Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The authors provide an updated clinical review of Horner syndrome. The most current recommendations regarding pharmacologic diagnosis and radiographic evaluation are highlighted. Important issues regarding the evaluation of Horner syndrome in children are also reviewed.
|
• Horner syndrome is caused by an interruption of the oculosympathetic pathway that extends from the hypothalamus through the brainstem, upper spinal cord, paraspinal region, chest, neck, skull base, and orbit. | |
|
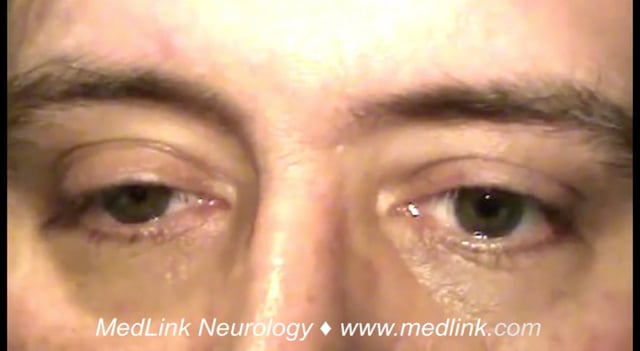
• Ipsilateral ptosis and miosis are the principal clinical manifestations, but both abnormalities are minimal and often overlooked; anhidrosis is difficult to assess. | |
|
• When the cause is not obvious, pharmacologic testing with topical apraclonidine 0.5% should be performed to confirm the diagnosis of Horner syndrome, as there are other causes of ptosis and miosis. | |
|
• Topical cocaine 10% is used as a diagnostic test in preference to apraclonidine in children aged 2 years or younger because apraclonidine may cause cardiopulmonary side effects, but the diagnostic endpoint is not as clear as for apraclonidine. | |
|
• Topical hydroxyamphetamine, formerly used to localize Horner syndrome, is no longer recommended. | |
|
• Common causes of Horner syndrome are neck, chest, and upper spinal procedures, trauma, and tumors, as well as carotid dissection and cluster headache. | |
|
• In isolated Horner syndrome without preceding trauma or procedures, the cause will remain indeterminate in up to 70% of cases even after adequate imaging evaluation. | |
|
• When the cause of Horner syndrome is clinically obvious—after internal jugular vein cannulation, neck, chest, or spinal surgery, or a known cavernous sinus lesion—imaging is not necessary. | |
|
• When there are localizing clues to a Horner syndrome, imaging can be targeted to that location. | |
|
• When there are no localizing clues, MRI/MRA or CT/CTA should scan from the skull base to the mid-thoracic region. |
Horner syndrome consists of the clinical triad of ptosis, miosis, and anhidrosis. It is caused by interruption of the ipsilateral oculosympathetic pathway. That pathway courses from the hypothalamus through the brainstem, upper spinal cord, paraspinal region, chest, neck, and skull base and orbits to reach the Müller muscle elevator of the upper lid and the dilator muscle of the iris.
The syndrome is named after a Swiss ophthalmologist named Johann Friedrich Horner, but he was not the first to describe it. In 1727, Pourfour du Petit reported ptosis, miosis, and enophthalmos after cutting the sympathetic nerve trunk in dogs (47). Edward Selleck Hare reported similar signs in a published letter to the London Medical Gazette in 1838 (22; 15).
Rabbit experiments in 1852 by the French physiologist Claude Bernard led to the connection between the cervical sympathetic fibers and Horner syndrome (45; 48), so that the French typically call it the “Bernard-Horner syndrome.”
In 1864, Mitchell described ipsilateral ptosis, miosis, and reduced facial sweating in an injury of the sympathetic trunk from a gunshot wound to the neck. In 1869, Horner published a report of the more complete syndrome, which included ptosis, miosis, enophthalmos, increased skin temperature, and dryness of the ipsilateral face; the patient had poor dilation of the affected pupil following instillation of atropine and preserved pupillary constriction to the parasympathomimetic agent calabar (23).
Ptosis. The ptosis of the upper lid is caused by impaired innervation of Müller muscle, a small band of sympathetically innervated smooth muscle originating on the underside of the levator palpebrae and inserting on the tarsal plate of the upper lid. Ptosis in Horner syndrome ranges from 0.5 to 4 mm, with a mean of 1.6 mm (50). In fact, ptosis may rarely be absent (36). In some cases, denervation of the lower lid retractors causes mild elevation of the lower lid margin so that it covers the lower border of the cornea, a phenomenon called “upside-down ptosis” (43).
