
Balo concentric sclerosis
Jul. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
White blood cells use adhesion molecules to attach to CNS endothelial cells, then penetrate the blood-brain barrier, and then cause inflammatory demyelination in multiple sclerosis. Antibodies that target VLA-4 adhesion molecules prevent exacerbations and progression of multiple sclerosis. Some patients on this therapy natalizumab have developed progressive multifocal leukoencephalopathy (PML). The author discusses the role of adhesion molecules in immune activation, penetration of the blood-brain barrier, and provocation and prevention of PML. The most recent information on monitoring for PML and clear definitions of the risk of PML are detailed.
|
• Natalizumab binds to very late activation antigen-4 (VLA-4) on immune cells. These drug-coated cells can no longer bind to CNS endothelium nor cross the blood-brain barrier. | |
|
• The drug lowers the number of CSF T cells by at least 10-fold and CSF B cells by 6-fold, and prevents attacks and progression of multiple sclerosis. | |
|
• With less immune surveillance in the brain, and perhaps activation of the virus by the treatment, the ordinarily innocuous JC virus can cause progressive multifocal leukoencephalopathy (PML). | |
|
• Quantitating serum antibodies to JC virus allows better estimation of risk for progressive multifocal leukoencephalopathy. | |
|
• Extending the natalizumab dosing interval from 4 to 6 weeks reduces the risk of PML. |
Natalizumab changes the ecology of immune cells in the blood and brain. This suggests new principles about multiple sclerosis therapy, T cell/endothelial cell interactions, CNS viral infections, and CNS immunology.
|
Topics addressed in this article include the following: |
|
(1) VLA-4 affects lymphocyte adhesion, migration, and organ development |
|
(2) VLA-4 and integrins control adhesion between cells in the bone marrow and thymus; adhesion is modified by the sympathetic nervous system |
|
(3) Integrins control immune cell homeostasis in the periphery and CSF |
|
(4) Variable integrin expression and affinity on immune cells in multiple sclerosis |
|
(5) Immune costimulation and adhesion are targets for therapy of multiple sclerosis; natalizumab may enhance peripheral immune activation |
|
(6) VLA-4 and interferon beta affect T cells and endothelial cells and modify penetration of the blood-brain barrier |
|
(7) Brain immune regulation is unique and differs from immunity in blood |
|
(8) Anti-VLA-4 therapy for multiple sclerosis |
|
(9) Adverse effects of anti-VLA-4 therapy and possible unexpected benefits |
|
(10) Type I interferons potentiate the effects of natalizumab but can also prevent JC virus infection |
|
(11) Other therapies may potentiate the clinical benefit of natalizumab |
|
(12) PML pathology, MRI, and clinical profile: JC virus, target cells, and why PML develops with natalizumab therapy |
|
(13) Direct detection and monitoring of JC virus and other polyoma viruses |
|
(14) Calculation of risk of PML in multiple sclerosis based on prior treatments and antibodies to JC virus (STRATIFY). American and European guidelines for monitoring |
|
(15) Treatment of PML and immune reconstitution inflammatory syndrome (IRIS) |
|
(16) Can PML be predicted during natalizumab therapy, and can PML be prevented by natalizumab “drug holidays” or extended dosing intervals? |
|
(17) Natalizumab withdrawal provokes exacerbations of multiple sclerosis; strategies to prevent multiple sclerosis reactivation |
Natalizumab is a human IgG4 kappa monoclonal antibody. Short complementarity-determining region segments of a potent murine anti-VLA-4 antibody comprise the antigen recognition site of this mouse-human hybrid antibody. The IgG4 isotype does not bind complement and persists for a long time in the circulation. Natalizumab binds to the alpha4-chain of VLA-4 glycoprotein (alpha4,beta1 integrin; very late activation 4 antigen) on immune cell membranes. Antibody-bound VLA-4 no longer interacts with vascular cell adhesion molecule 1 (VCAM-1) on endothelial cells to prevent adhesion and also immune cell activation. After an impressively short trip from lab bench to bedside, natalizumab was approved by the U.S. Food and Drug Administration in 2004 for the treatment of relapsing-remitting forms of multiple sclerosis. It is also effective in moderately to severely active Crohn disease and possibly in rheumatoid arthritis.
Adhesion molecules allow immune cells to attach to other immune cells and to endothelial cells. Lymphocyte function-associated antigen (LFA-1) attaches to intercellular adhesion molecule (ICAM-1), and very late activation antigen-4 (VLA-4) binds to VCAM-1.
VLA-4 is present on chronically activated T cells, and on B cells, monocytes, eosinophils, and basophils, but not on polymorphonuclear neutrophils (PMNs). VLA-4 is an alpha4beta1 integrin (CD49d plus CD29 subunits) that binds to vascular cell adhesion molecule (VCAM-1), mucosal addressin CAM (MAdCAM), fibronectin, osteopontin, and thrombospondin. These ligands are present on vascular endothelial cells, including those in the blood-brain barrier, on macrophages and microglia, in gut and genital mucosa, and in the extracellular matrix of the brain (astrocytes and neurons). VLA-4 ligands induce neurite outgrowth. Blocking VLA-4 in the brain could theoretically have adverse consequences on developing or regenerating neurons.
A related integrin, alpha4beta7 (LPAM-1), binds MAdCAM and fibronectin. VLA-4 has slightly lower affinity for MAdCAM than for VCAM. MAdCAM is a mucosal and CNS addressin that directs immune cells to the gut and brain. In Crohn disease, the VLA-4/MAdCAM interaction enhances migration into the gut by T cells and especially Th17 cells, which express more beta7 than Th1 cells. PMNs do not use alpha4 integrins for migration, suggesting that VLA-4 blockers could have less effect in IL-17/PMN-mediated diseases such as Crohn disease, the East Asian form of multiple sclerosis, and neuromyelitis optica. VLA-4/MAdCAM is important for T cell migration in chronic experimental allergic encephalomyelitis and possibly in chronic multiple sclerosis.
Four steps allow immune cells to adhere to vascular endothelium:
(1) Short-term “tethering” slows the otherwise rapid movement of lymphocytes through the post-capillary venules. L-selectin glycoprotein on leukocytes tethers to E- and P-selectins on endothelial cells to slow the cells.
(2) “Rolling” is weak and transient and further slows immune cells. L-selectin, P-selectin, and PNAd on T cells that bind to endothelial cells. In addition, alpha4beta1 and alpha4beta7 on T cells bind to VCAM and MAdCAM on endothelial cells. PSGL1/CLA, on Th1 cells, but not on Th2 cells, is increased in multiple sclerosis and binds to E- and P-selectins on endothelial cells. The plasma membrane is 5 to 10 nm thick, and the extended form of the integrin extracellular domain is approximately 15 nm, forming a significant net for adhesion.
(3) “Activation” changes the conformation of VLA-4 within milliseconds, causing clustering of these adhesion molecules, and transforms them from a bent/closed conformation to a high-affinity state on lamellipodia or filopodia at the leading edge of the immune cell (43). CXCR4 chemokine receptors and intracellular Rap1 co-localize in filopodia and aid in structural activation of VLA-4.
When VLA-4 is activated, the molecule extends and opens like a jackknife. The VLA-4 tails then separate in the membrane, and adhesion increases 100-fold (14).
(4) “Strong adhesion” is necessary for penetration through the vessel wall. Here, VLA-4 binds to VCAM-1 or MAdCAM, and LFA-1 binds to ICAM-1, leading to “outside-in activation.” Natalizumab blocks strong, stable adhesion but not the initial steps of contact, rolling, or capture.
VLA-4 affinity is increased by mechanical stretching during adhesion, by reducing agents (cellular isomerases that break disulfide bonds), and by Ca++ and Mn++ ions (“outside-in” activation). In culture, Mn++ causes 8-fold more adhesion of human T cells to human umbilical vein endothelial cells and pericytes.
Affinity, and not simply VLA-4 expression, determines adhesion and migration. Immune cell subsets express VLA-4 of various affinities, so simple quantitation of VLA-4 expression may not reflect the ability to bind and migrate. In the blood, VLA-4 is expressed in a low-affinity conformation by most cells, including almost all T cells; however, VLA-4 is in an active conformation on some B cells, NK cells, and monocytes (Table 1). With activation, VLA-4 on T and B cells becomes high-affinity, even though the amount of VLA-4 does not change. Activation with phorbol esters increases VLA-4 affinity on human memory T cells but not on naïve T cells.
There are multiple activation sites on the extracellular portion of the VLA-4 molecule. VLA-4 is activated from “inside-out” by intracellular second messengers that are induced by phorbol esters, the T and B cell receptors (ie, activated immune cells), prolactin, complement, LTB4, membrane-bound chemokines (acting through G-protein coupled receptors, GPCR), and fever. Soluble chemokines do not cause extension of the VLA-4 molecule. However, endothelium-expressed chemokines activate RhoA and cause a nearly instantaneous extension of LFA-1, which is then immediately activated by adjacent ICAM-1 ligand. Some anti-VLA-4 antibodies (eg, TS2/16) are even more potent than these physiological activators (14). Mn++ plus an activating MAb (TS2/16) increases binding affinity 1000-fold.
In contrast, pentoxifylline decreases integrin affinity and inhibits integrin-mediated adherence to activated endothelial cells. Pentoxifylline blocks CXCR12 chemokine-activated G proteins that activate Rap1 and redistribute and activate VLA-4. Pentoxifylline and other cAMP inducers (beta-adrenergics, prostaglandins) could potentiate the effects of anti-VLA-4 antibodies.
|
T cells |
B cells |
NK cells |
Monocytes |
PMN | |
|
Resting whole blood cells |
3% |
10%* |
10% (50% in humans) |
13% |
0% |
|
Whole blood activated with 1mM Mn++ |
35% (60% in human blood; largely memory T, few naïve T cells) |
75% |
ND |
ND |
ND |
|
Bone marrow |
35% | ||||
|
Thymus |
5% -- immature CD3low thymocytes | ||||
|
Spleen |
7% |
7% | |||
|
Lymph nodes |
0% |
0% | |||
|
Peritoneum |
25% |
28% | |||
|
VLA-4 expression values are from mouse blood and lymphoid organs (31); human values are in parentheses. *There is more high-affinity VLA-4 on immature B cells and B1 cells in mice than in humans (31). | |||||
CNS and peripheral vascular endothelium differ in adhesion properties. The blood-brain barrier endothelium is highly specialized and less permeable to solutes and immune cells. Secondly, there is little selectin on parenchymal CNS vessels, but selectin is highly expressed in the choroid plexus. Consequently, no lymphocyte rolling (step 2) is seen in the spinal cord microcirculation, using window preparations. Instead, encephalitogenic VLA-4-positive T cell blasts interact almost exclusively with VCAM-1 (step 4). Thus, blockade of VLA-4 adhesion will have profound effects on immune cell homing to the CNS.
Multiple sclerosis microvessels are activated, unlike those in normal brain. Endothelial LFA-1 assists T cells in migrating through multiple sclerosis vessels within 3 to 8 hours. Microvessels with activated endothelial cells bind to activated T cells in a positive feedback loop.
After breaching the blood-brain barrier, T cells and monocytes pause in the brain perivascular Virchow-Robin space. Only a few T cells will enter the parenchyma. These are usually activated, antigen-specific encephalitogenic cells (0.01 cells per gram of tissue per 100 injected cells; 100-fold less than in spleen or lung) (19). They persist in the perivascular space if they recognize brain antigens expressed on antigen-presenting cells. VLA-4 on T cells also interacts with VCAM-1 on pericytes.
The choroid plexus and meninges form a different type of barrier. P-selectin in the veins of these structures allows immunosurveillant central-memory T cells to enter the human brain and perform. This multistep process contrasts with the rapid adhesion of activated anti-brain T cells to parenchymal endothelium through VLA-4/VCAM-1 and LFA-1/ICAM-1 (19).
Monocytes, NK and B cells, and especially immature B cells just released from the bone marrow, express VLA-4, enhancing traffic into the CNS. T cells need to be activated and then express “very late activation 4” antigen to penetrate into the CNS.
Conversely, interactions between VLA-4 and VCAM-1, and between alpha4beta7 and MAdCAM-1, control the exit of developing T cells from the thymus and B cells from bone marrow. High-affinity VLA-4 retains lymphocytes in thymus and bone marrow. With immune cell maturation, VLA-4 affinity and expression are reduced, and the cells migrate to the periphery. Antibodies to VLA-4 mobilize hematopoietic progenitor egress from the bone marrow by 200-fold within 72 hours of infusion. (See increased peripheral B cells in blood, and JC virus, below.)
Granulocyte colony-stimulating factor (G-CSF) acts through the sympathetic nervous system to loosen the embrace of stem cells by osteoblasts. This myeloid cytokine upregulates dopamine receptors and beta2-adrenergic receptors on immature CD34 cells and increases their motility, proliferation, and secretion of matrix metalloproteases. It also generates type 2 dendritic cells that enhance the development of Th2 cells. Note, CD34 stem cells are mobilized into the blood within 2 days of an ischemic stroke.
Anti-VLA-4 antibodies are even more potent than granulocyte-colony stimulating factor (G-CSF) in causing the release of CD34 stem cells from bone marrow, and preventing blood CD34 cells from migrating back to the bone marrow. Natalizumab-induced CD34 cells have lower levels of adhesion molecules and secrete less matrix metalloprotease-9 than G-CSF-mobilized hematopoietic stem cells. Together, G-CSF and anti-VLA-4 antibodies synergize to increase the mobilization of stem cells by a further 10-fold. This synergy is important if natalizumab is to be combined with chemotherapy, bone marrow transplantation, or possibly erythropoietin. Drug interactions that affect stem cell mobilization are important because CD34 stem cells are reservoirs for the JC virus that causes progressive multifocal leukoencephalopathy.
Genes involved in B-cell activation and differentiation are upregulated by natalizumab therapy. Natalizumab also elevates the numbers of circulating basophils, eosinophils, lymphocytes, and reticulocytes.
Natalizumab’s ability to increase peripheral white blood cells and release stem cells from bone marrow is reminiscent of a stress response influenced by the sympathetic nervous system. The sympathetic nervous system innervates bone marrow and releases vasoactive intestinal polypeptide, neuropeptide Y, and norepinephrine, which reduces CXCL12 on osteoblasts. Low CXCL12 causes bone loss but also allows hematopoietic stem and progenitor cells to be mobilized from their usual close association with osteoblasts. Beta-adrenergic agonists enhance stem cell mobilization; beta blockers inhibit mobilization. There is strong evidence for sympathetic nervous system disruption in multiple sclerosis, presumably caused by hypothalamic and spinal cord plaques in sympathetic pathways. This disruption causes denervation hypersensitivity, with increased expression of beta2-adrenergic receptors on immune cells (38), and possibly on CD34 cells, osteoblasts, and osteoclasts.
Sympathetic input inhibits osteoblasts to cause osteopenia. Osteoclasts produce chemokines and present antigen to CD8 cells; they also induce suppressor/regulatory CD8 cells. Multiple sclerosis patients have low bone density, disrupted sympathetic outflow, and low regulatory CD8 T cells. Thus, it is likely that the bone marrow milieu in patients with sympathetic nervous system damage may be more sensitive to common drugs such as terbutaline (beta agonist; enhances mobilization) and propranolol (beta blocker; inhibits stem cell mobilization). These agents could modify the stem cell release provoked by VLA-4 blockers. Natalizumab effects on osteoclasts, derived from the monocyte lineage, and associated bone-resorbing dendritic cells are also possible.
The abundance of white blood cells in the circulation is kept constant by “immune homeostasis” regulated by feedback between the number of lymphocytes and their absorption of the serum cytokines, IL-7 and IL-15. More IL-7 is available when lymphocytes are missing; the IL-7 bloom then induces new lymphocytes, especially CD8 memory T cells, as well as CD4 regulatory and CD4 memory T cells. Some of these peripherally expanded cells are poor suppressors and autoreactive because their tolerance is not controlled by development in the thymus. Peripheral immune expansion and poor tolerance are especially relevant in multiple sclerosis, where thymic output falls to that of a person approximately 30 years older. IL-4, IL-2, IL-21, B-cell-activating factor, and type I interferons also enhance homeostatic expansion. In multiple sclerosis, there are fewer recent (naïve) thymic emigrants (TREC+); polymorphisms in the IL-7 receptors are linked to the onset of multiple sclerosis; oral antigens, gut microflora (39), parasites, and other environmental factors may influence the onset and relapses of multiple sclerosis.
