





Neuroimmunology
Balo concentric sclerosis
Jul. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Neuromyelitis optica spectrum disorder (NMOSD), historically known as Devic disease, is an autoimmune neurologic disorder associated with antibodies against aquaporin-4, a water channel expressed on astrocytic endfeet. The pathologic hallmark of neuromyelitis optica spectrum disorder is damage to the astrocytes by the anti-aquaporin-4 (AQP4) antibody through complement-dependent cytotoxicity, subsequently leading to demyelination and neuronal loss. Neuromyelitis optica spectrum disorder is clinically characterized by symptoms of optic neuritis, transverse myelitis, area postrema syndrome, as well as brainstem, cerebellar, and cerebral syndromes. Neuromyelitis optica spectrum disorder is associated with devastating clinical disability without proper and early initiation of treatment. In the past 5 years, there have been significant advances in the understanding of the pathophysiology of neuromyelitis optica spectrum disorder, which have led to the approval of four disease-modifying treatments (DMTs) for this condition by the U.S. Food and Drug Administration (FDA).
|
• There is some overlap in the clinical phenotype of patients who have auto-antibodies against myelin oligodendrocyte glycoprotein (anti-MOG) and those with anti-AQP4 antibodies, but these are distinct pathophysiological and clinical entities. | |
|
• Anti-AQP4 antibody testing by a cell-based assay (CBA) in the serum is required for the diagnosis of antibody-positive neuromyelitis optica spectrum disorder. | |
|
• Aggressive treatment at the onset of a relapse, including steroids, plasma exchange, and early initiation of prophylactic therapy, leads to better recovery and mitigation of long-term disability in patients with neuromyelitis optica spectrum disorder. | |
|
• Pregnancy is an important consideration when treating women with neuromyelitis optica spectrum disorder. Unlike multiple sclerosis, disease activity does not decrease during pregnancy, and relapses during pregnancy can lead to severe disability if prompt, acute treatment is not initiated. | |
|
• There are four FDA-approved drugs for patients with anit-AQP4 positive neuromyelitis optica spectrum disorder. Treatment approach should be individualized based on patients’ age, comorbidities, level of baseline disability, and risk of adverse events, particularly infections in the older and more severely disabled subjects who have a history of recurrent UTIs, indwelling catheters, and pressure sores. |
Thomas Albutt is given credit for initially describing the syndrome now called neuromyelitis optica (03). Albutt observed that some patients with myelitis had coexistent funduscopic abnormalities, and these patients had a more severe disease course. Eugene Devic, a French physician, was initially interested in studying typhoid fever. Devic’s eponymic distinction later emerged after he summarized the known cases of neuromyelitis optica along with his own observations in 1894 and presented them at the French Congress of Medicine in Lyon, coining the terms “neuro-myélite” and “neuroptico-myélite” (17). In 1907, the term Devic disease was first used, and since then, Devic’s name has been associated with this disease.
In 2004, Lennon and colleagues discovered the antibody associated with the syndrome termed neuromyelitis optica immunoglobulin G (NMO-IgG), ie, anti-AQP4 antibody. That discovery has changed the way we diagnosed and treated neuromyelitis optica. In 2007, the term neuromyelitis optica spectrum disorder was coined to encompass all the different manifestations associated with this disease.
|
• Neuromyelitis optica spectrum disorder is a destructive, inflammatory disease of the central nervous system historically characterized by selective involvement of the spinal cord and optic nerves. However, other distinct syndromes were later described in patients with anti-AQP4 antibody, including regions such as the diencephalon, area postrema, cerebrum, and midbrain (36). |
Symptoms onset is usually subacute, occurring over days to weeks, although a rapid progression of symptoms has been noted. The first manifestation of neuromyelitis optica spectrum disorder is optic neuritis in 56% to 76% of patients, transverse myelitis in 13% to 39%, and simultaneous optic nerve and spinal cord involvement in 4% to 11% (51; 106; 27). The interval between visual and spinal cord symptoms can be days (51; 106) or years (61; 73). It is not essential that optic neuritis and myelitis occur concurrently in neuromyelitis optica spectrum disorder. One cohort of patients in Brazil had a mean interval of 18 months between optic nerve and spinal cord symptoms. Maximum neurologic deficits occur within 2 to 4 weeks after onset (01). Prior to the advent of highly efficacious therapy for neuromyelitis optica spectrum disorder, an expanded disability scale score (EDSS) of 3 occurred at a mean of 6 months, and an EDSS of 6 was reached at a mean of 7 years (27). This is twice as fast as that seen in multiple sclerosis. However, recent studies suggest that with highly effective treatment, a large percentage of patients would stay stable or improve with time (64).
Optic neuritis. Patients may present with visual loss, pain with eye movement, and dyschromatopsia. In neuromyelitis optica spectrum disorder, the visual loss is often severe, with visual acuity worse than 20/200 at nadir, and may be unilateral or bilateral at onset. Bilateral optic neuritis is more common in neuromyelitis optica spectrum disorder patients than in multiple sclerosis. Optic nerve involvement is longitudinally extensive, affecting two thirds or more of the nerve. Either central scotomas or peripheral visual field defects can occur. The optic disc is edematous when acute inflammation extends past the lamina cribrosa (papillitis) but appears normal in retrobulbar optic neuritis. Recovery can be poor at follow-up, with residual blindness found in a large proportion of patients (60% to 69%) (42).
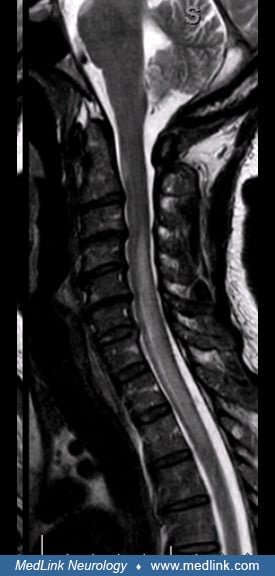
Transverse myelitis. Spinal cord involvement presents as a rapidly ascending sensory deficit with a sensory level, paraplegia or quadriplegia, and gait disturbance. Myelitis is usually longitudinally extensive, expanding over three or more vertebral segments.
Variants such as Brown-Sequard syndrome or syringomyelia are rare (94; 51). Autonomic instability results from dysfunction of sympathetic neurons in the spinal cord. Bladder dysfunction usually presents at onset, mainly with urinary retention. Most patients require an indwelling urinary catheter that is maintained for several weeks. Some may require a permanent suprapubic catheter.
Painful tonic spasms can be seen in patients following attacks of transverse myelitis (91). Neuromyelitis optica spectrum disorder relapses are a medical emergency. Acute respiratory arrest may occur in high cervical lesions. Complications of paralysis, including dysautonomia, decubitus ulcers, urinary tract infections, and sepsis, are the usual causes of death.