During natalizumab therapy, the peripheral blood lymphocyte count doubles. This lymphocytosis correlates with improved clinical and MRI response. After treatment, there are twice as many circulating B cells and seven times more immature B cells, but they produce less immunoglobulin. The lymphocytosis is likely from the release of B-cell and T-cell precursors from the bone marrow and thymus, evidenced by more immunoglobulin kappa and T-cell receptor excision circles; both are under VLA-4 control. Secondly, natalizumab prevents lymphocyte migration into the CNS. It is possible that “excess” memory T cells, formerly destined to enter the brain, are tolerized or gradually deleted from the blood. Similarly, T cells are deleted by apoptosis during interferon-beta therapy. Monoclonal anti-VLA-4 (antibody 9C10) causes apoptosis of activated T cells, but other anti-VLA-4 antibodies do not (antibodies PS/2 and R1/2). Nonetheless, all of these antibodies block VLA-4/VCAM-1 interaction.
Most CSF immune cells, especially T cells, disappear during natalizumab therapy. IL-7 levels are low in CSF during multiple sclerosis relapses, but the influence of low IL-7 on the rare immune cells in multiple sclerosis CSF is unknown.
CSF oligoclonal bands, described as an unchanging “fingerprint” (Tourtellotte 1998, personal communication), do not change with most multiple sclerosis therapies. In four of six natalizumab-treated patients, however, bands disappeared after 10 infusions, and local IgG production was decreased.
Natalizumab binds to the alpha4 component of alpha4beta1 (VLA-4) and to alpha4beta7 (LPAM-1), inhibiting migration through brain endothelial cells by the vast majority of T cells and by 75% of monocytes. Therapy halves soluble VCAM in the serum, preventing soluble VCAM from activating blood-brain barrier endothelial cells that would otherwise generate reactive oxygen species and matrix metalloprotease activity, which are required for leukocyte migration. Natalizumab reduces the number of VLA-4-positive T cells from 92% to 3%, depending on time since infusion (50).
VLA-4 expression is twice as high on blood B cells and monocytes as on T cells, twice as high on CD8 as on CD4 T cells, higher on memory T and B cells than on naive cells, and low on FoxP3 regulatory T cells (50). Cells with low VLA-4 expression have difficulty migrating through fibronectin in a 2-compartment Boyden chamber, a model of the blood-brain barrier (without VCAM) (50). Anti-VLA4 therapy prevents entry of B cells and activated Th1 cells into the CSF. Natalizumab therapy has differential effects on peripheral lymphocyte subpopulations. Natalizumab therapy reduces the expression of VLA-4 on all MNC; the effect is twice as pronounced on CD4 and CD8 T cells (50% of their original VLA-4 expression) as on B cells and monocytes (30% reduction) within 90 minutes of the infusion. There are two cautions regarding this report of VLA-4 fluctuation. VLA-4 expression rebounds to normal in blood before the next monthly infusion, yet CSF immune cell percentages are not affected. Secondly, affinity was not measured, even though the high-affinity form of VLA-4 drives adhesion.
VLA-4 expression on naive and memory T cells is debatably low in multiple sclerosis (reports vary). Subnormal VLA-4 expression in multiple sclerosis is surprising, as chronic antigenic exposure elevates alpha4 integrin and osteopontin mRNA (eg, after bee venom immunotherapy to treat bee-sting hypersensitivity). Already low VLA-4 levels on peripheral immune cells would suggest that anti-VLA-4 therapy in multiple sclerosis will be potent. VLA-4 expression is greater on lymphocytes in secondary progressive multiple sclerosis than in primary progressive multiple sclerosis and relapsing-remitting multiple sclerosis, suggesting that therapeutic benefit could vary with disease state. Note that these studies measure expression, not binding affinity.
Costimulatory interactions that enhance immunity include (1) CD80 (B7-1) and CD86 (B7-2) on antigen-presenting cells paired with CD28 (stimulatory) and CTLA-4 (inhibitory) on T cells, (2) CD40 on antigen-presenting cells with CD154 (CD40L) on T cells, and (3) LFA-3 on endothelial cells with CD2 on T cells. Blockers of costimulation and adhesion have been tested in multiple sclerosis.
CD80 expression is increased on B cells and on some activated T cells during multiple sclerosis exacerbations (25). Serum interferon-gamma and TNF-alpha increase during exacerbations; they induce CD80 and upregulate CD86 on endothelial cells. This is important because CD80 potently enhances antigen-specific B cell responses. Potentially part of its broad therapeutic effects, interferon-beta downregulates CD80. However, CTLA-4-Fc constructs that block the interaction of CD80 with its ligands had no clinical benefit.
CD40/CD40L costimulation is increased in multiple sclerosis immune cells as part of a positive feedback loop that generates IL-12 and interferon-gamma, induces chemokine secretion, and enhances VCAM-1 and ICAM-1 expression by human endothelial cells. Trials to block this interaction in multiple sclerosis are in progress.
Based on animal studies, CD2/LFA3 is much less important than VLA-4/VCAM-1 in T cell penetration of the blood-brain barrier. CD2 expression on lymphocytes is abnormal in multiple sclerosis and is less capable of transducing activation signals. Trials with blockers of this interaction are contemplated in multiple sclerosis.
Adhesion blockade has been ineffective as a therapy for multiple sclerosis apart from anti-VLA-4 antibodies. Anti-LFA-1 antibodies that block adhesion to ICAM-1 had no clinical effect on multiple sclerosis in a well-designed trial, although oral infections were more frequent.
VLA-4 interactions enhance immune activation in multiple sclerosis and in its disease models. Interaction of VLA-4 with its ligands is costimulatory and enhances activation, proliferation, and immune reactivity of T and B cells. T cells transfected with CD49d (the alpha4 component of VLA-4) proliferate strongly to self-cells and to otherwise suboptimal levels of antigen.
The importance of VLA-4 in CNS inflammation was established in experimental allergic encephalomyelitis, an animal model of multiple sclerosis. Anti-MBP T cell clones expressing high levels of VLA-4 were better at inducing experimental allergic encephalomyelitis than clones expressing low VLA-4. Several nonactivating anti-VLA-4 Abs significantly blocked T cell and monocyte adhesion to brain endothelial cells and reduced the severity of experimental allergic encephalomyelitis.
Certain anti-VLA-4 Abs (P4G9, HP1/7, and PS/2) can activate immune responses; others may block B cell activation. The PS/2 antibody induces an active conformation of the VLA-4 molecule and enhances costimulation. Consequently, this antibody activates Th1 cells and interferon-gamma secretion and induces CD4 migration into the CNS. The PS/2 antibody, given in the first week after induction, ameliorates experimental allergic encephalomyelitis. However, if PS/2 is given at the peak of disease or during remission, it worsens experimental allergic encephalomyelitis. VLA-4 also enhances the ability of myelin basic protein-reactive T cell induction of pro-inflammatory cytokines in microglia. VLA-4 tethers to VCAM-1 on B cells and enhances B cell signaling. In parallel, the B cell receptor enhances tight adhesion.
Ninety minutes after infusion of natalizumab into multiple sclerosis patients, their immune cells are refractory to stimulation. Natalizumab reduces VLA-4 expression on T cells by 49%, on B cells by 29%, and on monocytes by 25%, but the numbers return to baseline before each infusion (50). After the first natalizumab infusion, there is lymphocytosis and a mild increase in cells that produce IL-2, IL-17, and interferon-gamma in vitro.
Anti-VLA-4 therapy induces RNA coding for a large number of B-cell activation and differentiation markers. Some of these may enhance JC virus replication, such as the Spi-B transcription factor, which elevates the neurotrophic JC virus variants in B cells and stem cells (section 12). Natalizumab also induces T cell and monocyte expression of Tbet, a Th1 and possibly Th17 marker that induces both inflammatory and anti-inflammatory cytokines in serum.
The postcapillary venule is the site of T-cell penetration through blood vessels. Past the capillaries, blood flow slows, allowing more time for attachment. Post-capillary high-endothelial venules have a unique morphology and overexpress adhesion molecules that beckon tissue-specific immune cells.
The lesions of multiple sclerosis correspond to the paths of CNS veins, reflecting the importance of lymphocyte adhesion to postcapillary venules. Ninety-four of 95 plaques surround veins on MR venography (63). Venous anatomy, adhesion molecules, and flow dynamics suggest why multiple sclerosis lesions predominate in periventricular white matter. Cerebral blood volume is highest in the periventricular white matter of the brain (ie, many white blood cells pass through this area) compared to the centrum semiovale and intermediate white matter regions. The blood flow rate in the periventricular white matter in multiple sclerosis is more than 2-fold slower than in the normal brain, allowing more time for adhesion. MRI visualizes relatively large venules downstream from the activated high endothelial venules. However, the lesions are likely to start at small-lumen postcapillary sites with activated high endothelial venules (see figure, Lymphocyte penetrating blood-nerve barrier).
The interaction between T cells and endothelial cells is dynamic and mutually reinforcing. Activated T cells and monocytes secrete cytokines that induce more MHC class II and costimulatory molecules, as well as P- and E-selectins, ICAM-1, and VCAM-1 on endothelial cells. Contact with CD8 and NK cells induces MHC class II on endothelial cells, independent of cytokines. Activated endothelial cells secrete chemokines, which induce the active conformation of VLA-4 on leukocytes and stronger adhesion by T cells. Interferon-beta rapidly disrupts the positive feedback loop between white blood cells and endothelial cells; gadolinium-positive MRI lesions disappear within days of starting interferon-beta therapy.
Penetration of the blood-brain barrier is a two-step process. Cells from inside postcapillary venules must first cross the endothelial cell/tight junction of the blood-brain barrier. Many assume this occurs by breaking the strong connection between two endothelial cells. Compared to peripheral junctions, unique “tight” junctions in the brain endothelium do not express VCAM-1 and are nearly impervious. One route into the brain may be through endothelial cells.
In this model, immune cells bind strongly to VCAM-1 and ICAM-1 on the surface of endothelial cells. They are then internalized and penetrate transcellularly, moving directly through endothelial cells (“emperipolesis” = “wandering around inside”). This was first seen with lymph node endothelial cells, and then demonstrated in experimental allergic neuritis (EAN) (03) and experimental autoimmune encephalitis. In EAN, the immune cells appear to be activated, but the postcapillary venules are not obviously activated and are not pseudo-columnar (unlike high endothelial venules).
The second step above, crossing the basal lamina from the perivascular space, requires gelatinases. These matrix metalloproteases secreted by T cells and monocytes degrade dystroglycan and lyse the dense subendothelial lamina. Natalizumab may not affect this step. However, interferons prevent matrix metalloprotease secretion, suggesting potential synergy with natalizumab in preventing cells from entering the CNS.
Immune cell subsets differ in their ability to penetrate the blood-brain barrier:
(1) Fluor-labeled activated human cells injected into mouse carotid arteries adhere to lipopolysaccharide-activated venules using intravital microscopy of mouse brain. CD8 memory and effector cells, but not CD4 cells, from patients with exacerbations display excessive rolling and arrest. P-selectin and endothelial selectin binding is predominant in CD8 cells (and possibly more important in mice than humans), whereas VLA-4 and VCAM-1 interactions are most important for CD4 adhesion. Preserved selectin binding by CD8 cells could account for the higher CD8/CD4 ratio in CSF during natalizumab therapy.
(2) ConA mitogen-activated CD4 T cells are more efficient than CD8 T cells and B cells at migrating through a monolayer of cytokine-activated rat brain vascular endothelial cells. B cells and CD8 cells adhere more avidly, but CD4 cells migrate better, especially after they or the endothelial cells are activated. In models (1) and (2), anti-VLA-4 antibodies would selectively decrease CD4 cell penetration.
(3) In a model using adult human cells, supernatants from microglia and astrocytes encourage firm junction formation between brain endothelial cells. B cells and monocytes migrate more easily than T cells. B cells use VLA-4/ICAM, but not VCAM (01). Monocytes have the most high-affinity VLA-4 molecules (Table 1), but also use CCL-2 and metalloproteases for penetration. As they penetrate the blood-brain barrier, monocytes increase its permeability to soluble molecules and to migrating T cells.
Th2 lymphocytes migrate twice as well as Th1 cells. Th2 cells express more VLA-4 and LFA-1 (Antel J 2005, personal communication), and natalizumab inhibits Th2 migration more than Th1 migration. Only Th2 cells express the chemokine receptor, CCR2, which recognizes the chemokine, MCP1, secreted by endothelial cells. In contrast, PSGL1/CLA on Th1 cells, not on Th2 cells, binds to E- and P-selectins on endothelial cells. Because Th2 cells migrate better, this suggests that VLA-4 and CCR2 are critical molecules at the blood-brain barrier, and the selectins are not. Supernatants from Th1 cells, but not Th2 cells, upregulate VCAM-1 and ICAM-1 on endothelial cells; this demonstrates the dynamic interaction between Th1 and endothelial cells. Conversely, preincubation of these human brain endothelial cell cultures with interferon-beta reduces migration of Th1 cells, but not Th2 cells, implying that it can enhance CNS immune suppression by favoring Th2 over Th1 migration. CD8 suppressor cell entry has not been studied.
(4) In experimental allergic encephalomyelitis, myelin oligodendrocyte glycoprotein (MOG)-specific Th1 cells express high levels of alpha4 integrin and are dependent on VLA-4 for entry into the spinal cord. Th17 cells have low levels of alpha4 integrin, but more alpha4,beta7, another natalizumab target. Antigen-specific Th17 cells, even in mice whose immune cells do not express VLA-4, are able to enter the brain by using LFA-1 and alphaL,beta2.
(5) Concomitant medications can alter these dynamics. Glatiramer therapy induces IL-4, a Th2 cytokine that inhibits VLA-4 expression on CD8 cells. Glatiramer in combination with natalizumab could prevent anti-JC virus CD8 cells from reaching the brain.
Natalizumab blocks immune cell entry into the CSF in multiple sclerosis. With therapy, CD4 T cells, CD19 B cells, and CD138 plasma cells are essentially gone from the CSF; CD8 T cells fall to one fifth of pretherapy levels. The ratio of CD4/CD8 falls, perhaps because CD8 cells continue to express more VLA-4 than CD4 cells. Th17 cells are less affected than Th1 cells in mice. Entry of CD4+FoxP3+ regulatory T cells (Tregs) is also blocked, comparable to other subsets; their function in multiple sclerosis remains ineffective during natalizumab therapy. B cells in the CSF are reduced, as are the IgG index and sometimes oligoclonal IgG bands.
The brain is an “immunologically privileged” site. A tight blood-brain barrier and high levels of P-glycoprotein transport by the endothelium block serum proteins from leaking into the surrounding tissue. There is reduced immune activation in the CNS – less leukocyte trafficking, significant endogenous immunosuppression (glial production of neurosteroids, prostaglandins, TGF-beta, and IL-10), microglia that are poor at presenting antigens, low MHC class I and minimal or no MHC class II expression on CNS cells, and restricted lymphatic flow out of the brain (54). Lymphatic fluid flows from CNS perivascular spaces (glymphatic drainage; glial plus lymphatic) to cribriform plate, to nasal lymphatics, and then to cervical lymph nodes. This induces a Th2 immune bias to CNS antigens in the periphery. Less inflammation against CNS antigens should ameliorate multiple sclerosis and possibly neurodegenerative diseases.
In multiple sclerosis, endothelial cells and microglia are activated, astrocytes are activated and hypertrophied, and inflammatory cytokines disrupt oligodendroglia and neurons. The brain loses its immune privilege, and there is intermittent immune activation and brain cell death.
Adhesion molecule expression on normal brain endothelial cells and within the brain parenchyma is lower than in other sites. Even during experimental allergic encephalomyelitis, VLA-4 and LFA-1 are downregulated on T cells after they enter the CNS environment. VCAM-1 is undetectable on normal human endothelial cells but appears on microglia and monocytes at sites of inflammation in multiple sclerosis. Low-level expression of VCAM-1 on blood vessels and microglial cells and of VLA-4 on immune cells in perivascular cuffs and parenchyma in acute plaques markedly increases in chronic active and chronic silent multiple sclerosis plaques. VCAM-1 is the main adhesion molecule on endothelial cells in the CNS and on pericytes in plaques; selectins and ICAM have minimal expression. This matches the therapeutic failure of anti-LFA-1 (the ICAM partner) in multiple sclerosis, compared to the pronounced benefit from natalizumab (VLA-4).
“Pioneer lymphocytes” arrive in the CNS within 2 hours after infusion of anti-myelin T cells in mice (passive transfer experimental allergic encephalomyelitis). These cells penetrate into the CNS by binding to P-selectin and ICAM-1 expressed on cells in the choroid plexus. These cells do not use VLA-4 for adhesion and would not be blocked by anti-VLA-4 therapy. In multiple sclerosis brains, P-selectin is found on large venules in the meninges and choroid, but not small venules – the site of T cell penetration (40), so P-selectin may not be involved in immune breach of the blood-brain barrier. During experimental allergic encephalomyelitis, choroid plexus epithelial cells upregulate VCAM-1 and ICAM-1, and begin to express MAdCAM-1. These molecules are not seen on the choroid vascular endothelium or on parenchymal endothelium. In contrast, MBP-specific CD4 T cells adhere to VLA-4 on subcortical and periventricular endothelial cells 24 hours after injection. The later influx of memory T cells is likely to be blocked by natalizumab.
Osteopontin is a ligand for the activated form of alpha4beta1 integrin and several other integrins expressed on monocytes, so it could modify responses to anti-VLA-4 Abs. Osteopontin is a matrix glycoprotein named for its stimulatory effects on bone osteoclasts. It is expressed by macrophages and astrocytes in and around multiple sclerosis plaques, and by microglia, astrocytes, neurons, and choroid plexus in experimental allergic encephalomyelitis lesions. The biological effects of osteopontin in multiple sclerosis, including changes in VLA-4 function, are complex.
T cells and monocytes secrete osteopontin. In sarcoidosis, lymphocyte expression of osteopontin correlates with granuloma maturity, T cell chemotaxis, T cell adhesion, and T cell costimulation. Osteopontin prevents apoptosis of activated T cells and some monocytes. It is induced by interferon-gamma, and in turn, osteopontin induces IL-12 and more interferon-gamma-cytokines that are detrimental in multiple sclerosis. Osteopontin enhances Th1-type immunity, triggers relapses of EAE, and increases CNS inflammation. Osteopontin knockout mice have a shift to Th2 immunity, milder experimental allergic encephalomyelitis, and less neurodegeneration in models of Parkinson disease and stroke. Twelve hours after ischemia to the brain, osteopontin appears in microglia and induces astrocyte chemotaxis and activation through integrin signaling. It also induces proliferation of oligodendrocyte precursors and formation of myelin. In multiple sclerosis, osteopontin serum levels are slightly elevated and correlate with a generalized shift to Th1 immunity. In mononuclear cells from untreated patients, the SPP1 (osteopontin) gene is elevated 200-fold in stable multiple sclerosis and 900-fold during exacerbations (20).
Interferon-beta inhibits osteoclast differentiation and actually increases bone formation, in contrast to osteopontin’s osteoclast-stimulating activity. Regulatory T cells secrete TGF-beta plus IL-4 and also block osteoclast differentiation. Interferon-beta-1b therapy does not elevate plasma osteopontin levels in relapsing-remitting multiple sclerosis. However, interferon-beta reduces RNA expression by 450-fold in stable disease compared to untreated stable multiple sclerosis, by 1700-fold in active multiple sclerosis (20). Thus, interferon therapy may synergize with natalizumab on multiple biological effects.
In the phase IIb Antegren/natalizumab study, six monthly infusions reduced relapses by 57% in 213 relapsing-remitting and secondary progressive (one third of total) patients (46). (Later observational studies also showed benefit on cumulative clinical damage in secondary progressive multiple sclerosis, but there was no benefit in primary progressive multiple sclerosis.) The therapeutic effect lasted 2 to 3 months after the final infusion in both forms of multiple sclerosis.
Full responders, with no clinical or MRI activity during natalizumab therapy, have less active disease at baseline. Counterintuitively, pre-study clinical activity does not predict rebound activity after withdrawal of natalizumab (56). Based on these studies, inactive disease may also rebound after natalizumab cessation.
In a 25-month phase III multiple sclerosis trial of 942 relapsing and remitting patients (randomized at two natalizumab-treated to one placebo; AFFIRM study), monthly infusions of 300 mg natalizumab reduced new and enlarging T2 lesions by 83%, Gd-enhancing MRI lesions by 92%, T1 black hole development by 40% (15% with natalizumab compared to 25% with placebo), and relapse rate by 66%. Brain atrophy was greater in year 1 but less in year 2; the fall at one year may be from anti-inflammatory effects, as seen with interferon-beta and glucocorticoids. Long-term natalizumab therapy has minimal benefit on brain atrophy. However, “brain atrophy” may be pseudoatrophy due to less CNS inflammation. After switching to other therapies, MRI T2 lesion volume expanded by 0.66 ml/year in newly-untreated and by 1.98 ml/year with monthly intravenous methylprednisolone versus continued natalizumab (49). Brain volume was -0.24 ml/year with continued natalizumab, but increased by +0.13 ml with no therapy and deceased by -0.74 with intravenous methylprednisolone. Thus, withdrawal of natalizumab therapy may provoke inflammation and increase brain volume.
Progression was slowed by 36% at 1 year and 42% at 2 years of therapy. Post hoc analysis showed that treated patients were more likely than placebo patients to have “no evidence of disease activity” on clinical (64% free in treated vs. 39% in placebo), radiological (58% vs. 14%), or both measures (37% vs. 7%). Therapy reduced the risk of cognitive worsening by 43% compared to placebo. Natalizumab therapy reduces neurofilaments in CSF by 3-fold, down to healthy control levels, suggesting amelioration of damage to CNS nerves. In a post hoc analysis of the 116-week AFFIRM study, out of 620 available subjects from the original 942 patients, 25% had clinically defined improvement (CDI), and 20% had progression (CDP) (51).
Another trial evaluated 1171 patients who had had at least one exacerbation in the prior year during therapy with weekly intramuscular interferon-beta-1a, ie, they were weighted toward suboptimal interferon-beta-1a responders. Patients were randomized into interferon alone or interferon plus natalizumab (SENTINEL study). Natalizumab in combination with interferon reduced new and enlarging T2 lesions by 83%, Gd-enhancing MRI lesions by 89%, relapse rate by 55%, and progression by 24% at two years, compared to interferon alone. The superiority of natalizumab versus placebo was not studied. Natalizumab also abolished MRI and clinical activity in five patients with aggressive multiple sclerosis who had only a partial response to cyclophosphamide plus autologous stem cell transplantation. On average, starting this therapy de novo is more effective in suppressing relapses than interferons, glatiramer, dimethyl fumarate, teriflunomide, and possibly S1P modulators.
The annual relapse rate is reduced by 0.92 when the expanded disability status scale (EDSS) rating is less than or equal to 3.5, by 0.70 for EDSS of 4 to 6, and by 0.57 for EDSS greater than or equal to 6. In most patients, the onset of benefit is rapid, regardless of baseline disease activity. Therapy restores visual and sensory evoked potentials in one third of patients but does not improve magnetic evoked potentials. EDSS scores improve in 69% of patients over two years versus placebo; this correlates with quality of life.
Quality of life on physical and mental subscales improved with natalizumab in two trials; subjects on placebo or interferon-beta-1a alone declined, and patients often reported feeling better. There was 43% less cognitive decline than with placebo; the benefit was equivalent to intramuscular interferon-beta-1b. Fatigue is reduced in most studies. Pain from multiple sclerosis is reduced in 56% of patients, but 15% have more pain with therapy (Foley J 2009, personal communication). Migraine severity decreases from 14 to 10.5 on the migraine disability assessment scale. This therapy increases productivity by 3.3 hours per week.
Subcutaneous natalizumab is therapeutically equivalent to the intravenous drug (REFINE study). It reduces costs by 63%. It is preferred by 88% of patients because it saves time.
Forty-nine patients of African ancestry in two large studies had benefit from natalizumab on clinical (60% fewer relapses), cognitive, and MRI measures; benefit was equivalent to that seen in Caucasian patients. These patients have excessive T-dependent B cell responses and more serum anti-neuronal antibodies. In 19 children with multiple sclerosis, the drug improved EDSS and prevented Gd-enhancing lesions. Pregnancy registries show no adverse outcomes. Benefit on relapses and progression is equivalent to anti-CD20 therapy and to hematopoietic stem cell transplants, with fewer side effects than with transplants.
Low serum vitamin D levels correlate with more attacks while on natalizumab. The annualized relapse rate is 0.31 with vitamin D levels of less than 50 nmol/L but improves to 0.10 with levels of 50 nmol/L or higher. This suggests that vitamin D-induced shift from Th1 to Th2 immunity enhances natalizumab benefit, as with interferon-beta and S1PR modulator therapy (22).
Occasional patients have worsening of multiple sclerosis symptoms after the first one or two infusions. In highly active patients, lesions active at baseline persisted longer in the first two weeks after starting natalizumab. There was a rebound increase in relapses in this subgroup during months 3 to 5 after the second of two infusions (38% vs. 9%, p < 0.005). Early multiple sclerosis activity might be expected in a treatment that is not 100% effective, or it may be from elevation of pre-B cells (7-fold increase) and mature B cells (3-fold increase) in blood, fewer regulatory T cells, or activated T cells that produce inflammatory cytokines (interferon-gamma, TNF-alpha, and IL-17). However, there were no therapy-linked exacerbations in a Danish nationwide study or in a post hoc analysis of the highly active subgroup of several large studies. Every 6-week infusions reduce the risk of PML, without provoking exacerbations.
Circulating B and cytotoxic NK cells increase, but CD8+CD28- and CD4+ regulatory T cells do not. However, the few cells that penetrate into the CSF are anti-inflammatory, such as CD4 Treg and IL-10+ PD-1+ CD8 Treg. CSF white cells express more IL-10 and less interferon-gamma, IL-1beta, IL-6, IL-23, osteopontin, chemokines, and matrix metalloprotease-9, and there is a 3-fold reduction of neurofilament light chains to normal levels. Lower osteopontin correlates with cognitive improvement. DNA variants in the WNT pathway, potentially affecting BBB penetration, myelination, and neuro-repair, are predicted to control responses to natalizumab (15).
A third of patients have recrudescence of multiple sclerosis symptoms 24 to 28 days after each infusion (“wearing off” phenomenon). This is more common early in the course of treatment, as trough natalizumab serum levels climb and the disease quiets over time. The half-life of natalizumab after a single 3 mg/kg dose is 4 to 5 days (57). After monthly infusions of 4.5 mg/kg, the half-life becomes 11 days. This half-life is shorter than that of many other monoclonal antibody therapies. Natalizumab-VLA-4 complexes are internalized and presumably degraded. Receptors (VLA-4) are over 80% saturated for at least 28 days after a single 300 mg dose; binding falls to 50% at 2 months and 40% at 3 months. The drug reduces the expression of VLA-4 on immune cells by approximately 10%.
Expected loss of drug activity can be based on kinetic markers after drug withdrawal. Serum natalizumab level is less than 1 µg at 84 days, immune cell surface saturation with anti-VLA-4 is less than 20% at 42 days, and white blood cell count falls back to normal levels at 98 days. This suggests that a prolonged half-life, plus a slow off-rate, should lead to a therapeutic effect that lasts for several months after drug discontinuation (see extended dosing to prevent PML, section 16). In support, there is persistent depression of the CSF white cell count after natalizumab therapy for at least 6 months (reduced CD4 and CD8 T cells, CD19 B cells, and CD138 plasma cells; CSF changes are greater than changes in the blood; additional therapy was not detailed). A rise in the CSF cell count and more neuroaxonal damage correlate with anti-JC virus serum antibodies.
However, in another series of patients on no post-natalizumab therapies, the rebound of CSF white blood cells after discontinuation was faster, with low CSF white blood cells at 50 days, but a rebound to normal after 100 days (32). Cells remained low for 100 days after treatment with the combination of natalizumab plus interferon or glatiramer. CSF cell counts return to baseline at 14 months in patients treated with other therapies (62). The risk of secondary progression is 2.7-fold if no other therapy is begun. There have been cases of PML 1 to 6 months after the drug is stopped, before CNS immunity is restored.
Natalizumab has effects on other diseases. It ameliorates active psoriasis (07), Crohn disease, and, possibly, rheumatoid arthritis. Case reports show improvement in Susac syndrome, Rasmussen encephalitis, and multiple sclerosis-associated intermediate uveitis (13). Anti-VLA-4 Abs prevent insulitis in spontaneous diabetic NOD mice.
Small-molecule VLA-4 antagonists prolong rat cardiac transplants and inhibit experimental autoimmune uveitis. In multiple sclerosis, firategrast, an oral anti-alpha4beta integrin molecule, reduced Gd-enhancing MRI lesions at high doses, but increased lesions at the low dose. With its 3.5-hour half-life, serum levels were variable, and high doses increased bladder infections and caused vomiting.
In summary, anti-VLA-4, an antigen-nonspecific therapy, ameliorated MRI lesions, exacerbations, cognitive loss, and progression, and it markedly reduced CSF white blood cells in multiple sclerosis. Side effects were minimal, except for PML, and the combination with interferon was superior to interferon-beta alone. It should not be forgotten that this drug and all other multiple sclerosis therapies are not perfect, and there are still some breakthrough exacerbations and progression.
There are few rare allergic reactions to natalizumab. In patients with mild to moderate infusion reactions, some infusion centers premedicate with antihistamines (diphenhydramine 50 mg, loratadine 10 mg), acetaminophen (1000 mg), and oral steroids (prednisone 50 mg).
Natalizumab increases peripheral lymphocyte counts in 50% and causes eosinophilia in 20%. Infusion reactions are 10-fold more common in those with high eosinophils. The rise in the number of B, NK, and T cells is a marker of better efficacy; those with relapses on therapy are four times more likely to have a minimal rise in lymphocytes.
There was no tuberculosis in the pivotal trials, and no statistically significant increase in cancer, melanoma, CNS lymphoma, or infections. In contrast, VLA-4 blockade is potentially therapeutic in cancer, as is interferon-beta. Integrins expressed on cancer cells enhance metastasis and tumor invasion; the block of integrins by natalizumab could potentially prevent metastasis. However, natalizumab does decrease NK cell killing of melanoma cells.
Neutralizing antibodies to natalizumab increase the chance of an infusion reaction. Hypersensitivity reactions are 5% overall, but with re-exposure after prolonged absence of therapy, they are 24%. The effect of VLA-4 polymorphisms on neutralizing antibody generation and on the efficacy of natalizumab therapy is unknown. Smokers have a 2.5-fold higher rate of neutralizing antibody formation than nonsmokers, in addition to the reality that smoking promotes exacerbations and progression and cancels the benefit of multiple sclerosis therapies.
Serum antibody titers to cytomegalovirus, Epstein-Barr, and JC virus, and myelin-oligodendrocyte basic protein (MOBP) are increased in multiple sclerosis, but viruses other than the JC virus are not reactivated. There is a risk of PML, which is dependent on prior immunosuppression and serum anti-JC virus antibody titers (see Section 12). During embryonic development, VLA-4/VCAM-1 interactions are important in chorioallantoic fusion, skeletal development, and formation of the septum between the aorta and pulmonary trunk, as well as sympathetic cardiac innervation. Fibronectin and integrins, including VLA-4, are important in early angiogenesis in the CNS. Embryonic defects appear in murine VLA-4 knockouts and with small molecule antagonists of VLA-4. A small molecule alpha4 integrin antagonist, oral CDP323, had no clinical benefit in multiple sclerosis, but pregnancies were not reported. Importantly, in multiple teratogenicity studies of anti-VLA-4 antibodies, defects have not been seen because antibodies are too large to cross the placenta.
Pregnancy registries for natalizumab show it is safe. Of 35 German women who received natalizumab during early pregnancy, 28 had normal infants, one had hexadactyly, and the others had miscarriages or elective termination, suggesting there was no adverse effect during pregnancy. There was no rebound in multiple sclerosis activity with drug withdrawal in these pregnancy studies, but another study shows there is a rebound (52).