Incomplete recovery after a transverse myelitis attack is more common in neuromyelitis optica spectrum disorder patients compared to multiple sclerosis, especially in untreated patients. In the long term, 37% to 44% of patients with AQP4+-NMOSD will need ambulatory aid (08).
Area postrema syndrome (APS). This syndrome is the third most common core clinical presentation of neuromyelitis optica spectrum disorder, characterized by intractable nausea, vomiting, or hiccups. It’s associated with lesions of the dorsal medulla/ area postrema. Area postrema syndrome may be present at onset or during subsequent relapses, isolated or associated with other symptoms.
Other brainstem or cerebral syndromes. Patients with neuromyelitis optica spectrum disorder may have other symptoms, including encephalopathy, diplopia, nystagmus, trigeminal neuralgia, facial numbness, or hearing loss (88). In addition, patients with neuromyelitis optica spectrum disorder can also present with fevers, hypothermia, hypersomnia, or narcolepsy due to involvement of the diencephalon (81; 91; 88). Patients may present with an encephalopathy, similar to acute disseminated encephalomyelitis (ADEM) or posterior reversible encephalopathy syndrome (PRES), especially in pediatric cases (98).
A series of 32 patients with neuromyelitis optica spectrum disorder were followed, and after 6 months, MRI was normal in only 16. Fourteen had nonspecific MRI abnormalities, two had cervical lesions extending into the brainstem, and two actually had an MRI appearance similar to multiple sclerosis (79).
Diagnostic criteria. At the time of its original description by Devic, there was considerable debate over whether neuromyelitis optica was a variant of multiple sclerosis. Early on, neuromyelitis optica was recognized as a separate clinical entity (28; 94). But then, later studies considered it a variant of multiple sclerosis (21; 84; 59; 51). To establish neuromyelitis optica as a distinct entity from multiple sclerosis, several authors proposed specific diagnostic criteria (61; 19; 73; 22; 111; 108).
In 2006, the Mayo group proposed diagnostic criteria reflecting the understanding of the serologic and radiographic characteristics of neuromyelitis optica spectrum disorder at that time (111).
In 2015, due to better understanding of the clinical and radiologic presentations of neuromyelitis optica spectrum disorder, new diagnostic criteria were set forth by the International Panel for NMO Diagnosis (108). The group recommended expansion of the diagnostic term from neuromyelitis optica to neuromyelitis optica spectrum disorder to incorporate an expanding clinical phenotype. The group also recommended dividing neuromyelitis optica spectrum disorder into two groups: an anti-AQP4 antibody-positive group and an anti-AQP4 antibody-negative group. Patients with neuromyelitis optica spectrum disorder who test positive for the anti-AQP4 antibody need one core clinical characteristic and no better explanation for their presentation. The core clinical features include the following:
|
• Optic neuritis |
However, patients with neuromyelitis optica spectrum disorder who test negative for the anti-AQP4 antibody or for whom the antibody test is unavailable require at least two of the core clinical characteristics, in addition to all of the following:
|
• One episode must be optic neuritis, transverse myelitis with LETM, or area postrema syndrome | |
|
- Optic neuritis with normal or nonspecific white matter lesions OR T2 hyperintense lesion involving the optic nerve OR T1 gadolinium positive lesion extending for greater than half the length of the optic nerve or involving the optic chiasm | |
As with anti-AQ4 antibody positive patients, there should be no better explanation for the patient’s symptoms.
Classifying patients with neuromyelitis optica spectrum disorder who are negative for the anti-AQP4 antibody can be challenging. For anti-AQP4 antibody-negative patients, the term neuromyelitis optica spectrum disorder is applied to patients who fulfill clinical and radiological criteria in terms of recurrence (at least two episodes) of longitudinally extensive transverse myelitis, optic neuritis, or brainstem syndromes. In contrast to patients who are anti-AQP4 antibody positive, patients who are anti-AQP4 antibody negative tend to be Caucasian, do not have a female preponderance, have simultaneous presentation of optic neuritis and transverse myelitis at onset, have a monophasic presentation, and typically have less severe long-term visual impairment (90). The detection of other pathogenic autoantibodies may further help define this anti-AQP4 seronegative subset of patients, comprising about 30% of the neuromyelitis optica spectrum disorder population. About 25% of such patients are antimyelin oligodendrocyte glycoprotein (MOG) antibody positive (75) and are now considered to have a separate disease called MOG antibody associated disease (MOGAD).
Disease course. Most patients with AQP4+ NMOSD would relapse if the disease is untreated. In the relapsing form, the attack rate is 1.3 per year (range 0.1 to 5.5) (27). Relapses occur within 6 months of the initial attack in 55% of patients, within 18 months in 27%, and after 5 years in 18% (106). The mean interval until a second attack is 17 months (27). In contrast to multiple sclerosis, neuromyelitis optica spectrum disorder does not have a progressive form (64).
Risk factors for having future relapses include anti-AQP4 antibody, female sex, short between-attack interval, and systemic autoimmunity (106; 110; 105; 55). Respiratory failure occurs more in relapsing patients, especially those with high cervical spinal cord lesions that compromise the phrenic nerve. Historically, the 5-year survival rate in neuromyelitis optica spectrum disorder was 90% for monophasic and 68% for relapsing patients (110). This study has not been repeated, but survival rates are much better now given earlier recognition of the disease, anti-AQP4 antibody testing, and aggressive treatments, especially with FDA-approved agents with high efficacy. In contrast to adult disease, pediatric neuromyelitis optica spectrum disorder has a lower incidence of relapses and has less clinical burden of disease (41; 05).
Breakthrough neuromyelitis optica spectrum disorder disease activity in otherwise stable patients should prompt an evaluation for infections, notably urinary tract or gastrointestinal infections. Experiments in mice have shown there is cross-reactivity between aquaporin Z (found in E coli bacteria) and human aquaporin 4. Thus, a urinary tract infection may cause a relapse due to molecular mimicry (86). Another lab has shown that the T cell epitope within aquaporin-4 receptors shares homology with a sequence from the adenosine triphosphate-binding cassette transporter permease of Clostridium perfringens. Thus, patients presenting with a Clostridium infection could also experience a neuromyelitis optica spectrum disorder relapse (101).
Historically, neuromyelitis optica spectrum disorder had high morbidity and mortality. Five-year mortality rates in the past were as high as 30% in the relapsing forms (110). However, this number has greatly improved due to earlier recognition and aggressive treatment. One study suggested that 5 years after the initial presentation, more than 50% of patients with neuromyelitis optica spectrum disorder were blind in one or both eyes or required ambulatory help (04). High morbidity results from complications associated with transverse myelitis, such as respiratory failure from high cervical cord lesions, infections, autonomic dysfunction, and bladder retention. Younger patients and males tend to have a better outcome than elderly and female patients. Factors that predict worse prognosis include older age at onset, high relapse rate in the first year after the diagnosis, severe first attack, poor recovery after the first attack, elevated CSF IL-6, and a high number of brainstem lesions (100). A multicenter retrospective analysis of 182 patients with anti-AQP4-positive neuromyelitis optica spectrum disorder found several factors that were associated with increased disability as measured by the expanded disability scale score. These factors included a delay in diagnosis and treatment, older age at first attack/disease onset, length of spinal cord lesion, and presence of symptomatic brain/brainstem lesions (66). Studies show better prognosis in disability resulting from earlier diagnosis and high-efficacy treatments (64).
An 18-year-old Black woman with a past medical history of sickle cell trait and asthma presented 1 month postpartum with acute triparesis with bladder involvement and a T4 sensory level. She was found to have a longitudinally extensive transverse myelitis from the medulla to C7. Her brain MRI was normal. Acutely, she was treated with intravenous steroids and plasma exchange. She was placed on a prolonged steroid taper and azathioprine for prophylaxis. The following year, she developed two episodes of right optic neuritis and another episode of transverse myelitis. MRI showed enhancement of the right optic nerve as well as enhancement and increased T2 signal from C3-T11 on MRI. Three years later, she developed optic neuritis affecting the optic chiasm and both optic nerves. The following year, she developed another transverse myelitis. Two years later, she had recurrent thoracic transverse myelitis. She was treated each time with intravenous steroids, IVIG, plasma exchange, or IVIG, with minimal recovery. Her clinical course was complicated by neurogenic bladder, constipation, and frequent urinary tract infections, which often preceded her relapses. Prophylactically, she had been on azathioprine, mycophenolate mofetil, mitoxantrone, IVIG, and rituximab. Eleven years after her initial presentation, she was wheelchair-bound due to paraplegia, blind in the right eye, and had severely decreased visual acuity in the left eye.
This case illustrates the severity of neuromyelitis optica spectrum disorder associated with high morbidity and poor recovery with each relapse. Many of the relapses were precipitated by urinary tract infections, which is common. The patient was treated with several immunosuppressive therapies but continued to have high disease activity. Fortunately, prognosis may improve with the newer FDA-approved disease-modifying treatments for anti-AQP4-positive neuromyelitis optica spectrum disorder (AQP4+ NMOSD).
The etiology of neuromyelitis optica spectrum disorder remains unknown, as with other autoimmune diseases. About 10% to 20% of patients with neuromyelitis optica spectrum disorder have another coexisting autoimmune disease, notably Sjögren disease (39), myasthenia gravis, systemic lupus erythematosus, and systemic sclerosis. A greater percentage (approximately 50%) have autoantibodies, such as antinuclear antibodies, extractable nuclear antigens, Sjögren syndrome antigen A/B (SSA/SSB), antithyroglobulin, antithyroid microsomal, antigastroparietal cell, and p-antineutrophil cytoplasmic antibodies (some of which can be disease-causing) (73; 43; 110). It appears that neuromyelitis optica spectrum disorder occurs as a separate comorbid condition in patients with preexisting systemic autoimmune disease. Patients with neuromyelitis optica spectrum disorder rarely have autoantibodies to paraneoplastic disease markers, such as acetylcholine receptors, glutamate decarboxylase 65, voltage-gated potassium channels, and voltage-gated calcium-channels (80). The association of neuromyelitis optica spectrum disorder with a variety of autoantibodies suggests widespread B cell polyclonal activation (58).
Neuromyelitis optica spectrum disorder has been associated with infections, such as herpes viruses (human herpesvirus 6, HSV-2, and varicella), Mycoplasma pneumoniae, tuberculosis, Epstein-Barr virus, and HIV (44; 107; 45; 12; 63; 93; 02; 68; 07). Neuromyelitis optica spectrum disorder could be triggered by these chronic infections in hosts who have preexisting vulnerability or by B-cell activation.
Genetics. The role of genetic factors in neuromyelitis optica spectrum disorder is unknown. There are reports of familial occurrence, but no conclusive evidence exists (65; 10; 113). There is evidence that neuromyelitis optica spectrum disorder is associated with human leukocyte antigen alleles DR1*801, DP1 501, and DPA1 202, but not with DRB1*1501, which is associated with multiple sclerosis (30). A 2018 study looking at neuromyelitis optica spectrum disorder and healthy controls using whole genome sequencing suggested that certain genetic variants in the MHC region play a role in the pathology of neuromyelitis optica spectrum disorder (18).
Pathophysiology. The anti-AQP4 antibody is directed against aquaporin-4 (AQP4), which is a water channel predominantly expressed in the CNS (53). Indirect immunofluorescence shows a distinctive antibody binding in the pia and subpia, Virchow-Robin space, and microvessels in the white and gray matter of the cerebellum, midbrain, and spinal cord. Anti-AQP4 binds to the luminal face of cerebral microvessels. Dual labeling with glial fibrillary acidic protein reveals juxtaposition of aquaporin-4 to astrocytic foot processes. Anti-AQP4 also binds to distal urine-collecting tubules in the renal medulla and to the basolateral membranes of gastric parietal cells (53). The proposed pathway of astrocyte destruction involves a 2-step process. First, there is opening of the blood-brain barrier, likely by a T-cell-dependent mechanism (71; 101; 56). Then, circulating anti-AQP4 antibodies bind to aquaporin-4 expressed on the astrocyte foot processes. Next, internalization of aquaporin-4 down-regulates pathways for glutamate homeostasis, which promotes further blood-brain barrier permeability (31). Binding of anti-AQP4 to AQP4 in the presence of complement leads to complement activation, chemo-attraction of granulocytic white blood cells, and astrocyte injury via antibody-dependent cytotoxicity (89; 34). During an acute attack, serum IL-6 levels are elevated, which is a growth factor for plasmablasts (25). In turn, this leads to an enhanced production of anti-AQP4 antibodies (34).
Pathology. Lesions in neuromyelitis optica spectrum disorder show perivascular and parenchymal leukocyte infiltration, deposition of IgG, IgM, and complement, loss of GFAP and AQP4 staining, hemorrhagic exudation, edema, capillary proliferation, vascular fibrosis, hyalinization, tissue necrosis, and cavitation (38). Both white and gray matter are affected. Lesions are more similar to necrotizing vasculitis than to the predominantly demyelinating lesions of multiple sclerosis or acute disseminated encephalomyelitis.