There are rare reports of immune thrombocytopenic purpura (ITP), anemia, primary CNS lymphoma, herpes encephalitis, neuroborreliosis, cryptococcal meningitis, and ocular toxoplasmosis. However, there is no apparent increase in these diseases over background levels in over 142,000 treated patients (July 2015), and latent tuberculosis is not activated. Six cases of drug-induced liver injury have been reported.
Rebound of multiple sclerosis after discontinuation of natalizumab is discussed in topics 8 and 16.
In neuromyelitis optica, 7 months of natalizumab slightly increased the exacerbation rate and significantly increased the EDSS score in five patients (41).
Unexpected benefits of natalizumab. GM-CSF and natalizumab both trigger the release of cells from the bone marrow and are potentially useful in harvesting cells for immune cell transplants (topic 2).
Natalizumab does not change immunoglobulin G responses with COVID or tetanus toxoid vaccinations but reduces responses to some other antigens to 70% of normal. It also reduces the expanded T-cell receptor repertoire of multiple sclerosis in the blood, and more so in CSF. There is re-expansion of the repertoire, perhaps virus-specific, during natalizumab withdrawal while treating PML. COVID-19 severity is reduced with natalizumab in comparison to untreated patients, perhaps because of a shift to Th1/Th2 immunity (66).
Antibodies to VLA-4, and small molecules that block VLA-4, will block VLA-4 binding to fibronectin. In rats with tendon injuries, anti-VLA-4 improves healing by preventing adhesion formation, a major impediment to healing (35).
Natalizumab disrupts myeloma binding to stroma and inhibits stroma- and vascular endothelial growth factor (VEGF)-induced signaling and tumor growth; it sensitizes multiple myeloma to proteasome inhibitors. Similarly, anti-VLA-4 disrupts the immune escape of B-cell lymphoma cells by preventing their binding to bone marrow stroma.
The combination of interferon plus natalizumab in the pivotal trials was linked to two cases of PML. The apparent synergy in disease induction between interferon-beta-1a and natalizumab is not statistically significant, with only two PML cases. However, the two agents could theoretically synergize or block JC virus pathology in multiple ways.
Interferon-beta induces the immunosuppressive Th2 cytokine, IL-10, in T cells and generates CD8 suppressor T cells. These CD8 cells are responsible for some of the therapeutic benefits of interferons, glatiramer acetate, S1P1 modulators, and anti-CD20 antibodies (24), but could potentially suppress CNS immunity during natalizumab therapy. VLA-4 blockade of trafficking by CD8+CD28- regulatory cells has not been studied.
Interferon-beta therapy reduces expression of VLA-4, ICAM-1, and VCAM-1, and other adhesion molecules on endothelial cells (53), on T cells, and on monocytes. Interferon therapy reduces VLA-4 mRNA in mononuclear cells from patients who respond clinically to interferon-beta but has no effect on nonresponders. Second, during interferon-beta therapy, VCAM-1 is shed from vascular endothelial cells (likely from peripheral endothelial cells, but possibly from activated CNS endothelial cells). Serum VCAM could block VLA-4 on immune cells and, in turn, block T cell-endothelial cell interactions. Nevertheless, circulating VCAM may have little impact because the binding affinity of soluble VCAM is 100 to 1000 times less than that of membrane-bound VCAM.
Anti-VLA-4 antibodies selectively decrease CD4 cell penetration of the blood-brain barrier, frustrating CNS antiviral responses. Preincubation of human brain endothelial cell cultures with interferon-beta reduces migration of Th1 cells, but not of Th2 cells, favoring (inhibitory) Th2 over Th1 migration. The combination could synergize with a block of blood-brain barrier adhesion by natalizumab.
Interferon-beta reduces barrier permeability through a direct effect on endothelial cells, without any change in tight junction proteins. It downregulates interferon gamma-induced adhesion molecules on endothelial cells and counteracts the increased microvascular permeability induced by lipopolysaccharide (53). Passage of T cells into the CNS through a transcellular route (emperipolesis), and not through tight junctions, predominates in experimental allergic encephalomyelitis. Passage through cells depends on the interaction between LFA-1 and ICAM-1. Type I interferons also increase CD73 on endothelial cells. This protein converts extracellular AMP into adenosine, a potent anti-inflammatory molecule that reduces permeability through umbilical vein endothelial monolayers. Type I and II interferons impair endothelial cell pinocytosis, already low in blood-brain barrier endothelial cells, and this may further block positive feedback between endothelial and immune cells.
Finally, matrix metalloproteases, reduced by interferons, can alter the surface charge of endothelial cells and allow passage directly through endothelial cells into the perivascular space and then facilitate passage through the basement membrane into the brain. Low serum MMP9 levels before therapy predict the development of PML (23).
Immune cells in multiple sclerosis secrete excessive levels of matrix metalloproteases, which degrade tight junctions. Activation of lymphocytes via VLA-4 induces production of matrix metalloproteases. However, natalizumab reduces levels of MMP9 in serum and CSF. Interferon-beta markedly reduces matrix metalloprotease secretion by T cells, inhibiting penetration of monocytes and lymphocytes through the blood-brain barrier. This suggests the two therapies could synergize in inhibiting MMP production.
Interferon-beta plus anti-VLA-4 therapy could have unexpected consequences. Type I interferons block the secretion of chemokines, which induce G proteins to activate VLA-4. The relative effects of 6 MU weekly interferon-beta versus more frequent interferon-beta on interactions with natalizumab are unknown. High-dose interferon-alpha interferes with neuronal function, but interferon-beta does not (see MedLink Neurology article “Multiple sclerosis”). Finally, inhibition of beta1 integrin signaling may inhibit myelination.
Interferon-beta could indirectly affect serotonin 5HT2A receptor expression and increase susceptibility to JC virus infection. Interferon-beta induces the enzyme indoleamine 2,3-dioxygenase, which decreases tryptophan and serotonin. When tryptophan is reduced, there is a compensatory increase in 5HT receptor expression. Early reports suggested that elevation of serotonin 2A receptors on neurons, astrocytes, and oligodendrocytes should enhance JC virus entry, although this is disputed. Interferon-beta has not been associated with the development of PML. However, Sjögren syndrome and lupus, which have high endogenous type I interferon levels and other immune abnormalities, are linked to JC virus infection (21).
Type I interferons enhance T cell responses against polyoma viruses. Toll-like receptors (TLR) on immune cells recognize single-stranded (TLR7) and double-stranded RNA viruses (TLR3) and DNA viruses (TLR9). Activation of these receptors by viruses causes B cell proliferation (TLR7, TLR9) and induces type I interferon secretion by plasmacytoid dendritic cells and interferon-stimulated genes, including type I interferons.
In several reports, interferon-beta had only a suggestive benefit in human PML. Interferon’s antiviral actions should prevent PML but may not be curative because little interferon is able to cross the blood-brain barrier. It is believed that only 0.1% of serum interferon-beta enters the normal CNS (53). Despite this, a strong interferon signature in the mouse brain 6 hours after interferon-beta injection indicates there is biologically significant penetration into the CNS (27). CSF interferon-beta levels have not been studied in multiple sclerosis, but blood-brain barrier leakage from CNS inflammation may increase interferon penetration of the blood-brain barrier.
In contrast to the arguments above, interferon-beta inhibits JC virus infection and spreading in cultured SV40-transformed human fetal glial cells. In vivo, interferons reduce levels of serum JC virus DNA. In a study with much higher serum JC virus DNA levels than in other reports, serum is positive for JC virus in 29% of healthy controls, 46% of untreated multiple sclerosis patients, and 14% of interferon-beta-treated patients. Finally, type I interferons actually upregulate their own interferon responses.
Statins interfere with leukocyte-endothelial cell adhesion by reducing the inflammatory response generated through lipid mediators in venules. Statins also reduce CD11b on monocytes and VCAM-1 expression on endothelial cells.
PML during therapy with other multiple sclerosis disease-modifying drugs is rare, through 2023 to 2024: 53-61 fingolimod, three siponimod, one ozanimod, three ocrelizumab (fewer than expected after many switched from natalizumab), zero ofatumumab, 12 dimethyl fumarate (1 of 50,000), zero teriflunomide, one interferon-beta, zero glatiramer acetate, and zero cladribine.
Fingolimod/FTY720 causes lymphocyte retention in lymph nodes and, thus, prevents them from reaching the CNS. Lymphocyte egress from lymph nodes is blocked when FTY720 binds to the sphingosine 1-phosphate receptor-1 (S1P1). Interferon beta also causes lymphocyte retention in lymph nodes by inducing the lectin CD69, which in turn binds to and negatively regulates S1P1. FTY720 and interferon beta would be expected to synergize by reducing lymphocyte penetration of the blood-brain barrier. FTY720 could enhance antiviral activity by retaining the virus and immune cells within secondary immune organs. Conversely, it could prevent cytolytic CD8 cells from entering the CNS. Dimethyl fumarate lowers the lymphocyte count; low counts are linked to PML cases.
Glatiramer does not cause PML even though it causes a Th1 to Th2 shift that may cause CNS immune suppression in some patients. IL-4, a Th2 cytokine, inhibits VLA-4 expression on CD8 cells. Synergy with natalizumab has not been tested.
In other life-threatening disorders, PML is associated with many drugs used as multiple sclerosis treatments, including glucocorticoids, autologous bone marrow transplants, cyclophosphamide, mycophenolic acid, azathioprine, methotrexate, intravenous immunoglobulin, mitoxantrone, and monoclonal antibodies (adalimumab, one case; alemtuzumab, 14 cases; bevacizumab, three cases; cetuximab, one case; efalizumab, eight cases (this drug has a black box warning for PML); radioactive ibritumomab tiuxetan, five cases; infliximab, four cases; and rituximab, 114 cases (1 of 30,000), in WHO, Canadian, and PubMed databases. Efalizumab, an anti-CD11a (LFA-1) used to treat psoriasis, caused PML in 1 in 500 patients and was withdrawn from the market because the risk-to-benefit ratio was too high. In non-multiple sclerosis patients, at least 114 cases of PML have occurred with rituximab, an anti-CD20 MAb. The incidence of PML is 1 of 100,000 with rituximab used to treat rheumatoid arthritis, but 1 of 5000 in systemic lupus erythematosus. Many of these immunosuppressive drugs cause the release of bone marrow cells. “Demyelinating disease” after anti-TNF antibody infusions could be confused with PML.
Chemotherapy suppresses immune responses. The combination of natalizumab with other drugs, such as fludarabine and cladribine, and also rituximab, leflunomide, statins, glatiramer, and even beta-adrenergic agonists and pentoxifylline, poses theoretical risks (05). Prior treatment with chemotherapy increases PML risk 4-fold during natalizumab therapy.
Before 2004, when natalizumab was approved to treat multiple sclerosis, PML had never been seen in untreated multiple sclerosis patients. Why did PML appear during natalizumab therapy, without any increase in other infections or CNS cancers? What is the relative risk of PML with natalizumab alone, when combined with interferon-beta, or with immune suppression?
In the pre-AIDS era, PML caused visual deficits (40%; typically retrochiasmal homonymous hemianopsia or cortical blindness, not from optic nerve lesions), motor weakness and hemiparesis (30%), dysarthria and myoclonic seizures (20%), and change in mentation and personality (33%), plus occasional apraxia, abulia, and aphasia. PML rarely involves the optic nerves or spinal cord (11). Onset is usually insidious over weeks to months but is sometimes rapid and is occasionally indolent for over 1 year. In contrast to multiple sclerosis, PML is subacute, with continuously progressive symptoms. PML in multiple sclerosis patients on natalizumab therapy presents with cognitive problems (48%), motor abnormalities (37%), language disturbance (31%), and visual deficits (26%) (04). Approximately 25% of multiple sclerosis patients with PML die from PML; 75% survive with noticeable or severe disability.
A third of patients with PML have seizures; 90% of these PML lesions have a cortical MRI signal, compared to 50% without seizures. EEG may show slowing before MRI becomes diagnostic.
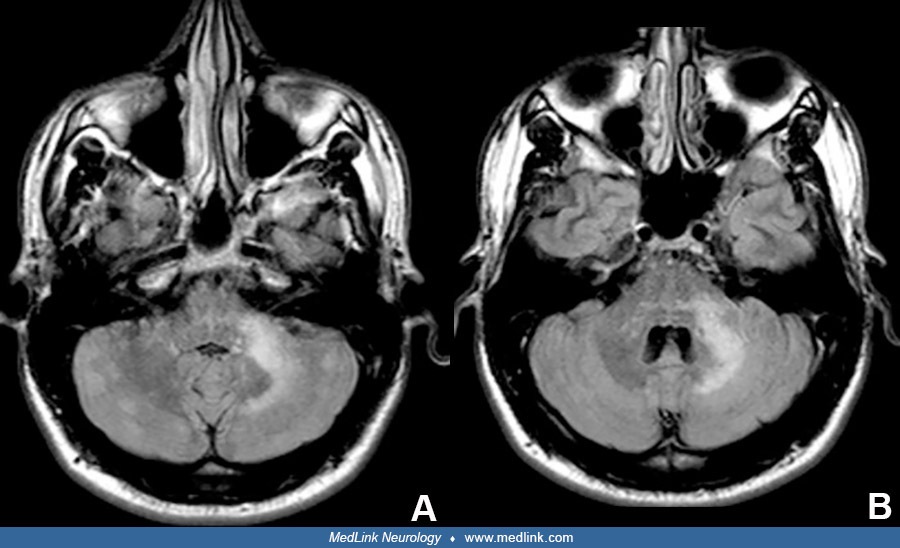
MRI is almost always positive during PML (Table 2). Widespread MRI lesions are associated with fatality. Lesions are more common in frontal and parieto-occipital areas. The lesions are often large, monofocal, asymmetric, and diffuse in the white matter; usually have no mass effect; and seldom regress. They show hypointense T1 and high-signal T2 (“ground glass”), FLAIR, and DWI lesions. Lesions enhance in approximately 40% of patients (vs. 10% in HIV), diffusely or with a punctate, perilesional “Milky Way” appearance. Transient punctate gadolinium-positive areas in the parietal, frontal, and thalamic areas can precede T2 lesions by 6 months and are present in 90% of patients with PML. Lesions are often in the centrum semiovale with a sharp border and sometimes extend up to the subcortical U fibers, forming a scalloped, often ill-defined border there. PML lesions are somewhat more likely to appear at the gray-white junction than in periventricular spaces, suggesting local tropism in affected areas of the CNS or differences in local CNS immune regulation. Adjacent to the subcortical lesion, 60% have a hyperintense cortical signal. Lesions also occur in the basal ganglia, thalamus, and brainstem (“hot cross bun sign,” “across the pons”). Crescentic cerebellar lesions (“shrimp sign” or a “seahorse sign” white matter lesions that abut but spare the dentate nucleus) are seen in one fourth of PML cases, but not in multiple sclerosis, with 85% specificity. Widespread MRI lesions are associated with fatality. Rare reports show insidious PML with no MRI changes.
PML lesions differ from multiple sclerosis plaques (61). New MRI lesions are smaller and are in periventricular regions, optic nerve, and cord; and usually enhance with gadolinium. Cortical subpial lesions appear in multiple sclerosis but not in PML. Lesions in the spinal cord and optic nerve are exceedingly rare and are uncommon in the brainstem in PML, unlike in multiple sclerosis. Giant “tumefactive” multiple sclerosis plaques can be confused with PML. The relative balance for lesions in PML versus multiple sclerosis is as follows: large confluent T2 (74%/2%:PML/multiple sclerosis), deep gray matter (31/7), and crescentic cerebellar (23/0) (11). Elevated myoinositol and lipid/creatine and low N-acetylaspartate (NAA) on magnetic resonance spectroscopy indicate inflammation.
|
PML (in Multiple Sclerosis) |
Multiple Sclerosis |
Stroke | |
|
T1 |
Decreased |
Normal or decreased |
Decreased |
|
T2 FLAIR |
Increased* |
Increased |
Increased |
|
Diffusion-weighted imaging (DWI) |
Increased |
Normal |
Increased strong |
|
Apparent diffusion coefficient (ADC) |
Normal or low at the edge, higher in the center |
Normal |
Low |
|
Gd-enhancing |
40% |
100% of recent and in some reactivated lesions** |
Not typical |
|
Borders |
Sharp |
Vague deep, sharp near cortex |
Sharp |
|
Character |
Periventricular |
Larger, centrum |
Varies |
|
Magnetic resonance spectroscopy (MRS) |
Low NAA/Cr (I< NI) |
Low NAA/Cr |
Low NAA/Cr -- |
|
Cho/Cr = choline/creatine; mI = myo-inositol; Lip1&2 = lipid/lactate & lipid/macromolecule; NAA = N-acetylaspartate; I< NI = IRIS<Non-IRIS. | |||
JC virus-associated cerebellar granule cell neuronopathy causes cerebellar symptoms and atrophy. Several cases have been reported with natalizumab therapy of multiple sclerosis; others have appeared with HIV, CD40 ligand deficiency, following immune therapy for non-Hodgkin lymphoma, and sarcoidosis ± immune therapy. PML in untreated sarcoidosis appears with onset of sarcoid (60%, 1/3 of all cases have lymphopenia), is predominantly male (70%), has a CD4 T cell count over 200/uL (60%), and has a high mortality (60%). JC virus encephalopathy from infected cortical pyramidal cells and JC virus meningitis from infected leptomeningeal cells have been reported in non-multiple sclerosis patients. Linkage to the MRI “shrimp sign” is unexplored.