Two autopsy studies on patients with neuromyelitis optica spectrum disorder found signs of sublytic astrocytopathy in addition to the known lytic lesions discussed earlier (97; 29). A study by Takai and colleagues performed histopathological analysis on eight autopsied cases of anti-AQP4-positive neuromyelitis optica (97). They found four stages of astrocytopathy based on astrocyte morphology and immunoreactivity. Stage one included astrocyte lysis with an extensive loss of astrocytes with fragmented particles. Stage two included progenitor recruitment with a loss of astrocytes except for small, nucleated GFAP-positive fiber-forming processes. Stage three included protoplasmic gliosis with the presence of star-shaped astrocytes with abundant GFAP-reactive cytoplasm. Stage four included fibrous gliosis with lesions composed of densely packed mature astrocytes. The group found that stages one and two were seen within 2 months of clinically acute relapse. The complement degradation product (C3d) was present in all four stages.
Another study by Guo and colleagues described 23 neuromyelitis optica autopsy cases (clinically or pathologically confirmed) using brain, spinal cord, and optic nerve lesions (29). They analyzed astrocytic morphology, immunohistochemistry, AQP4 RNA transcripts, and demyelinating activity. They found suggestions of a global astrocytopathy with lytic and sublytic lesions. Several features that accompanied degeneration included dystrophic astrocyte morphology, prominent cytoplasmic vacuolation, lysis, and Rosenthal fiber deposition. They found emperipolesis of invading leukocytes into astrocytes under immune attack in which astrocytes contained peripheral immune cells; the biological significance of this is unknown. Astrocyte reactivity did not require anti-AQP4 IgG binding at the individual cell level. Astrocyte reactivity was also seen in nonlesional areas that retained AQP4 protein. Of note, the anti-AQP4 antibody serostatus was missing in 14 subjects. Both studies suggest a spectrum of lytic to sublytic astrocytopathy in the setting of neuromyelitis optica spectrum disorder.
Neuromyelitis optica spectrum disorder was historically grouped with other demyelinating diseases, such as multiple sclerosis and MOGAD. However, neuromyelitis optica spectrum disorder is distinct from demyelinating diseases. It is an astrocytopathy rather than an oligodendrocytopathy. Table 1 shows demographic, radiological, and pathological differences between neuromyelitis optica spectrum disorder and the other two common primary demyelinating diseases, multiple sclerosis and MOGAD.
|
Characteristics |
Neuromyelitis optica |
Multiple sclerosis |
MOGAD |
|
Age (median, estimate) years |
40 |
30 |
28 to 30 |
|
Sex F:M |
9:1 |
3:1 (relapsing) |
1:1 |
|
Ethnicity |
Greater incidence in East Asians and Blacks |
White > Blacks |
No difference so far |
|
Clinical course | |||
|
Monophasic |
30% |
NA |
20% to 70% |
|
Relapsing |
70% |
80% |
30% to 80% |
|
Progressive (secondary and primary |
No |
10% to 15% primary from onset; more than 50% become secondary progressive |
No |
|
MRI features | |||
|
Brain lesions |
Peri ependymal, dorsal medulla, brainstem, diencephalon, cerebral peduncles |
Oval, usually perpendicular to ventricles |
ADEM, cortical, brainstem, cerebellar |
|
Spinal cord lesions |
Longitudinal extensive > 3 vertebral body length |
Punctate lesions, < 3 vertebral body length |
Transverse myelitis < 3 vertebral body length† |
|
Optic nerve lesions |
Can be bilateral, poor recovery |
Usually unilateral with good recovery |
Can be bilateral with good recovery |
|
CSF findings | |||
|
White blood cell |
Polymorphonuclear cells >> lymphocytes |
Primarily lymphocytes |
Lymphocytes > polymorphonuclear cells |
|
Protein |
Elevated |
Typically normal |
Elevated |
|
Oligoclonal bands |
Usually absent‡ |
> 90% present |
Usually absent‡ |
|
MMP-9 |
Decreased |
Elevated |
Normal (serum elevated) |
|
IL-6 |
Increased |
Normal |
Increased |
|
Pathology | |||
|
Gray matter |
Necrosis |
Demyelination |
Demyelination |
|
White matter |
Necrosis |
Demyelination |
Demyelination |
|
Lesion immunopathology | |||
|
Macrophages |
+++ |
+++ |
+++ |
|
Coexisting autoimmune diseases |
30% to 50% |
Infrequent |
Infrequent |
|
Disability progression |
Attack related |
Can be attack independent |
Attack related |
|
| |||
|
* relapses occur based on ethnic background and duration of steroid taper | |||
Neuromyelitis optica spectrum disorder is a rare disease. In a systematic review from 2021, the worldwide incidence of neuromyelitis optica spectrum disorder in adults varies from 0.029 to 0.7 per 100,000 persons-years. The prevalence was estimated to be 0.34 to 10 per 100,000 persons 0.06 to 0.22 per 100,000 in children. The highest incidence and prevalence are found in persons of African heritage followed by Asains (74). A 2016 comparative study investigated the incidence and prevalence of neuromyelitis optica spectrum disorder and anti-AQP4 IgG seropositive status in patients with a prior diagnosis of inflammatory demyelinating disease. These patients were either residents of Olmsted County, Minnesota (82% Caucasian) or residents of Martinique (90% Afro-Caribbean). The authors found the prevalence in Martinique to be 10 per 100,000 but in Olmsted County only 3.9 per 100,000 (23). The prevalence is estimated to be 3.42 per 100,000 in Japan (70), 3.56 per 100,000 in South Korea (52), and 3.31 per 100,000 among Chinese in Malaysia (32). In multiple recent nationwide studies, the prevalence of neuromyelitis optica spectrum disorder among Caucasian has been approximately 1 out of 100,000 (33).
The disease can be seen in young adolescence to late adulthood, and the median age at onset is around 40 years. Neuromyelitis optica spectrum disorder occurs in the pediatric population with an average age of onset of 7 to 14 years (41; 57). Neuromyelitis optica spectrum disorder is more common in women, accounting for more than 80% of cases, although in children there is no difference in gender.
Patients suspected of having neuromyelitis optica spectrum disorder should be treated immediately with immunosuppressive therapy to prevent relapses and decrease disability. Because neuromyelitis optica spectrum disorder relapses are unpredictable, prolonged prophylactic therapy with immunosuppressive medications is highly recommended. There is no consensus on the duration of therapy, and this continues to be a topic of discussion and research. For other autoimmune diseases such as systemic lupus erythematosus and myasthenia gravis, long-term immunosuppression with mycophenolate mofetil, azathioprine, or rituximab is tolerated for many years with proper monitoring, without major safety concerns.
In most cases, neuromyelitis optica spectrum disorder presents with optic neuritis and longitudinal myelitis and less frequently with diencephalon, brainstem, and cerebral involvement. A large group of diseases can also affect these sites (see Table 2). A thorough history, physical examination, serum and CSF testing, and imaging studies can reliably distinguish neuromyelitis optica spectrum disorder from other diseases listed in Table 2.
|
Demyelinating and dysmyelinating diseases | |
|
MOGAD | |
|
Connective tissue diseases, vasculitides, and autoantibody syndromes | |
|
Sjögren disease | |
|
Infections | |
|
Viral: varicella zoster virus, Epstein-Barr virus, HIV, cytomegalovirus, human T-cell lymphotrophic virus, hepatitis B, arborviruses (West Nile virus), enteroviruses, Herpes simplex viruses | |
|
Bacterial: Lyme disease, syphilis, tuberculosis, mycoplasma | |
|
Toxic and nutritional | |
|
B12 deficiency | |
|
Neoplastic | |
|
Lymphoma | |
|
Vascular | |
|
Stroke | |
Neuromyelitis optica spectrum disorder is associated with several connective tissue diseases, most notably Sjögren disease and systemic lupus erythematosus. About 30% to 50% of patients with neuromyelitis optica spectrum disorder have either elevated autoantibodies or autoimmune diseases such as Sjögren disease, systemic lupus erythematosus, Hashimoto thyroiditis, pernicious anemia, connective tissue diseases, ulcerative colitis, or primary sclerosing cholangitis (110). For neuromyelitis optica spectrum disorder, the odds ratio for having another autoimmune disease is increased 10-fold (104). Sjögren disease co-occurred with neuromyelitis optica spectrum disorder in 16 out of 20 patients in a review, whereas systemic lupus erythematosus and systemic sclerosis co-occurred with neuromyelitis optica spectrum disorder in only a few case reports (24). Myasthenia gravis coexisted with neuromyelitis optica spectrum disorder in 2% of cases, often with the diagnosis of myasthenia gravis preceding neuromyelitis optica spectrum disorder (24).
There is evidence that optic neuritis and longitudinal myelitis could be the presenting manifestations of an underlying connective tissue disease, even before the diagnosis of connective tissue disease is made. In a large case series of primary Sjögren disease with neurologic involvement, 56 out of 82 patients had CNS disease (15). CNS disease preceded the diagnosis of Sjögren disease in 45 of these patients (80%). More importantly, SSA/SSB was positive only in 43% of the cases but a positive salivary gland biopsy was evident in 95% (39). Hence, a positive salivary gland biopsy is a more reliable indicator of underlying Sjögren disease than serological testing.
Patients with certain cancers could have the neuromyelitis optica spectrum disorder clinical phenotype and anti-AQP4 antibodies; however, such cases are rare. These malignancies include breast, lung, thymus, lymphoma, and carcinoid. There are rare reports of monoclonal gammopathy as well (Pittock and Lennon 2008). Neuromyelitis optica spectrum disorder phenotype in the presence of malignancies raises the possibility that dysregulated immunity associated with cancers may lead to nonspecific B-cell activation and enhanced auto-antigen presentation.
Diagnostic workup for neuromyelitis optica spectrum disorder requires a careful history, clinical presentation, characteristic MRI features, spinal fluid analysis, and extensive blood workup.
History and physical exam | |
History including travel and vaccinations | |
Basic studies | |
Complete blood count | |
Infectious studies | |
HIV | |
CSF workup | |
Cell count and differential | |
Rheumatologic workup | |
Erythrocyte sedimentation rate | |
Special serology | |
Anti-MOG and NMO-IgG | |
MRI studies | |
Contrast-enhanced MRI of the brain with fat-saturated orbital views of the optic nerves | |
Evoked potentials | |
Visual evoked potential | |
Special studies | |
Minor salivary gland biopsy (see text) | |
Serology. A serum marker for neuromyelitis optica spectrum disorder, the anti-AQP4 antibody (aka NMO-IgG antibody), is available commercially. Since Lennon and colleagues discovered the anti-AQP4 antibody with a tissue-based assay in 2004, further laboratory tests have been developed with increased sensitivity and specificity (54). Current available lab tests include cell-based assays, tissue-based assays, and ELISA (a protein-based assay). A study compared the different assays for AQP4-antibodies (82). Cell-based assays using fluoroimmunocytochemistry or flow cytometry have a median sensitivity of greater than 90% and a specificity of 100%. Tissue-based assays using immunohistochemistry have a sensitivity of 78% and a specificity of 100%. Lastly, ELISA has a median sensitivity of 60% and a specificity of 97%. Thus, patients with a high clinical suspicion for neuromyelitis optica spectrum disorder should be tested with a cell-based assay (82). It should be noted that CSF testing for the anti-AQP4 antibody has less diagnostic sensitivity (60). It is the authors’ recommendation to use high-sensitivity, cell-based assays for detecting anti-AQP4 and anti-MOG antibodies.
MRI. Radiographically, there are distinct differences between multiple sclerosis and neuromyelitis optica spectrum disorder (09). In comparison to multiple sclerosis, optic neuritis in neuromyelitis optica spectrum disorder is often longitudinal and extensive, involving up to two thirds of the optic nerve. Lesions predominantly affect the posterior aspects of the optic nerve (unilaterally or bilaterally) and may involve the optic chiasm (46).
Acute spinal cord lesions are typically longitudinally extensive, centrally located, edematous, involve gray matter, are hypointense on T1, and show enhancement with gadolinium. Chronic lesions are associated with cord atrophy and cavitation. Short transverse myelitis lesions can be found in a small number of neuromyelitis optica spectrum disorder patients. Like LETM lesions, they are centrally located and involved more than half of the cross-sectional area of the spinal cord (46; 108).
Brain lesions, often of a nonspecific type (46), can be seen in nearly 50% of patients with neuromyelitis optica spectrum disorder at disease onset, occur with higher frequency over time, and may appear multiple sclerosis-like in 10% of cases (95). In the brain, typical neuromyelitis optica spectrum disorder lesions preferentially affect the area postrema, the periependymal areas of the ventricles and periaqueductal gray, splenium of the corpus callosum, the diencephalon, long corticospinal tracts, and brainstem (46; 108). On MRI, these lesions have cloud-like enhancement and display vasogenic edema (100).