The histopathologic triad in PML is multifocal demyelination, oligodendroglia with enlarged hyperchromatic nuclei, and bizarre enlarged astrocytes with hyperchromatic lobulated nuclei. Demyelination is from JC virus–induced dysfunction and cytolytic destruction of oligodendroglia (lysis is necrotic, not apoptotic), with secondary loss of myelin. Oligodendrocyte nuclei become large, hyperchromatic, and basophilic with intranuclear inclusions comprised of crystalline arrays of virus particles (“fried egg” appearance). Virus can spread within oligodendrocyte extensions or along myelin in white matter, but not grey matter. As oligodendrocytes die, virions are slowly released. Budding extracellular vesicles also spread the virus. Astrocytes in areas of demyelination are hypertrophied (giant, bizarre, ballooned), with multiple mitotic figures that can be confused with astrocytomas. In mice transgenic for the JC virus large T antigen, there is dysmyelination with minimal expression of RNA for myelin basic protein and hyperproliferating astrocytes.
Lesions in AIDS-associated PML have more extended foci, more often involve gray matter and infratentorial regions, have fewer atypical astrocytes, and more often have perivascular infiltrates than in non-AIDS forms.
CD8 cytolytic T cells eliminate MHC class I +, JC virus-infected target cells. Anti-JC virus T cells control the latent form of the virus in healthy people. In a natalizumab-treated patient with multiple sclerosis who developed PML, autopsy showed few cells in the cerebral perivascular/Virchow-Robin spaces, no CD4 cells, rare dendritic cells, or macrophages (elevated without natalizumab), but some CD8 cells and increased MHC class I, indicating an attempt at an anti-viral T cell response. In relapsing-remitting multiple sclerosis with natalizumab-associated PML with an IRIS reaction, the JC virus was present in white matter and neocortex, and lesions had sharp borders containing CD4 and CD8 T cells. The older multiple sclerosis plaques were separate and distinct from adjacent PML lesions and were hypocellular, with myelin loss, axonal preservation, and gliosis—but no JC virus.
The CSF of PML cases in AIDS and multiple sclerosis patients is usually nearly normal, with occasional mild pleocytosis (fewer cells during immunotherapy or AIDS), slightly increased protein, and, rarely, increased IgG. Brain biopsy with histopathology and electron microscopy, using in situ hybridization or DNA polymerase chain reaction for JC virus, is the gold standard but is not perfect. Laboratories vary in their ability to detect CSF JC virus by polymerase chain reaction. The limit of detection is 500 virus DNA copies in many hospital labs (in which only approximately 75% of multiple sclerosis PML cases are positive), 50 copies in commercial tests, and five copies in the E Major/A Nath NIH lab (99% detection).
Virus infections and perhaps cancer are less prevalent than expected in multiple sclerosis, possibly from hyperactive immunity (see MedLink Neurology article Multiple sclerosis: neuroimmunology). PML was usually not suspected in multiple sclerosis and had not been reported before natalizumab-associated cases, although white matter lesions may have been misdiagnosed. There are no cases of PML during two million patient years of interferon-beta therapy for multiple sclerosis, often in combination with steroids and chemotherapy. However, two cases appeared in the combination of interferon plus natalizumab in the pivotal trial.
Pathogenesis of PML. PML arises from a defect in CNS immune surveillance, ie, few T cells in the brain, possibly in conjunction with peripheral replication and activation of the JC virus. It is likely that natalizumab (1) induces activation of the JC virus, (2) causes redistribution of cells infected with the JC virus, or (3) interacts with interferons or immunosuppressive drugs to affect CNS immune surveillance. Other immune-privileged sites (testes, ovaries, and eye) are apparently unaffected. The dearth of other infections during therapy with natalizumab, plus the usual absence of PML in multiple sclerosis, even after prolonged glucocorticoids, chemotherapy, and stem cell transplantation, suggests a unique phenomenon in multiple sclerosis, and abrogation of immune surveillance is not the only cause.
PML was first detailed in association with leukemia and lymphoma, then in autoimmune disorders, transplants, sarcoidosis, idiopathic CD4 lymphocytopenia, connective tissue disease, and, more recently, in AIDS. Earlier reports go back to Hallervorden in 1930. Approximately 5% of cases were "primary” PML, with no associated immunosuppression, but PML patients are usually immunosuppressed. PML rate is 1.24 of 1000 per year after heart or lung transplants and also appears after bone marrow transplants. It is assumed that PML in connective tissue disease is from chronic immunosuppressant therapy. However, 40% of PML cases in systemic lupus erythematosus had minimal or no prior immunosuppressive treatments. Other factors in systemic lupus erythematosus and perhaps neuromyelitis optica, such as a strong “interferon signature” (21) and DNA variants (HLA—section 15, C8B and FUN3—complement activation, LY9—costimulatory and binds hepatitis C, STXBP2—release of cytolytic NK cell granules), could contribute to predisposition if high levels of interferons synergize with other factors (see “Type I interferons can potentiate the effects of natalizumab”) or alter immune regulation. There has been a large shift in the epidemiology of this formerly rare disease with the spread of HIV. Five percent of untreated AIDS patients developed PML. The percentage fell with widespread use of highly active antiretroviral therapy (HAART) for HIV, but AIDS is still the most common cause of PML.
JC virus and its target cells. PML is caused by the JC virus. This nonenveloped polyomavirus has a small 5000 bp circular double-stranded DNA genome, with only six genes, a microRNA, and a noncoding regulatory region. It is closely related to the human BK virus and to primate SV40, a neurotrophic polyomavirus that causes CNS tumors. Primary infection and the chronic quiescent state are asymptomatic.
Evidence of prior infection with the JC virus is present in 10% of 5-year-olds and up to 80% of elderly patients. In the multiple sclerosis age range, serum antibodies to the JC virus are present in approximately 55% of all people. The incidence of infection increases by approximately 1% per year (33). The incidence is high in Korea, greater in Europe than Japan, and lowest in the United States. Males are affected slightly more than females. There are 14 different JC virus phenotypes based on variation in the coding region. Phenotypes can be used as markers of human migration. Types 1 and 4 predominate in Europe, and types 3 and 6 in Africa. Infections with two different virus genotypes can occur. Genotypes have not yet been linked to virulence.
Transmission and the portal of entry are unknown but may be fecal oral (oropharyngeal--tonsil stromal cells, gastrointestinal epithelial cells) or respiratory. The virus is typically passed from parents to children. Tonsillar stroma, bone marrow, and B cells contain JC virus DNA. The JC virus is often latent in renal tubular epithelial cells and is excreted in urine, especially in the urine of people more than 40 years old. Nonetheless, rearranged variants are hard to detect in urine. JC virus prevalence in semen and urine doubles in infertile compared to fertile males. There are up to 1000 viral particles per ml in sewage water from divergent geographical areas (10). JC virus in public and private swimming pools is likely.
CD34 precursor B cells in the bone marrow contain latent JC virus. In culture, the JC virus binds to all B cells but few T cells. Surface-bound JC virus seldom leads to infection or JC virus mRNA transcription (< 1% of B cells). Nonetheless, low-level productive infection is sustained by a reservoir of B cells in the spleen, tonsils, and bone marrow.
JC virus is an extremely cell-associated virus, although serum occasionally contains free virus. A virus in extracellular vesicles could allow viral receptor-independent cell entry. It is not shed at high levels, suggesting cell-to-cell interactions are important for virus spread. JC virus has restricted tropism. It infects oligodendroglia (reducing myelin gene expression), astrocytes (at low levels), tonsillar stromal cells, B cells, granulocytes (a frequent reservoir), CD34+ stem cells in bone marrow and blood, and possibly choroid plexus cells. The archetype JC virus predominates in kidney uroepithelial cells: 30% of the population has JC virus in urine. One lab found roughly equivalent JC virus DNA levels in CD34, B cells, monocytes, T cells, PMN, and NK cells; all cells contained much more virus DNA than the serum. Another lab found that JC virus DNA is absent in CD3+ cells and is restricted to CD19+ and CD34+ cells, which is relevant because natalizumab triples the number of CD34+ cells in blood. In vitro, JC virus infects vascular endothelial cells, kidney epithelial cells, amnion cells, and the Jurkat T cell leukemia line, but not primary T cells. JC virus is usually latent and quiescent in B cells, bone marrow cells, the genitourinary tract, and occasionally in the brain.
In the Virchow-Robin spaces and germinal center-like areas within the meninges, B cells with surface-bound JC virus are held in check by T cells. JC virus DNA fragments present in normal brain oligodendrocytes and astrocytes indicate that the archetype and the pathogenic JC virus–Mad form both have full access to all regions of the brain. In the brain, B cells in inflammatory multiple sclerosis lesions do not harbor replicating JC virus. Brain presence of JC virus is altered little by immunosuppression. This indicates that the normal, and even the partially compromised, immune system efficiently and inevitably clears all forms of this virus.
After peripheral infection or virus activation, B cells may act as “Trojan horses” to carry the virus from the blood into the brain. Infected B cells can transmit the virus to human fetal glial cells in culture. Fetal glial cells and B cells share common DNA-binding factors that bind to the JC virus regulatory sequence and allow more efficient replication of the virus than mature B cells. JC virus infection of astrocytes enhances transcription of hundreds of genes and suppresses a third of that number. These regulated genes affect cell proliferation, signaling, and inflammation.
Natalizumab activates the virus in white blood cells, and then it becomes neurotrophic. B-cell recombinases could rearrange the virus genome. The JC virus archetype is present in normal brains at low levels. Pathogenic mutations can arise in several brain areas from separate JC variants, based on rearranged noncoding control regions (NCCRs). To become pathogenic, the NCCR of the archetype virus rearranges to the neurotropic Mad strain, with multiple mutant variations. The NCCR is a bidirectional element that contains promoter and enhancer elements and the origin of viral replication. Viral replication in oligodendrocytes and astrocytes is normally inhibited by the SF2/ASF splicing factor (SRSF1 gene), which blocks JC virus RNA splicing and suppresses the NCCR. SF2 is unable to bind to some Mad mutations. The VP1 coding region also frequently mutates in PML, allowing escape from immune control by anti-VP1 T cells and antibodies. Natalizumab-treated and non-treated patient groups have similar rearrangements.
Natalizumab therapy induces many B-cell activation and differentiation markers (44), so it is possible that high levels of some of these transcription factors (POU domain family, Spi-B, NF-1X, Oct1, and Oct2) will enhance JC virus replication; levels are highest in natalizumab-treated patients who develop PML. NF-1X and Spi-B are upregulated in glial, hematopoietic progenitor, and B cells. Spi-B binds to TATA boxes in the JC virus Mad-1 and -4 mutants, but not to this site in the (non-PML) archetype. After the Mad mutations appear, susceptibility is not from changes in receptor binding but rather is molecular, at the level of these intracellular transcription factors (Major E 2009, personal communication).
Viruses related to JC bind to several cell surface proteins for internalization. The VP1 protein of human BK virus binds to gangliosides GT1b and GD1b with alpha-(2,8)- and alpha-(2,3)-linked sialic acids, but not to VLA-4. Murine polyoma virus binds to alpha-(2,3)-linked sialic acid, followed by virus binding to VLA-4, and then is retained on the cell surface or internalized through non-clathrin-coated vesicles. Unique to the JC virus, the VP1 capsid protein attaches to alpha(2,6)-linked sialic acid on lactotetrasaccharide series C (LSTc) (37). 5HT2 receptors are present on neurons and are also highly expressed on brain microvasculature and on the abutting astrocytes at the blood-brain barrier. There are also 5HT2 receptors in the area postrema and the choroid plexus. Interferon-beta could indirectly affect serotonin 5HT2A receptor expression by inducing indoleamine 2,3-dioxygenase, an enzyme that decreases tryptophan and serotonin. Low tryptophan levels cause a compensatory increase in 5HT receptor expression. This potentially enhances JC virus binding to neurons, astrocytes, and lymphocytes, as all express 5HT2A receptors.
Internalized virus activates the MAP kinases, ERK1, and ERK2. B cells, some T cells, and oligodendroglia and astrocytes express alpha-(2,6) sialic acids on glycans. A later study found sialic acids on microglia, monocytes, choroid plexus epithelium, meninges, and renal distal tubules and collecting ducts, but none on astrocytes and oligodendrocytes. Thus, oligodendrocytes and astrocytes internalize JC virus through an unknown pathway. Once internalized, the JC virus could spread to other cells through extensive astrocyte syncytia and gap junctions.
The JC virus large T antigen is detected in oligodendrocytes, astrocytes, and selected neurons such as cerebellar granule cells, but is seldom detected in the kidney or microglia. JC virus T antigens hijack cell cycle machinery, block p53 and Rb, and force cells into an S-phase-like state. Infected cells are maintained in an immature, virus-producing state, avoiding apoptosis. Large amounts of T antigen are a marker for viral reactivation and lytic infection. Certain CNS cells have high concentrations of the DNA-binding proteins that are necessary for lytic infection. The transcription factor Tst-1 (aka Oct-6) is found only in oligodendrocytes and Schwann cells; it stimulates transcription of early and late virus genes. NF-1X must be in excess for virus replication. Other cells that are somewhat susceptible to lysis are astrocytes and CD34+ and CD19+ B cells.
There are two types of JC virus regulatory region: the nonpathogenic archetype found in kidney cells and the rearranged neurotrophic form with repeated activation sequences. The kidney-resident archetype regulatory sequence has only one 98 bp sequence plus 23 and 68 bp insertions. The archetype is linked to nonlytic latency and possibly dissemination, is present in B cells and kidney cells, and is associated with a better clinical outcome. Inside the cell, c-Jun and NF-1 class X protein, NF-1X (high in tonsillar stromal cells and glia, sites of JC virus infection) bind to adjacent sites on the JC virus regulatory region. In the prototype Mad-1 variant, the 200 base-pair JC virus regulatory region contains a 98 bp tandem repeat with one c-Jun and two NF-1 binding sites and two TATA sequences. Spi-B also binds only to Mad-1 and Mad-4 variants. The neurotropic Mad variants appear in infected brain or tonsil tissue, and their presence correlates with poor clinical outcome. Transfection of NF-1X into neurons makes the neurons susceptible to JC virus infections. Spi-B binding sites in the tandem repeats are increased in all virus isolates from cases of PML. Spi-B is upregulated in B and CD34+ B cells of multiple sclerosis patients treated with natalizumab, before they enter the CNS (45). Spi-B is also increased in CD4 and CD8 cells, which do not harbor the virus.
Anti-VLA-4 therapy mobilizes JC virus-infected B cells and progenitor cells in large numbers and provokes release from bone marrow into the peripheral blood. JC virus is activation by some anti-VLA-4 antibodies (ibid). Mobilization is followed by the ingress of the activated virus into the CNS. Reduced penetration of anti-JC virus cytolytic and memory T cells into the CNS during VLA-4 blockade may set the stage for PML. The cells that do penetrate the blood-brain barrier are anti-inflammatory.
Mechanisms for activation of the JC virus by HIV and other viruses. Five percent of patients with AIDS develop PML. There are approximately 7500 AIDS/PML cases per year in the United States. This incidence is higher than in other disorders with immunosuppression, suggesting that HIV has unique interactions with the JC virus.