Cerebrospinal fluid analysis. CSF analysis in neuromyelitis optica spectrum disorder shows a unique pattern in comparison to other demyelinating diseases (see Table 1). In neuromyelitis optica spectrum disorder, CSF shows pleocytosis of 30 to 50 cells/µL with high percentage of granulocytes and eosinophils (104). This contrasts with multiple sclerosis, where CSF pleocytosis is mild (usually less than 10 cells/µL) and more than 90% of the cells are lymphocytes. In neuromyelitis optica spectrum disorder, CSF protein is usually elevated and oligoclonal bands are typically absent. About 30% of patients with neuromyelitis optica spectrum disorder could have positive oligoclonal bands during an acute attack. However, if oligoclonal bands are present, they tend to disappear over time, especially during disease stability (06). This is in sharp contrast to multiple sclerosis, where oligoclonal bands persist regardless of disease status. CSF immunoglobulin G synthesis rate is not routinely elevated in neuromyelitis optica spectrum disorder, and if so, is at a low level. The albumin CSF/serum ratio, which is a marker of blood-brain barrier breakdown, is elevated in more than half of patients studied with neuromyelitis optica spectrum disorder. Elevation of certain CSF markers correlates with the length of transverse myelitis during acute relapses, including albumin CSF/serum ratio, total protein, and L-lactate (35). Other uniquely elevated measures in acute attacks of neuromyelitis optica spectrum disorder compared to multiple sclerosis include IL-6 and glial fibrillary acidic protein (90; 100). Oligoclonal bands are present in less than 13% of patients with anti-MOG syndrome, and there is an elevated CSF/serum albumin ratio in 32% (37).
Lip biopsy. In 2008, our lab found a high prevalence of positive lip biopsies in patients with neuromyelitis optica spectrum disorder or longitudinally extensive transverse myelitis with clinical suspicion for Sjögren disease (40). Between 2004 and 2007, we evaluated 25 patients who met diagnostic criteria for neuromyelitis optica spectrum disorder (16 patients) or longitudinally extensive transverse myelitis (nine patients) who had sicca symptoms. None of these patients had clinical or laboratory evidence of other connective tissue disorders, such as systemic lupus erythematosus, rheumatoid arthritis, or scleroderma. All patients had serologic testing for SSA/B antibodies and anti-AQP4 antibody. Twenty patients had biopsies of their minor salivary glands. Biopsy results were considered positive if there was evidence of severe inflammation (a “focus score” of 3 or higher on a scale of 0 to 4, with higher scores indicating more and denser inflammatory infiltrate). Of 25 patients with sicca complaints, only four patients (16%) fulfilled diagnostic criteria for Sjögren disease, based on the European or International Sjögren’s Disease criteria. Of the 20 patients with sicca complaints who had a salivary gland biopsy, 16 patients (80%) met criteria for severe inflammation of the minor salivary glands. This included nine of 12 patients with neuromyelitis optica spectrum disorder and seven of eight patients with longitudinally extensive transverse myelitis. Our findings suggest that there may be subclinical evidence of Sjögren disease in patients with neuromyelitis optica spectrum disorder who have sicca complaints. This also suggests that there will be additional systemic autoimmune diseases in patients with neuromyelitis optica spectrum disorder due to overlapping pathogenic mechanisms (40).
Optical coherence tomography. Optical coherence tomography is a noninvasive method of evaluating the thickness of the retinal nerve fiber layer. In multiple sclerosis, there is atrophy of the retinal nerve fiber layer in eyes affected by optic neuritis as well as eyes unaffected by optic neuritis, reflecting chronic, subclinical axonal loss (83). The retinal nerve fiber layer in patients with neuromyelitis optica spectrum disorder is much thinner than in patients with multiple sclerosis. This is consistent with observations that recovery from optic neuritis is poor in neuromyelitis optica spectrum disorder (34). After the first attack of optic neuritis, some authors have proposed a cutoff of 15 µm thinning in the retinal nerve fiber layer versus normal as suggestive of neuromyelitis optica spectrum disorder rather than multiple sclerosis (85). In neuromyelitis optica spectrum disorder, the retinal nerve fiber layer thickness correlates not only with visual acuity but also with the expanded disability scale score (16; 69).
|
• Treatment of neuromyelitis optica spectrum disorder consists of acute treatment of attacks and chronic treatment to prevent further attacks. | |
|
• Four mechanism-based drugs have been FDA approved for AQP4+ neuromyelitis optica spectrum disorder. |
Patients presenting with acute attacks of neuromyelitis optica spectrum disorder require urgent care. Patients with brainstem lesions, especially medullary, and high cervical cord lesions need close monitoring due to potential risk for cardiovascular and breathing complications.
Treatment for neuromyelitis optica spectrum disorder is immediate and also long-term. Acute management of neuromyelitis optica spectrum disorder attack typically involves high dose intravenous steroids. With severe presentations, plasma exchange can be performed. However, the decision to use plasma exchange is individualized and left to the treating physician. An alternative to plasma exchange is intravenous immunoglobulin (IVIG). Some physicians have used IVIG after plasma exchange in more severe cases of neuromyelitis optica spectrum disorder.
|
Methylprednisolone |
1 g intravenously for 5 to 7 days followed by slow prednisone taper (6 to 8 weeks) |
|
Plasma exchange |
Can be given concurrently with methylprednisolone five to seven exchanges every other day of 2 to 3 L over 1 to 2 weeks |
|
Intravenous immunoglobulin |
0.4 g/kg for 5 days or 2g/kg total dose divided over 3 days, as tolerated |
For acute attacks, first-line treatment is methylprednisolone 1 gram intravenously for 5 to 7 days, followed by a slow steroid taper over 6 to 8 weeks. Often, plasma exchange (PLEX) is started at the same time as steroids. Plasma exchange is usually given as five to seven treatments of 55 mL/kg given every other day. Clinical improvement after plasma exchange occurred in four of six patients unresponsive to steroids (110). IVIG has been used as an alternative in acute relapses. Based on their experience, the authors have observed better outcome using PLEX over IVIG for the treatment of acute relapses. A retrospective cohort study based on the registry of the German Neuromyelitis Optica Study Group found that initiation of plasma exchange early in the attack improves clinical outcomes, especially for patients who are anti-AQP4 antibody positive (49). Aggressive treatment at the onset of a relapse, including steroids, plasma exchange, and early initiation of prophylactic therapy, has led to better recovery and lower disability in patients with neuromyelitis optica spectrum disorder.
Historically, the maintenance therapies for neuromyelitis optica spectrum disorder have included chronic immunosuppression with mycophenolate, azathioprine, methotrexate, cyclophosphamide, and mitoxantrone, the latter three agents less commonly used. One common off-label disease-modifying treatment for neuromyelitis optica spectrum disorder is rituximab. A meta-analysis of rituximab data showed relapse freedom in about 63% of patients and adverse events in 16% (26). In a small prospective study (randomized, double-blinded, placebo-controlled) involving 19 patients, 37% of patients on placebo had relapses versus none in those treated with rituximab over 72 weeks (96).
During the period June 2019 to August 2020, three disease-modifying therapies were approved by the FDA for curtailing relapses in patients with neuromyelitis optica spectrum disorder who are positive for the anti-AQP4 antibody: eculizumab (Soliris), inebilizumab (Uplizna), and satralizumab (Enspryng) (approvals in June 2019, June 2020, and August 2020, respectively). Ravulizumab was approved by the FDA in March 2024 for AQP4-positive neuromyelitis optica spectrum disorder.
Eculizumab. Eculizumab is a monoclonal antibody that binds to complement protein C5 and inhibits its cleavage to C5a and C5b products, thereby preventing the formation of the terminal multiprotein complement complex C5b-9, also known as membrane attack complex. Because anti-AQP4 antibody is IgG1 subclass and has a high affinity for complement, inhibition of membrane attack complex formation prevents antibody-induced deposition of the membrane attack complex and astrocyte destruction (103). Eculizumab is administered intravenously at a dose of 900 mg intravenously once a week for 4 weeks, 1200 mg intravenous on week 5, then 1200 mg intravenous every 2 weeks thereafter. In terms of efficacy, eculizumab reduced the number of neuromyelitis optica spectrum disorder relapses by 94% over the 48-week course of the trial. Adjudicated relapses occurred in 3% of patients in the eculizumab group and 43% in the placebo group, with annualized relapse rate of 0.02 in the eculizumab group and 0.35 in the placebo group. The mean change in the EDSS score was -0.18 in the eculizumab group and +0.12 in the placebo group. The infusions were well tolerated. Upper respiratory tract infections and headaches occurred more in the eculizumab than the placebo group. There was one death from pulmonary empyema in the eculizumab group (77).
Prior to starting eculizumab, immunization with meningococcal vaccine is necessary at least 2 weeks prior the infusion. If there is urgency in starting eculizumab, then a 2-week course of appropriate antibiotic may be given while receiving the vaccinations series. The Advisory Committee on Immunization Practices recommends receiving both quadrivalent meningococcal conjugate (MenACWY) and serogroup B (MenB) meningococcal vaccines. Patients taking eculizumab should be given a 2-dose primary series with MenACWY at least 2 months apart and be revaccinated every 5 years. They should also be given MenB with either a 2-dose series of MenB-4C at least 1 month apart or a 3-dose series of MenB-FHbp at 0, 1-2, and 6 months (see package insert for full details).
Between both the original PREVENT trial data and the open-label extension, 137 patients were treated with eculizumab for a median of 133.3 weeks (5.1–276.9 weeks). The open-label extension period did not raise any new or unexpected safety issues. Regarding efficacy at 192 weeks, 94.4% of patients remained relapse free (109).
Ravulizumab. A phase 3 trial of ravulizumab for anti-AQP4-positive neuromyelitis optica spectrum disorder was completed in 2022 (76). Ravulizumab is also a monoclonal antibody that binds C5. Ravulizumab has a longer half-life than eculizumab (8 weeks versus 2 weeks), thus, allowing for extended dosing intervals. Among the ravulizumab-treated patients (n=58), there were no relapses during 84 patient-years of treatment. Compared to the placebo group from the PREVENT trial, ravulizumab reduced relapse risk by 98.6%. The median study time for ravulizumab-treated patients was 73.5 weeks. The safety profile was consistent with eculizumab. Two patients receiving ravulizumab developed meningococcal infections; both recovered with no sequelae. Ravulizumab is used as a weight-based intravenous infusion. The first maintenance dose is administered 15 days after the loading dose, followed by 1 dose every 8 weeks. Because of the risk of serious meningococcal infections, ravulizumab and eculizumab are only available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). The same precautions mentioned above for eculizumab apply also for ravulizumab.
Inebilizumab. Inebilizumab is a monoclonal antibody that binds to the B cell surface antigen CD19, which is expressed on pre-B and mature B lymphocytes and plasmablasts. It causes depletion of B cells and plasmablasts by antibody-dependent cellular cytolysis. It is administered intravenously, with initial dose of 300 mg given 2 weeks apart followed by 300 mg every 24 weeks. In the N-MOmentum trial, inebilizumab reduced the risk of a neuromyelitis optica spectrum disorder relapse in anti-AQP4 antibody positive patients by 77% compared to the placebo group during the 197-day study. In the anti-AQP4-IgG-seropositive subgroup, 11% of subjects receiving inebilizumab had an attack compared to 42% receiving placebo. In the anti-AQP4-positive patients, there was a significant benefit on EDSS with inebilizumab (EDSS worsening from baseline 34% placebo vs. 16% inebilizumab). Infusions were generally well tolerated with pretreatment regimens. Adverse events occurred in 72% of patients receiving inebilizumab and 73% receiving placebo. Serious adverse events occurred in 5% receiving inebilizumab and 9% receiving placebo (13). Prior to initiating inebilizumab, screening for hepatitis B virus, TB, and immunoglobulin levels is recommended. When prescribing inebilizumab, one should be aware of infusion-related reactions, development of hypogammaglobulinemia over time, and potential increased risk of infection – including progressive multifocal leukoencephalopathy and potential reactivation of hepatitis B and tuberculosis (see package insert for full details). Between both the original N-MOmentum trial and the open-label extension, 75 patients were treated with inebilizumab for over 4 years. There were no new or unexpected safety issues. The annualized attack rate was 0.052 attacks per person-year. Interestingly, the attack rate decreased in years 2 to 4 compared to year 1, suggesting enhanced efficacy over time (87).
Satralizumab. Satralizumab is a recombinant humanized antihuman monoclonal antibody against the interleukin 6 (IL-6) receptor. It causes inhibition of IL-6-mediated signaling through binding to soluble and membrane-bound IL-6 receptors. IL-6 is a pleotropic cytokine produced by lymphocytes, macrophages, and endothelial cells. It can act as a potent proinflammatory cytokine and is elevated in acute phase inflammatory responses. It also functions as a potent growth factor for plasmablasts, thereby increasing antibody production (11). Satralizumab is administered subcutaneously (subQ) with a loading dose of 120 mg at weeks 0, 2 and 4, then maintenance dosage of 120 mg subcutaneously every 4 weeks. The effectiveness of satralizumab in patients with neuromyelitis optica spectrum disorder positive for anti-AQP4 was demonstrated in two 96-week clinical trials. The SAkuraStar study (monotherapy trial) showed reduction in the number of neuromyelitis optica spectrum disorder relapses by 74% compared to placebo. Relapses occurred in 30% of patients in the satralizumab group and 50% in the placebo group. The rates of serious adverse events and infections were not different between the two groups (99). The SAkuraSky study (satralizumab added to baseline immunosuppressant therapy azathioprine, mycophenolate mofetil, or corticosteroids) showed reduction in the number of neuromyelitis optica spectrum disorder relapses by 78% compared to placebo. Relapses occurred in 11% of patients in the satralizumab group and 43% in the placebo group. The rates of serious adverse events and infections were not different between the two groups (114).