HIV could provoke PML through (1) severe AIDS immunosuppression and the paucity of T cells in the blood and CSF, (2) an increase in activated circulating (JC virus positive) B cells, CD34+ stem cells, and granulocytes, (3) erosion of the integrity of the blood-brain barrier, (4) activated CNS perivascular macrophages, which could increase adhesion molecule expression on adjacent endothelial cells, (5) upregulation of secreted cytokines that affect JC virus genes (not yet demonstrated), (6) HIV tat protein transactivation of the JC virus (59), and (7) HIV and natalizumab both binding alpha4beta7 gut integrins, potentially affecting barriers and immune cells.
(1) Lymphocytes are largely eliminated from the CSF during natalizumab therapy and during HIV infection. During AIDS, immune surveillance by T cells is lost in the brain, possibly paralleled by a rise in the number of JC virus-infected cells in blood and brain. JC virus–infected circulating B cells appear in 40% of immunosuppressed patients. Bone marrow cells are positive for JC virus in 47% of HIV-positive subjects, but only 13% of HIV patients. Moreover, infection with one virus changes the ecology of the CNS and may facilitate the passage of other viruses into the brain (02).
(2) VLA-4/VCAM-1 adhesion helps retain immune cells in the bone marrow. Blocking VLA-4/VCAM-adhesion mobilizes hematopoietic progenitors. JC virus-positive cells could increase in the blood if there is more release of cells from the bone marrow in HIV, or in multiple sclerosis after natalizumab.
(3) Serum proteins accumulate in white matter glia and neurons, indicating blood-brain barrier breakdown in AIDS. There is seldom gadolinium MRI enhancement in AIDS PML. This suggests that endothelial cells are not activated during HIV infection and that endothelial cell inflammation would not affect leukocyte migration.
(4) The perivascular space in multiple sclerosis contains large numbers of activated macrophages, which could affect the migration and adhesion of other immune cells to the endothelium. In PML lesions, perivascular B cells and surrounding astrocytes express JC virus DNA and protein, suggesting hematogenous spread is followed by infection of astrocytes. The exact sequence and interacting molecules are unknown.
(5) CNS inflammation in multiple sclerosis should enhance antiviral immune mechanisms, yet could also modify the JC virus life cycle.
(6) In glial cells, the HIV tat protein directly transactivates the tat-responsive cis-element in the JC virus regulatory region. In AIDS/PML, this noncoding control region loses sites for the SP1 transcription factor, but there is duplication of the up-TAR site that binds the HIV tat protein. HIV and JC virus generally infect separate cell lineages. Tat activation of the JC virus genome is not relevant in HIV-negative multiple sclerosis—unless endogenous retroviruses are a component of MS pathogenesis.
(7) The gp120 HIV envelope protein binds to integrin alpha4beta7 on T cells. This activates LFA-1 on T cells and facilitates cell-to-cell spread of HIV-1. Natalizumab, however, does not block replication of HIV in cultured T cells.
During AIDS-associated PML, JC virus is found in CSF (100% positive for JC virus DNA by nested polymerase chain reaction; 100% show active JC virus VP1 mRNA transcription, in some labs), urine (56% JC virus DNA/38% mRNA – ie, weak predictive value), blood plasma (24%/0%), B cells (20%/0%), and non-B cells (12%/ unknown%). However, JC virus DNA detection in other labs is less sensitive (76% with real-time polymerase chain reaction).
The SV40 large T antigen is a tumor promoter. The related large T antigen of the JC virus is potentially tumorigenic. Expression of the JC virus early promoter is higher in glial cells than in nonglial cells (59). JC virus is tumorigenic in primates and hamsters, causing astrocytomas, glioblastomas, medulloblastomas, and abdominal neuroblastomas, primitive tumors, and pinealomas. JC virus has been associated with human oligodendroglioma, glioma, oligoastrocytoma, colon cancer, and medulloblastoma, but there is no solid evidence for a causal role of JC virus in human tumorigenesis. Tumor development may be countered by the immune system when polyoma virus VP1 binds to sialic acid groups on immune cells and induces toll-like receptors to produce IL-12 and activate T cells.
Infections with viruses such as the Epstein-Barr virus could synergize with the JC virus. Epstein-Barr virus resides in B cells, activates B cells, and is linked to the development of multiple sclerosis. Sixteen of 23 patients with primary CNS lymphoma expressed DNA for the Epstein-Barr virus-transforming protein, LMP1; five also expressed JC virus proteins, including T antigen. In multiple sclerosis, B lymphoblasts, recombination between the JC virus and EBV can take place in the JC virus archetype noncoding control region. Cytomegalovirus infections activate the BK virus (related to the JC virus) and the JC virus. HHV-6 and cytomegalovirus can increase levels of the JC virus in JC virus–infected glial cells. Finally, HTLV-I tax protein activates the JC virus promoter.
Are other viruses perturbed by anti-VLA-4 agents? In 36 multiple sclerosis patients, BK virus in urine was reactivated from 8% at baseline to 22% on natalizumab therapy. JC virus also rose in urine, from 19% to 63% in 19 patients. In contrast, JC virus DNA in blood (28%) and urine (25%) was lower, and equivalent in interferon-beta-treated multiple sclerosis patients and healthy controls. With natalizumab treatment, however, the rate of becoming JC virus-positive increases 10-fold, suggesting there is more virus available to generate an immune response. Non-polyoma viruses are also affected by natalizumab. Human herpesvirus-6 is reactivated during natalizumab therapy, leading to elevated anti-HHV-6 antibodies in serum and occasionally HHV-6 DNA in CSF. Anti-VLA-4 antibodies (GG5/3), given 14 and 18 days after Borna disease virus inoculation, prevent most inflammation, but viral titer and Borna disease virus protein distribution in the brain are not affected. This suggests that anti-VLA-4 reduces T-cell migration to prevent inflammation. In Semliki forest virus-mediated demyelination in mice, anti-VLA-4 antibodies also decrease inflammation but do not change virus clearance. Finally, rotavirus, which causes childhood diarrhea, has viral capsid proteins that bind VLA-4, suggesting a role of rotavirus in Crohn disease-related PML. However, there is no clear link between rotavirus infection and Crohn disease, and natalizumab did not provoke significant gastrointestinal symptoms in Crohn disease trials.
PML and multiple sclerosis therapies are also extensively reviewed (47; 17).
Detection of JC virus in blood and brain depends on the technique used. Detection frequency for JC virus DNA in white blood cells is 25% with standard polymerase chain reaction, 55% with Southern blots, and nearly 100% with nested polymerase chain reaction. Detection of the presence of any JC virus DNA was equivalent between healthy controls and patients with HIV and PML, but amounts of viral genome were higher in PML. Lack of utility of JC virus DNA as a predictor of PML was confirmed in 13,000 samples from 1400 patients in natalizumab clinical trials, which showed 12 patients positive for JC virus in plasma, 0 positive in mononuclear blood cells, and 25% positive in urine before and after therapy. Twenty-five percent of adults will shed the virus in their urine during natalizumab therapy. JC virus DNA is found in genitourinary tissues in 75% of donors with high JC virus antibody levels, 13.3% with low levels, and 0% in seronegative donors. It is detected in 45% of other tissues when JC virus titers are high, 2.2% with low JC virus serostatus, and in 0 seronegative persons (06).
A positive CSF polymerase chain reaction for JC virus DNA strongly suggests the diagnosis of PML in the right clinical setting. Larger PML lesions yield more JC virus DNA, although the anatomical location is not linked to a positive test, and 16% of large PML lesions on MRI test negative for CSF JC virus DNA. Half of the patients with negative CSF testing will become positive within a month. Conversely, JC virus can be detected, absent PML; 9% of multiple sclerosis CSF samples were positive for JC virus DNA, yet no noninflammatory and inflammatory CSF controls were positive. Other authors have failed to find JC virus DNA in multiple sclerosis CSF, even during PML. During HAART for AIDS, PML stabilizes when CSF JC virus levels fall. Antibodies to JC virus are not elevated in untreated multiple sclerosis CSF or blood in the majority of reports. JC virus infections can generate oligoclonal bands specific for the JC virus VP1 capsid antigen and will elevate the CSF antibody index. CSF IgG to VP1 is present in 75% of PML cases, but only in 3% of controls. There is intrathecal synthesis of anti-VP1 antibody in 76% of HIV+ patients with PML, but only in 3% of other neurologic disease controls. VP1 is also a target for cytolytic T cells.
Serum antibodies to the virus indicate prior infection, and antibodies to the VP1 capsid protein are used for epidemiologic studies. The 2-step STRATIFY JC virus antibody test is described below.
DNA monitoring is better developed for the BK virus than for the JC virus. BK virus is a closely related polyomavirus that infects and is latent in the kidney. It also infects the bladder, prostate, cervix, vulva, lung, eye, liver, and brain. BK virus infects 90% of the healthy population at early ages; JC virus is acquired at about 1% per year. If a person is seropositive for BK virus serotype 1, he or she is less likely to be positive for JC virus, indicating that immunity against BK virus is partially protective against JC virus. The BK virus is found in some CNS tumors but does not cause PML. BK virus DNA increases in the urine during HIV infection and lupus; JC virus does not. BK virus causes nephropathy following renal transplants and immunosuppression (polyomavirus-associated nephropathy, PVAN). After kidney transplantation, BK viremia is the first sign of active virus replication and is present in 45% of renal transplant patients at 1 year. Viremia (15%) progresses to nephropathy (5% to 10%) and loss of the transplant (5% of all patients; 70% of patients with nephropathy) (12). Polymerase chain reaction is sensitive in monitoring for viruria, but a renal biopsy is the definitive test. Drugs used to prevent transplant rejection suppress the immune response to the BK virus, allowing viremia (Table 3). Lowering the dose of immunosuppressive drugs clears viremia in 95%, with an average time to clearance of 54 days.
BK virus studies suggest that reversing immunosuppression in PML could also clear the JC virus. For instance, removal of natalizumab with plasma exchange can trigger an immune response to the JC virus (see IRIS, below).
Serum samples during natalizumab therapy were positive for JC virus DNA in 2.3%, a rate comparable to that in normal controls. However, the anti-JC virus response by cytolytic CD8 cells in multiple sclerosis patients is much stronger than normal, even though virus DNA is not detectable in blood cells, plasma, and CSF.
|
Urine |
Blood | |
|
Cyclosporine A + Azathioprine |
44%* |
15% |
|
* Percent of transplant patients where DNA is detectable. BK virus DNA was measured by qualitative, followed by quantitative, real-time polymerase chain reaction (12). | ||
BK virus DNA can be used as a “best case” model for the detection of JC virus genomic DNA. Unfortunately, variability in assays for JC virus (antigen source, methods, polymerase chain reaction primers, and conditions) leads to differences in epidemiology, infection in multiple sclerosis, and prognostic indicators between studies. Moreover, there is no perfect primate or rodent model of JC virus infection.
There is no early warning assay that detects JC virus DNA before the development of PML, unlike the BK virus in urine. JC viremia, present in up to 2% of healthy subjects, is only weakly associated with PML, but does indicate there has been exposure to the virus. In most studies, natalizumab does not increase urine JC virus levels, but those with high anti-JC virus index do have higher titers. Anti-JC virus titers may reflect intermittent free virus, but not the complete antiviral picture. With help from CD4 cells, cytolytic CD8 T cells easily clear JC virus–infected cells in most people. Some clinicians routinely test for JC virus in serum or measure the CD4/CD8 ratio, but these tests can’t be recommended. The most reasonable peripheral tissue for JC virus detection may be white blood cells to capture putative JC virus–infected B cells and CD34+ stem cells. With PCR of flow cytometry-purified cells for JC virus T protein, 50% of CD34+ cells were positive in multiple sclerosis, and 20% were positive in healthy controls. Urine JC virus assays could be complementary to the more sensitive serum STRATIFY-2 assay (below). Virus in urine in antibody-negative patients, however, suggests that there is no immune response to systemic JC virus and that the brain is not yet at risk. Monitoring for JC virus and PML during natalizumab therapy is discussed in topic 16.
PML was not reported in multiple sclerosis until natalizumab therapy was introduced. The closest description was in a case of a woman with optic neuritis followed 12 years later by PML, after she developed AIDS. The optic neuritis had a rapid onset, lasted 1 week, resolved over 4 days, and was not followed by multiple sclerosis. Of interest, histopathology showed no optic nerve lesions, paralleling the case of PML after clinical optic neuritis in the pivotal trial of natalizumab therapy. MRI detection of PML is discussed in topic 16. Later reports include PML in a patient with common variable immunodeficiency plus multiple sclerosis during treatment with IM interferon-beta-1a.
Approximately 25% of natalizumab-treated patients with PML will die. The remainder is evenly split between severe, moderate, and mild neurologic sequelae. The inflammatory response in multiple sclerosis is greater than in HIV. This is relevant because good prognostic factors in HIV-associated PML are high CD4 cell count, cytolytic cells specific for JC virus T or VP1 antigens, Gd enhancement on MRI, and low JC virus load in the CSF. There is a 74-day delay from first symptoms to diagnosis.
Investigators and patients were aware of a potential risk of CNS lymphoma and systemic infections with this experimental drug, but concern had been allayed because there were minimal side effects over 5 years of human trials. After the PML two cases arose, some neurologists said this was expected, echoing the 2600-year-old philosophy of Epimenides of Crete, who declared that it was easy to predict past events. Administration of the drug was put on hold, as were important trials of natalizumab in primary progressive multiple sclerosis, Crohn disease, and rheumatoid arthritis, as well as other multiple sclerosis trials with small protein blockers of VLA-4/VCAM interactions. The drug was reintroduced in 2006 with a careful monitoring program (TOUCH program in the United States).
There have been 937 cases of PML during natalizumab therapy as of December 21, 2024; three of these had Crohn disease (08). In the pivotal trials, two cases were on concomitant interferon-beta-1a (after 28 and 37 natalizumab infusions). The increased risk was not statistically significant, but the combination is now avoided. (Possible synergy between these two therapies is described in section 10.)
PML during natalizumab therapy is not unique to multiple sclerosis. It developed in a Crohn disease patient treated with natalizumab in early trials, and there have now been three cases. The first patient had chronically deficient hematopoiesis and had received years of treatment with immunosuppressive drugs, some contemporaneous with three natalizumab infusions. After a temporary stop in therapy, five natalizumab infusions coincided with a rise in JC virus DNA and the development of PML. This infection in Crohn disease suggests that PML arose from chronic immunosuppression, possibly enhanced by natalizumab. Antibodies to more gut adhesion-specific alpha4,beta7 integrin, such as vedolizumab, are not associated with PML.
The risk of PML was roughly 1 of 1000 in all natalizumab-treated multiple sclerosis patients based on early data. With longer observation, the risk increased but is now slowly declining with monitoring of anti-JCV titers. As of February 2025, there were 924 cases of PML in 277,480 natalizumab-treated multiple sclerosis patients (1,245,981 patient years): 535 cases in Europe, 251 in the United States, and 154 in the rest of the world (Biogen Idec MedInfo, accessed January 5, 2026). The average global risk is 3.33 per 1000 patients (1 of 342) in all treated patients, weighted for shorter duration of infusion because the greatest number received 0 to 12 months. There are exceedingly few cases of PML in the first year of therapy: 0.05 per 1000 (1 of 25,000). However, the rate rises to 0.51 of 1000 during 13 to 24 months of infusion therapy (cumulative 0.56 of 1000), to 1.00 for 25 to 36 months (cumulative 1.56 of 1000), 1.27 for 37 to 48 months (cumulative 2.83 of 1000), 1.47 for 49 to 60 months (cumulative 4.10 of 1000), and 1.02 for 61 to 72 months (cumulative 5.12 of 1000 or approximately 1 of 195) JC virus-negative patients have no or very low risk after prolonged therapy (six cases). In 2015, European patients had more PML with prior chemotherapy (24% vs. 14% USA), but the frequency of PML in those with no prior chemotherapy was also increased 4-fold in Europe. Provocative chemotherapy was predominantly mitoxantrone, and to a lesser extent, azathioprine, cyclophosphamide, and methotrexate (17:5:5:5 cases, respectively, for these agents). Thus, given a PML incidence of 4.03 of 1000 treated per year (1 of 248) after 7 years of therapy, the risk of PML is 1 per 77 for U.S. patients with prior chemotherapy and 1 per 167 for chemotherapy-naive patients. The PML rate initially increased over time due to a slightly higher number of patients with prior chemotherapy and a greater proportion of patients on more than two years of therapy (see risk calculations, below) but has now declined with the use of the JC virus index.