Prior to starting satrilizumab, screening for hepatitis B virus and TB is recommended. Prior to initiating therapy, assess liver transaminases and serum bilirubin. Monitoring of ALT and AST levels is recommended every 4 weeks for the first 3 months of treatment, followed by every 3 months for 1 year, and thereafter as clinically indicated. Neutrophil counts should be monitored every 4 to 8 weeks after initiation of therapy and thereafter at regular intervals as clinically determined. See package insert for full details.
Open-label extension data for satralizumab are available. Safety data from patients receiving at least one dose of satralizumab included 75 patients from the SAkuraSky double-blind period and open-label extension, as well as 91 patients from the SAkuraStar double-blind period and open-label extension. Median treatment exposure was 4.4 years (0.1 to 7.0) in SAkuraSky and 4.0 years (0.1 to 6.1) in SAkuraStar. The open-label extension period did not raise any new or unexpected safety issues (115). Efficacy data from patients receiving at least one dose of satralizumab were evaluated, which included 49 patients from the SAkuraSky double-blind period and open-label extension as well as 62 patients from the SAkuraStar double-blind period and open-label extension. Median treatment exposure was 4.4 years (0.1 to 7.0) in SAkuraSky and 4.0 (0.1 to 6.0) in SAkuraStar. Of the SAkuraSky group, 71% remained relapse free, and 91% remained free of severe relapse. Of the SAkuraStar group, 73% remained relapse free, and 90% remained free of severe relapse. The annualized relapse rate was 0.12 in SAkuraSky and 0.08 in SAkuraStar. Long-term data demonstrate continued efficacy over time (50).
|
Eculizumab* |
900 mg intravenously once a week for 4 weeks, 1200 mg intravenous on week 5, then 1200 mg intravenous every 2 weeks |
|
Ravulizumab* |
Weight-based dosing; loading dose on day 1 then first maintenance dose on day 15, then maintenance dose every 8 weeks. |
|
Inebilizumab* |
300 mg given 2 weeks apart followed by 300 mg every 24 weeks |
|
Satralizumab* |
120 mg given subcutaneously at weeks 0, 2 and 4, then maintenance dosage of 120 mg subcutaneously every 4 weeks |
|
Rituximab |
loading dose of 500 mg to 1000 mg intravenous 2 weeks apart followed by 500 to 1000 mg intravenous Q 4 to 6 months, depending on the B cell recovery. Goal B cell count while on treatment is 0. |
|
Mycophenolate mofetil |
1.5 to 2 g daily, divided twice daily |
|
Azathioprine |
2 mg/kg divided twice daily (test patients for the enzyme thiopurine methyltransferase-TPMT- activity prior to starting therapy) and reduce dose if needed. |
|
|
Disease-modifying therapies for multiple sclerosis may be ineffective or even worsen neuromyelitis optica, making an accurate diagnosis critical (102; 34). Measurement of serum interferon I levels in patients with relapsing-remitting multiple sclerosis, neuromyelitis optica spectrum disorder, systemic lupus erythematosus, and healthy controls shows that compared to patients with relapsing-remitting multiple sclerosis, patients with neuromyelitis optica spectrum disorder have elevated type I interferon levels and increased in vitro responses of immune cells to IFN-beta stimulation (20). This may explain why treatment with interferons is not beneficial in patients with neuromyelitis optica. Other multiple sclerosis treatments that are associated with worsening of neuromyelitis optica spectrum disorder include glatiramer acetate, fingolimod, natalizumab, and alemtuzumab (48).
As neuromyelitis optica spectrum disorder is an entity that disproportionately affects women, pregnancy is a central factor to be considered during treatment. There is no conclusive evidence that neuromyelitis optica spectrum disorder influences fertility. However, the rate of miscarriage is increased in patients with neuromyelitis optica spectrum disorder, especially during the first trimester. Eclampsia also occurs at a higher rate in patients with neuromyelitis optica spectrum disorder, and comorbid autoimmune disease is an additional risk factor for eclampsia (72). A retrospective review of pregnant patients with neuromyelitis optica spectrum disorder suggests that there may be an increased relapse rate after delivery (47). Unlike multiple sclerosis, neuromyelitis optica spectrum disorder remains active during pregnancy.
For treatment of an acute relapse during pregnancy, steroids, plasmapheresis, or IVIG are considered relatively safe options. In cases of aggressive disease, immunomodulatory therapy may be considered throughout the pregnancy, such as azathioprine. In 2017, Shosha and colleagues published a retrospective analysis of the pregnancy literature in other autoimmune conditions and made recommendations about the use of certain immunosuppressant agents in pregnant patients with neuromyelitis optica spectrum disorder (92). In infrequent cases, the benefit of receiving these medications may outweigh the risk to the fetus. Both azathioprine and rituximab have been administered during pregnancy for other autoimmune conditions. Women with neuromyelitis optica spectrum disorder should be counseled to plan pregnancy so that their treatment can be optimized. One systematic review and case series investigated 102 pregnancies where rituximab was used within 6 months of conception in patients with multiple sclerosis and neuromyelitis optica spectrum disorder. The case series looked at 11 pregnancies (seven multiple sclerosis and three neuromyelitis optica spectrum disorder). Despite the limitations from the small sample size, rituximab appeared safe when used within 6 months of conception (14). A personalized approach to the treatment of neuromyelitis optica spectrum disorder during pregnancy is recommended.
Most maternal IgG transfer to the fetus happens in the second and third trimester. Thus, direct effects of anti-CD20 or anti-CD19 agents are most pronounced in the second and third trimesters of pregnancy. The elimination half-life of rituximab is about 18 to 32 days and the anti-CD19 agent inebilizumab about 18 days. Hence, by the second trimester, there is little if any rituximab or inebilizumab remaining in serum. Therefore, if conception is desired, it would be reasonable to wait 3 months after the last dose of anti-CD20 or anti-CD19 medication to pursue conception, and if conception is successful, to halt any further dosing until delivery. Because the pharmacodynamic effects of these agents are long-lived, more than 6 months, there is some protection against relapses offered by the B-cell depleting agents if used in this manner.
Because some patients with neuromyelitis optica spectrum disorder have autonomic dysfunction or high cervical cord and brainstem lesions, they require close monitoring.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Josephe Archie Honorat MD PhD
Dr. Honorat of the University of Chicago has no relevant financial relationships to disclose.
See ProfileAdil Javed MD PhD
Dr. Javed of the University of Chicago has no relevant financial relationships to disclose.
See Profile
Anthony T Reder MD
Dr. Reder of the University of Chicago received honorariums from Genentech, Genzyme, and TG Therapeutics for service on advisory boards and as a consultant and stock options from NKMax America for advisory work and an unrestricted lab research grant from BMS.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Child Neurology
May. 12, 2026
General Child Neurology
Apr. 24, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026