Antibodies to JC virus reflect the risk of developing PML during natalizumab therapy. Approximately 55% of the multiple sclerosis population has antibodies to the JC virus; those who do not carry the virus can’t develop PML. If a patient has a positive titer, risk is doubled to approximately 1 in 141 with prior chemotherapy but is 1 in 704 in chemotherapy-naïve patients. Antibody-negative patients have no risk unless tests are false-negative (2.2% chance with the 2013 second-generation ELISA; seen in rare cases of PML, perhaps with risk of 1 of 2000), but clinical data are vague; most or all convert to positive titers. In the USA TOUCH program, nine “titer-negative” at baseline developed PML, but all of eight tested had become JC-virus-positive for 14 to 62 months before PML arose. The rate of conversion is approximately 1% per year in untreated patients, but 10% per year in natalizumab-treated multiple sclerosis patients (33; 58). All risks calculated above are biased downward by the bulk of patients who have lower time of exposure. The estimates above are based on average risk that includes the first nearly safe year; the risk after 24 months of therapy rises from 2.84 (1 in 352) to 5.02 (1 in 199).
Anti-JC virus antibody titers in serum further stratify the risk of developing PML. Assays include STRATIFY from Biogen, and ImmunoWELL, also an ELISA, for titers in those treated with a biosimilar (natalizumab-sztn). High titers increase the chance of developing PML and are not a marker of protection from the virus. High anti-JC virus titers suggest an antibody response to ongoing virus exposure, perhaps to large numbers of the virus. Virus levels could be high from ineffective cell-mediated immunity or from direct activation of the virus by natalizumab.
The two-step STRATIFY JC virus antibody test is more sensitive than the hemagglutination assay and has 2.2% false negatives, an improvement over 2.8% in the prior assays. Titers of 0.2 to 0.4 are indeterminate and must be confirmed with a second assay. Titers above 0.4 are positive. High anti-JC virus titers increase the risk of developing PML by 10-fold compared to low titers. In patients never exposed to immunosuppressants at the 2-year point, anti-JC virus titers below an index value of 0.9 connote minimal increased risk (1 of 3000). From 0.9 to 1.5, the overall risk is 1 of 1000, and above 1.5 the risk is 1 of 120 per year (topic 13). Those with positive titers prior to immunosuppressant therapy, and for longer than 2 years of therapy, have a risk of 1 of 90. Titers in patients with prior immunosuppressant use were similar between patients with and without PML, suggesting different mechanisms in patients naive to or exposed to immunosuppressants. Another analysis estimates that after two years of natalizumab therapy, the PML risk with negative titers is 1 of 125 per year, but with positive anti-JC virus titers is 1 of 44. The risk in Europe is 2.5-fold greater than in the United States, so an index threshold of 0.6 has been suggested for Europe. The index does not change during pregnancy. Putting these numbers into perspective, daily life also has risks (33). The U.S. National Highway Traffic Administration states that car accidents happen to 1 of 62 people per year and cause death in 1 of 7575.
Risk per year is cumulative over time. For instance, a risk of 1 per 1000 over 5 years will become a risk of 5 per 1000 (1 per 200). Thus, calculations must be based on PML incidence by “treatment epoch.”
Cumulative risk estimates, published in MedLink for the prior five years, were confirmed with titer stratification (29). After six years of therapy, the risk was 1.6 of 1000 with a JC virus titer index of less than 0.9; was 8.5 for an index of 0.9 to 1.5; was 28 for an index of greater than 1.5; and was 27/1000 for patients with prior immunosuppressant use.
The risk of PML in multiple sclerosis is twice as high in Europe and the rest of the world versus the United States, perhaps because of more frequent use of immunosuppressants, different virus strains, or more virus exposure. Patients in the United States were 7 years older than those in Europe. Patients more than 50 years old are also at a higher risk, likely from immune senescence. The TOUCH monitoring program in the United States could aid in the early detection of PML symptoms. Risk stratification with anti-JC virus titers may lower the risk by removing many “at-risk” patients from therapy, but this effect may be obscured by a higher incidence of PML from prolonged duration of natalizumab therapy. PML is occasionally seen in connective tissue diseases, often on a background of prior chemotherapy, and in patients with lymphoid cancers or transient lymphopenia.
Patients are more likely than physicians to accept high risks of PML (28). However, there is a broad spectrum of patient and doctor behavior, influenced by different cultures.
The role of glucocorticoid therapy is not included in the PML risk calculations for multiple sclerosis. This commonly used intervention for exacerbations impairs JC virus-specific T cell responses, including cytolytic T cells. PML is more frequent in sarcoidosis during glucocorticoid therapy, likely amplifying T cell energy. Thus, steroids should not be used lightly for multiple sclerosis exacerbations during natalizumab therapy.
Drugs linked to the development of PML are natalizumab (reporting odds ratio [OR] of 35, likely accurate because of the intense TOUCH program) and fingolimod (Gilenya; no generic data), with 61 cases in 327,600 treated over 1,038,100 patient years. This is 18.6/100,000 or 1/5376 patients or 5.88 per 100,000 patient years. S1PR modulator-linked PML appears after 5.3 years of therapy (3.3 with natalizumab), in older patients, is more disseminated and Gd-enhancing on MRI, and has 8-fold more IRIS development, but also 6-fold more multiple sclerosis exacerbations after discontinuation (09). S1PR modulators and natalizumab deplete CNS immune cells; fingolimod also reduces cytolytic T cell numbers and function (30).
Dimethyl fumarate therapy is linked to PML in psoriasis, and there are 12 cases in multiple sclerosis patients, all associated with lymphopenia, but no cases with diroximal fumarate as of 3-1-2023. There is a possible case with teriflunomide after natalizumab therapy. Older patients are more likely to develop PML after switching from natalizumab to another therapy. There are three cases of PML with ocrelizumab. One followed a switch from fingolimod, with a preexisting PML lesion, then steroid therapy for “MS worsening.” Another was a therapy-naive 78-year-old man with progressive multiple sclerosis for 30 years, JC virus index of 2.5, and lymphopenia who developed PML 2 years after starting ocrelizumab. Other PML-linked drugs include efalizumab/anti-LFA-1 (OR 27, no longer on the market), rituximab/anti-CD20 (OR 23, likely to be over-reported), cyclophosphamide (8), azathioprine (6), tacrolimus (4), mycophenolate (3), methotrexate (2), eculizumab/anti-C5a (1), brentuximab vedotin/anti-CD30 and anti-mitotic, and ruxolitinib/JAK1/2 inhibitor.
Guidelines for PML monitoring. The American Academy of Neurology recommendations with respect to diagnosing PML are as follows. A diagnosis of PML can be made by brain biopsy or by a combination of clinical observations of a progressive course of new symptoms, PML-consistent white matter lesions on MRI, and JC virus in spinal fluid. Progressive neurologic symptoms should occur on a background of therapy with disease-modifying drugs. MRI lesions are high-signal intensity white matter lesions usually near the gray-white junction on T2 or FLAIR ± enhancement ± mild mass effect. The CSF JC virus DNA is detected with sensitive polymerase chain reaction assays.
In Europe, recommendations are to test for antibodies to the JC virus before therapy and every 6 months on therapy. MRI scans should be performed every 12 months when titers are negative, every 6 months when titers are less than 1.5, and every 3 to 4 months when titers are greater than 1.5. Additionally, MRI scans should be performed at the end of natalizumab therapy and three months after treatment. MRI costs are much lower in Europe than in the United States, and the cost and time in Europe are further reduced with a simple screening with FLAIR and diffusion-weighted MRI imaging. If PML is suspected, CSF JC virus polymerase chain reaction should be obtained, and it should be repeated if negative.
Serum neurofilament levels often rise with PML, on average 10-fold higher than baseline, and sometimes appear before clinical symptoms. There is a strong correlation between serum neurofilament levels and the size of the PML lesions. Serum neurofilament levels are a convenient and early marker of PML and, possibly, prognosis. Predictive algorithms are being generated. Although lower in those destined to develop PML, plasma MMP-9 levels increase in parallel to a rise in anti-JC virus antibodies (23). Neuroaxonal damage in multiple sclerosis is amplified by non-PML JC virus activity. Anti-JC virus titers parallel serum neurofilament levels (18), a potential confounder in studies of brain atrophy during this therapy. Natalizumab decreases intrathecal anti-JC virus antibodies, but not other antiviral antibodies; the relation to PML is unknown.
The risk of PML during natalizumab therapy must be balanced with its significant clinical benefit in multiple sclerosis.
Immune reconstitution inflammatory syndrome (IRIS) as CNS immune function is restored. To treat JC virus brain infections, the CNS immune response must be restored, ± direct therapeutic destruction of the virus. Immune reconstitution inflammatory syndrome (IRIS) appears as CNS immune function is restored. In a similar mechanism, progression of PML in patients with AIDS is slowed as the immune system is gradually reconstituted by highly active antiretroviral therapy (HAART). Young patients with high CD4 counts and low JC virus load who start HAART at the onset of PML do best. During AIDS-associated PML, gadolinium enhancement on MRI suggests there is activation of T cell-endothelial cell interactions and T cell penetration of the blood-brain barrier. Gd enhancement correlates with improved prognosis. Twenty percent of AIDS patients develop PML after HAART is started. Even though the MRI shows enhancement and symptoms temporarily worsen, these patients do better than AIDS patients who do not respond to HAART. Mechanisms of this paradoxical “immune reconstitution inflammatory syndrome” (IRIS) include (1) a blossoming immune rebound by cytotoxic CD8 cells against preexisting subclinical PML (also seen with reactions against mycobacteria; reduced with temporary steroid therapy), (2) activation of the virus by cytokines, or (3) changes in cell trafficking with a bolus of new precursor cells from the bone marrow. Enhancing MRI lesions are seen in half of AIDS and PML patients responding to HAART and in HIV-negative patients, but only rarely in HAART nonresponders.
Reconstitution of CNS immunity should also clear the virus in multiple sclerosis patients with PML. When natalizumab is stopped, serum natalizumab levels fall by 80% at 42 days and to less than 1 µg/ml serum at 84 days. Because some patients describe a “wearing off” of natalizumab benefit at 3 weeks past the last infusion, it can be assumed that immune cells begin to penetrate the CNS at that time. However, complete reconstitution of CNS immune surveillance is likely to take several months after discontinuing anti-VLA-4 therapy (34), unless the effects of natalizumab are reversed by plasmapheresis.
IRIS is seen in nearly all cases of PML in multiple sclerosis patients (16). It appears spontaneously in nearly half of the cases. After removal of the antibody with plasma exchange in the rest, IRIS blossoms at approximately 4 weeks (range: 11 days to 8 weeks). IRIS leads to abrupt clinical changes, reflecting damage at multiple sclerosis lesion sites or in new areas, and it sometimes causes seizures and fever. There is an extensive CD8 T cell, plasma cell, and macrophage infiltrate in lesions and the surrounding white and gray matter in four of five biopsies. Cytolytic CD8 cells are present in 91% of PML survivors, but in no PML progressors. Others find more CD4 T cells in PML lesions. CD4 T cells produce IFN-gamma, and occasionally both IFN-gamma and IL-4, and use a broad spectrum of strategies to aid the CD8 cytolytic cells in destroying JC virus-infected cells. MRI does not easily discriminate between ongoing PML and IRIS. More than half of multiple sclerosis PML IRIS cases show Gd enhancement on MRI (vs. 30% in HIV IRIS) (16).
Treatment of PML in multiple sclerosis. Multiple sclerosis patients have fewer viral infections and possibly less cancer than normal, possibly because of an overactive immune system and altered interferon regulation (see MedLink Neurology article Multiple sclerosis: neuroimmunology). Viral CNS infections are rare in multiple sclerosis patients, despite treatment with 11 families of disease-modifying drugs and chemotherapy. PML therapies have also been reviewed (37; 61; 26).
Natalizumab is one of the most effective therapies for relapsing-remitting multiple sclerosis. Long duration of therapy, prior immunosuppression or hematologic cancer, or concurrent interferon therapy could increase the risk of PML. Blood and CSF screening for the virus, as well as CD4/CD8 ratios, have not been helpful in predicting PML. If PML develops, natalizumab therapy should be discontinued, and the viral infection should be treated. A modest enhancement of CNS immunity is very effective.
Plasmapheresis (plasma exchange, PLEX) is often used, but is of questionable efficacy. Typically, there are three 1.5–blood volume sessions over 5 to 8 days; natalizumab concentrations are reduced by 92%, but PLEX also removes anti-JC virus antibodies. PLEX rapidly removes free natalizumab antibody and allows the return of functional VLA-4 to the lymphocyte surface, enabling the immune system to resume protection of the CNS. As proof of concept, after HAART is begun in AIDS, there is a rebound in the T cell count and partial improvement in immune function, and PML resolves to variable degrees. In multiple sclerosis, immune function is normal or above normal. Thus, complete removal of natalizumab should lead to protective (or even excessive--IRIS) immune function in multiple sclerosis/PML. In an early case, soon after natalizumab was discontinued, the MRI began to enhance in a multiple sclerosis/PML patient who later improved. In PML during natalizumab therapy, plasmapheresis halves the time of onset of IRIS (65) and doubles the duration of IRIS. However, in 219 natalizumab PML cases, PLEX had no benefit (Biogen 2024).
Leukocytapheresis could remove peripheral lymphocytes with bound anti-VLA-4 antibodies and mobilize bone marrow cells. This procedure needs to be tested for safety, monitoring whether it mobilizes bone marrow cells that contain the JC virus.
To be effective against PML, other agents should penetrate into the CNS, yet not suppress antiviral immunity.
Inhibitors of JCV infection and spread are detailed in (37).
In multiple sclerosis, blood-brain barrier disruption should allow penetration of some antiviral agents. Some therapies below provoke IRIS.
Interferon-beta effects against PML in multiple sclerosis remain uncertain, but it had a suggestive benefit PML in several reports. Interferon-beta is antiviral and prevents 95% of JC virus replication in human fetal glial cells. Type I interferons may not be curative because only 1 of 1000 of serum interferon-beta enters the normal CNS (53). However, a strong interferon signature in the inflamed EAE mouse brain, six hours after interferon-beta injection, indicates there is significant penetration into the CNS (27). CNS inflammation in multiple sclerosis may allow interferon to cross the blood-brain barrier. Anti-interferon antibodies enhance polyoma virus infections after virus is inoculated peripherally in mice. In vivo, interferons reduce levels of serum JC virus DNA. In a study with much higher virus DNA levels in serum than in other reports, serum was positive in 29% of healthy controls and in 46% of untreated multiple sclerosis patients, and it fell to 14% in interferon-beta-treated multiple sclerosis patients.
Interferon alpha, 5 MU three times per week, and interferon-beta have modest but erratic systemic benefit in HIV/PML. Intrathecal interferon-beta stopped decline and led to modest improvement in one case. Cidofovir and interferon alpha have anecdotal benefit, but cidofovir alone has no benefit in PML. Brincidofovir, a smallpox therapy with 100 times more CNS penetration, may be more effective (37).
Teriflunomide, a multiple sclerosis therapy, strongly interferes with BK replication, JC virus, astrocytes, and choroid plexus cells (37). Leflunomide, the parent compound of teriflunomide, reduces BK-virus induced renal transplant rejection, but is linked to PML (42). Cidofovir, a treatment for cytomegalovirus, has only occasional benefit in PML.
5HT2A receptor blockers are designed to cross the blood-brain barrier to treat depression. Risperidone at 4 mg twice daily, combined with paroxetine 40 mg twice daily to slow risperidone metabolism, may block JC virus entry into CNS cells and reverse PML in one patient. Olanzapine, ziprasidone, clomipramine, chlorpromazine, cyproheptadine, cetirizine, mirtazapine, and also clozapine, ketanserin, ritanserin, metoclopramide, and mianserin could potentially block virus binding to brain cells.
Chlorpromazine and clozapine synergistically block clathrin-dependent endocytosis of JC virus into glial cells. However, reported benefits in PML are underwhelming.
Ciprofloxacin decreases the load of the related BK polyomavirus in urine. It is unknown if this is a direct mechanism or is from this antibiotic’s ability to enhance immunity—a potential concern in multiple sclerosis, as Floxin antibiotics induce Th1 cytokines and can trigger multiple sclerosis exacerbations (Reder 1985). Cyclosporine and topiramate may have anti-JC virus activity (37).
Cytosine arabinoside (Ara-C) benefitted one third of cases of non-AIDS PML, but later studies were not as positive. Those who improved had residual deficits, and MRI improvement took more than 6 weeks. There was a dramatic response to Ara-C in one case of PML in multiple sclerosis. Of five immunosuppressed dermatomyositis patients who developed PML, four were treated with a reduction of immunosuppression plus Ara-C, and two survived. This drug has significant neurologic toxicity in cancer patients who had normal brains before therapy. Cidofivir, tenofivir, and topoisomerase I inhibitors (camptothecin and topotecan) are potentially helpful. Tenofivir alafenamide caused rapid but incomplete improvement of MRI lesions (64).
Filgrastim (granulocyte-colony-stimulating factor; G-CSF) promotes the growth of neutrophils and lymphocytes. It induced IRIS in 15 or 17 natalizumab PML cases, and 41% recovered to near baseline. G-CSF induction of multiple sclerosis exacerbations occurs in 20%. JC virus granule cell neuronopathy also improved in one report.
Fusidic acid (Fucidin) has reduced viruria and stabilized allograft function in a renal transplant patient with JC virus-associated nephropathy.
IL-2 therapy led to sustained recovery in cases of PML with myelodysplastic syndrome and Hodgkin lymphoma. IL-2, followed by pembrolizumab, a PD-1 blocker, has a possible additive effect. Larger studies in sarcoid with PML showed no benefit of IL-2.
IL-7 reconstitutes T cells. Immune-compromised patients (HIV, cancer) with greater than 50% IL-7 induction of lymphocytes and CD4 T cells (odds ratio = 5.9; ie, those who respond to IL-7) do better. IL-7 plus vaccination (below) with the VP1 JC virus capsid protein, plus imiquimod adjuvant, is safe and had surprising benefit in two patients.
IL-15, which stimulates NK and cytolytic CD8 T cells, is potentially beneficial.
Immune checkpoint inhibitors, such as pembrolizumab, have potential benefit (55). Reversing T cell exhaustion and immune suppression should activate antiviral CD8 cytolytic T cells.
Maraviroc blocks chemokine receptor type 5 (CCR5) and is used to prevent HIV binding to immune cells. CCR5 controls cell migration, and maraviroc could, therefore, affect cell migration. There are case reports of improvement of IRIS with maraviroc, but others see no benefit. Mefloquine is an antimalarial that inhibits endosomal acidification and inhibits JC virus infectivity in vitro. It has significant neuropsychiatric adverse effects and fails to treat PML.
High-dose steroids (1 g intravenously for 3 to 5 days, with slow oral taper) decrease symptoms and frightening MRI lesions of PML and IRIS, and are presumed to reduce disability. However, patients given prophylactic steroids fared worse than those taking steroids only when symptoms were severe (60). This Goldilocks phenomenon suggests a balance between treating severe inflammation and brain swelling versus inhibition of anti-JC virus cytolytic T cells--both inhibited by steroids.
Two other approaches are important.
JC virus vaccines, small interfering RNA, and small molecules that would disrupt natalizumab-ligand interactions are in the conceptual stage. Anti-BK virus and anti-JC virus prototype VP1 protein are potential targets. CRISPR/Cas9 can be used to induce mutations in the JC virus T antigen and to suppress viral replication.
Autologous or allogenic T cells specific for the JC and/or BK virus controlled PML in six of nine patients with hematologic cancers, and five were in good clinical condition (79% treated with directly isolated allogeneic virus-specific T cells stabilized or improved) (48). Rarely, patients improve spontaneously.
Therapy with natalizumab requires careful weighing of the risk–benefit ratio by well-informed patients and then close monitoring. This includes clinical vigilance, longitudinal neurologic exams by a multiple sclerosis expert, and a low threshold for any clinical signs of PML, plus a baseline MRI for comparison with later putative PML. In the U.S. TOUCH program, patients are quizzed for symptoms of PML before every natalizumab infusion.
PML is not predicted by peripheral white blood cell count, CD4/CD8 cell ratios, or JC virus genome (quantitative JC virus DNA) in blood, urine, or CSF. In untreated HIV patients, high JC virus DNA levels correlate with a worse prognosis. In HIV/PML, disease activity should be based on clinical, radiological, and virological criteria (CSF JC virus genome quantitation every 3 months).
Antibodies to the JC virus reflect immune recognition of virus protein or nucleic acid, and the titer of serum antibodies to the virus does determine the risk of developing PML. Elevated or rising titers suggest there is more virus present; this rise is missed by serum and CSF DNA assays. In patients never exposed to immunosuppressants after two years of natalizumab therapy, an index value less than 0.9 connotes minimal risk (1/3000), but from 0.9 to 1.5 the risk is 1 of 1000, and above 1.5 the risk is 1 of 120 per year.
There is a 10-fold increase in seroconversion from JC virus-negative to JC virus-positive during natalizumab therapy, in contrast to the usual 1% per year in untreated patients.
Other markers also predict the risk of PML but are not in wide use.
|
• In a difficult-to-perform assay, L-selectin (CD62 ligand, CD62L) was slightly lower on CD4 T cells in multiple sclerosis and lower still during natalizumab therapy. It was low on CD4 T cells in 5% of patients with a 55-fold increased risk of developing PML. Loss of L-selectin prevents the early rolling stage of cell migration through the blood-brain barrier. Other labs have been unable to confirm this finding, but the duration of cell storage and flow cytometry techniques were different. The European Medicines Agency (EMA) chose not to use this assay for predicting PML risk. | |
|
• Serum MMP9 is low in those destined to develop PML (23). | |
|
• The absence of lipid-specific IgM bands in the CSF, plus serum antibodies to JC virus, raises the odds ratio of developing PML to 60, from an odds ratio of 24 for antibodies to JC virus alone. Absent IgM bands also correlate with half as many white blood cells in the CSF. | |
|
• HLA-DRB1*15 haplotype reduces the odds of PML to 0.46, but HLA-DQB1*06:03 increases the risk of PML to 1.61, denoting a role for CD4 T cells in virus control. | |
|
• IL-10 rises in CSF with recent PML, suppressing antiviral immunity. | |
|
• Soluble VLA-4, osteopontin, and JC virus T-antigen protein are potential biomarkers of PML risk. | |
|
• Serum neurofilament light chains (section 14) | |
|
• PET imaging differentiates 96% of PML versus multiple sclerosis lesions—there is reticular accumulation of translocator protein on phagocytes in PML, but homogenous distribution in multiple sclerosis lesions. |
Some neurologists perform yearly MRIs to detect PML before clinical symptoms appear. Perhaps 90 of 678 (13.3%) cases of PML in multiple sclerosis were picked up by MRI before neurologic symptoms (Biogen Idec MedInfo). In many of these asymptomatic cases, however, the intensity of clinical monitoring and the diagnosis of PML itself is not clear. Conversely, other cases have been reported with clinical symptoms and a negative MRI. Nonetheless, MRI is essential to differentiate between multiple sclerosis and PML when symptoms appear.
Monitoring for PML with MRI is arguably not cost-effective. An MRI ± gadolinium at academic medical centers in the United States is approximately $7000, although FLAIR + DWI fast screening protocols of PML could be equally sensitive and much cheaper. Thus, the cost of detecting one case of PML by MRI a few weeks earlier than by clinical symptoms is $7000 x 1000 (1/1000 risk per year) x 12.5 (8% detected by MRI before clinical symptoms) = $87,500,000.
The posology or dosing of therapy could theoretically be adjusted to reduce the risk (not abrogate risk) of PML by less frequent infusions, pulsed therapy, or drug holidays. Some suggest that a drug holiday after 1 to 2 years might reduce the risk of if the drug is later restarted. However, drug holidays introduce the risk of exacerbations at 3 months. Extended interval dosing, from 28 to approximately 36 or 55 days, was safe in 300 or 600 patients. There was no increase in exacerbations, likely no excess of MRI lesions, no increase in serum neurofilaments, and over 2-fold fewer conversions to JCV-positive serum. Four cases of PML occurred; all were in the every-28-day group, with none in the extended-dosing cohort. Abstracts show a reduction in risk of one half to one third with 35- to 45-day intervals versus 30- to 31-day intervals. Patients stable for an average of 5.6 years and who discontinue natalizumab have a 7.2-fold increased risk of relapse within 2.5 years (36).
It is important to remember that these patients still have multiple sclerosis and that removal of effective therapy allows active multiple sclerosis to return. The same arguments apply to patients who have serum antibodies to JC virus. Those who do not have antibodies to the virus are at very low risk, and drug holidays for them are not logical. Low pretherapy disease activity, a fall in CSF B cells and immunoglobulin G and M synthesis during treatment, and higher natalizumab-induced blood lymphocyte counts, controlled by Akt-regulated apoptosis, predict that there will be fewer flares after the drug is stopped. Importantly, discontinuation of natalizumab to prevent the 1:100 to 1:000 chance of developing PML will almost certainly cause more multiple sclerosis activity unless another effective agent is started.
When natalizumab is discontinued, some physicians delay the start of therapies that reduce CSF immune cells (fingolimod) or suppress immunity (chemotherapy, glucocorticoids, possibly teriflunomide and fumarate). However, the risk of rebound multiple sclerosis exacerbations becomes more likely the longer a second therapy is delayed (discussed in topic 8). The risk of developing PML declines after cessation, and PML is very rare 6 months after the last dose.
A washout of other therapies before starting natalizumab must also balance the risk of transiently combining natalizumab with immunosuppression or with altered immune regulation, versus the risk of removing all therapy from a patient experiencing disease activity. A washout period of 3 to 6 months after chemotherapy and less than 6 weeks after interferon-beta was suggested by an expert panel. However, many patients contemplate a switch from other therapies because of disease activity but are still partially treated. This author has seen rapid clinical worsening within two weeks of withdrawing interferon-beta and glatiramer and recommends only several days or no gap in therapy. Immune compromise should be ruled out before starting natalizumab, with a complete blood count and evaluation for opportunistic infections.
A combination of natalizumab with interferon-beta or chemotherapy, or even glucocorticoids, may be unsafe. The two PML cases in the pivotal trials were in the interferon-beta-1a/natalizumab combination group, but the numbers are too small for confidence. Past and concurrent chemotherapy is a significant risk for PML. Glucocorticosteroids are immunosuppressive, but there is no obvious danger signal at this point in combination therapy with natalizumab.
Rebounds of multiple sclerosis may occur after stopping natalizumab. Angry immune cells are kept out of the brain by natalizumab, and activated white blood cells increase during therapy (topic 5). There was no rebound in the large pivotal drug trials in the subset that stopped therapy; clinical and MRI activity returned after 3 to 6 months, but only to near pre-natalizumab rates. Controlled drug holiday studies showed the same result. However, patients taken off the drug had much more multiple sclerosis activity (28%) than those who remained on therapy (0%). When the drug is withdrawn, cognition declines back to baseline by one year. CSF immune cells return in 3 to 6 months (34), and disease activity and/or MRI Gd-enhancing lesions sometimes reappear at 6 weeks. Many patients are highly active before therapy, so when they return to pretreatment levels of activity, a subset will seem to do poorly.
Switching to another drug prevents some rebound of multiple sclerosis activity. In the RESTORE trial, 175 patients who were stable on natalizumab were randomly divided into those who stayed on the drug (25%) and those who switched to placebo (25%) or other therapies (50%). Patients assigned to other therapies chose among the immediate start of glatiramer acetate (19%) or intramuscular interferon-beta-1a (19%), or a 3-month delay before starting monthly intravenous methylprednisolone (61%). Patients then switched back to natalizumab at week 28 or if they had an exacerbation. Those who remained on natalizumab were stable over the next 24 weeks (0% MRI lesions; 4% relapses). Those who switched had more activity: placebo (46%; 17%), methylprednisolone (40%; 15%), glatiramer acetate (53%; 27%), and intramuscular interferon-beta-1a (7%; 29%).
In the year after switching, another study found exacerbations in 22% of patients after subcutaneous interferon-beta and in 38% of patients after glatiramer acetate; 93% of recurrences occurred after two months. In a retrospective Swedish study, rituximab was more effective than fingolimod in preventing relapses in multiple sclerosis patients who switched from natalizumab due to positive JC virus antibody. Within 1.5 years of cessation of natalizumab, 1.8% (rituximab) and 17.6% (fingolimod) of patients experienced a clinical relapse (hazard ratio for rituximab = 0.10, 95% confidence interval [CI] = 0.02-0.43). Furthermore, contrast-enhancing lesions were found in 1.4% (rituximab) versus 24.2% (fingolimod). Twelve percent of switches from natalizumab to ocrelizumab have early clinical or MRI activity. Lastly, dimethyl fumarate may be effective in preventing multiple sclerosis rebound after discontinuing natalizumab. In a retrospective study, 39 patients were switched to dimethyl fumarate. There was no immediate rebound effect, but at 2 years, there were clinical relapses in five patients and MRI activity in eight.
Thus, (1) natalizumab is highly effective; (2) relapses recur 3 to 6 months after discontinuation, but on average are no more severe than the highly active multiple sclerosis state that preceded natalizumab therapy; (3) MRI lesions and relapses recur after switching to placebo, monthly methylprednisolone, subcutaneous interferon-beta, and glatiramer acetate (which has a slow onset of action), and to a lesser extent, intramuscular interferon-beta and ocrelizumab; and (4) the early start of a second drug is safer than a late start. Interferon-beta and glatiramer acetate may have a delayed therapeutic onset compared to fingolimod; these kinetics complicate prevention of disease rebounds, and residual symptoms after exacerbations may also differ between drugs. All told, the optimum time to start interferon-beta, glatiramer acetate, or fingolimod is within 0 to 2 weeks. In one study that measured time-dependent rebound of activity, exacerbations doubled with a 16-week wait compared to an 8- to 12-week wait before starting fingolimod. If feasible, when natalizumab is tapered down rather than abruptly, the multiple sclerosis relapse rate is reduced (67). Restarting natalizumab has also stopped rebound disease activity.
Understanding the mechanism of the development of PML in multiple sclerosis will lead to a better understanding of the JC virus life cycle, CNS immune surveillance by T cells on patrol, synergy between interferons and anti-VLA-4 agents, and the importance of VLA-4 in peripheral organs, the brain, and other immunologically privileged sites. A vaccine to the BK and JC viruses before renal transplant or natalizumab therapy could prevent renal and brain complications.
Natalizumab is highly effective in the treatment of multiple sclerosis. There are no more infections than expected (tuberculosis, fungal, bacterial, and viral), with the exception of PML. It is unknown if only the combination of natalizumab and interferon-beta predisposes to PML.
The dangers of natalizumab must be put in perspective. Discussion of other lifetime risks is important. There is excess mortality from multiple sclerosis and from airplane crashes (1/20,000), fire (1/1000), and automobile accidents (1/100). The author’s yearly risk of an accident while driving to work in Illinois is 1 of 10,000 per year (33). As with many medicines, there is a balance between risk and benefit. Anti-tumor necrosis factor therapy, anti-B cell therapy, and methotrexate therapy for rheumatic disease induce demyelinating disease, tuberculosis, lymphomas, and PML. In multiple sclerosis, mitoxantrone causes cardiac degeneration and lymphomas, and glucocorticoids have a plethora of side effects, without long-term benefit.
Reasonable suggestions using this therapy are as follows: (1) JC virus antibody-negative patients can be safely treated, with 6-monthly reassessment of their antibody status and clinical response; (2) JC virus–positive patients must weigh the risks described above. Perhaps those with prior immunosuppressive therapy and with high anti-JC virus titers should not be treated.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Anthony T Reder MD
Dr. Reder of the University of Chicago received honorariums from Genentech, Genzyme, and TG Therapeutics for service on advisory boards and as a consultant and stock options from NKMax America for advisory work and an unrestricted lab research grant from BMS.
See Profile
Francesc Graus MD PhD
Dr. Graus, Emeritus Professor, Laboratory Clinical and Experimental Neuroimmunology, Institut D’Investigacions Biomédiques August Pi I Sunyer, Hospital Clinic, Spain, has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Child Neurology
Apr. 24, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neuroimmunology
Mar. 29, 2026