Miosis. The affected pupil is smaller than the unaffected pupil, owing to denervation of the iris dilator, a radially oriented smooth muscle. Because the iris dilator muscle is relatively weak, anisocoria is minimal, ranging between 0.5 and 4.5 mm, with a mean of 1.7 mm (50). Both pupils constrict normally to light, so that the anisocoria essentially disappears in bright light. Thus, detection of the minimal anisocoria is best done in the least amount of light. Rarely, anisocoria may be utterly absent (54; 50) or occur intermittently (33; 40; 42).
In Horner syndrome, the miotic pupil dilates slowly when light is withdrawn (“dilation lag”), a phenomenon that can be observed clinically by quickly extinguishing room light. The degree of anisocoria should be greatest at 5 seconds after the light is withdrawn (29). However, this phenomenon is difficult to document without pupillography (09). When ptosis is minimal or absent, unequivocal pupil dilation lag favors a diagnosis of Horner syndrome over physiologic anisocoria, but pharmacologic confirmation of the diagnosis is essential.
Anhidrosis. Interruption of sympathetic fibers to the head and neck leads to loss of ipsilateral vasomotor and sudomotor function, impairing sweating and causing increased temperature and flushing of those regions. If the first-order or second-order neuron is interrupted, these signs will extend from the brow to the clavicle. If the third-order neuron is interrupted, these signs will be limited to a small patch on the medial brow. Vasomotor dysfunction is often inapparent in chronic lesions, as circulating adrenergic agents stimulate vasoconstriction. Anhidrosis is not commonly reported by the patient but can be assessed by using alizarin powder (65).
Iris heterochromia. Iris melanocytes require sympathetic innervation during prenatal development and infancy. Loss of that innervation leads to depigmentation of the ipsilateral iris (“hypochromia”) (25; 29). Hypochromia may also rarely develop in acquired Horner syndrome (12).
The ptosis and anisocoria of Horner syndrome are typically persistent, even if the cause of the Horner syndrome is relieved. The anisocoria causes no symptoms. The ptosis may cause impairment of vision and a cosmetic blemish. It can be relieved with topical instillation of apraclonidine or by a minor surgical procedure.
A 60-year-old woman presented with right eyelid droop of one day’s duration and mild new right facial pain. Right upper lid ptosis of 1.5 mm was present. In dim illumination, pupils measured 3 mm on the right and 4.5 mm on the left. Both pupils constricted briskly to light. The rest of the ophthalmic and neurologic examination was normal. Thirty minutes after binocular instillation of apraclonidine 0.5%, the ptosis had disappeared and the anisocoria had reversed (right pupil enlarged to 6 mm and left pupil remained at 4.5 mm in dim light), confirming a right Horner syndrome. MRI and MRA targeting the neck vessels disclosed a crescent-shaped precontrast T1 high intensity signal in the wall of the right cervical internal carotid artery immediately under the skull base indicative of methemoglobin, a blood breakdown product. The MRA showed narrowing of the lumen in that region. Based on a diagnosis of acute carotid dissection, she was treated with aspirin to prevent stroke. Six months later, she still had the Horner syndrome but no other neurologic manifestations. MRA showed recanalization of the dissected region. The aspirin was discontinued. For the lingering ptosis, she was successfully treated with topical apraclonidine once daily.
The cause of Horner syndrome depends on whether the first-order, second-order, or third-order neuron is affected (Table 1). The relative prevalence of each cause depends on the patient’s age, accrual circumstances, whether the Horner syndrome is the only pertinent abnormality (“isolated Horner syndrome”), and whether the Horner syndrome has been confirmed with pharmacologic testing (18; 36; 03; 10; 26; 05; 51; 50).
First-order (central) Horner syndrome. First-order neurons arise in the hypothalamus, descending through the brainstem in an uncertain location until they reach the medulla, where they are identifiable in the dorsolateral region and subject to damage in vertebral artery occlusion which leads to infarction of the dorsolateral medulla (Wallenberg syndrome) (36). In the C8-T1 segment of the spinal cord, the neurons synapse in the intermediolateral gray column (ciliospinal center of Budge-Waller).
First-order (central) Horner syndrome rarely occurs in isolation. It may rarely occur with hypothalamic lesions, but never in isolation (13; 58; 52). Horner syndrome is also very rare in lesions in the midbrain and pons, but the combination of Horner syndrome and contralateral trochlear nerve palsy localizes to the dorsal midbrain ipsilateral to the Horner syndrome (20). Horner syndrome is a common presence in dorsolateral medullary stroke (Wallenberg syndrome) but is often overlooked among the more prominent and disabling manifestations. However, in the setting of acute ataxia, nystagmus, skew deviation, swallowing difficulty, intractable singultus (hiccuping), and ipsilateral facial and contralateral extremity hypesthesia, the identification of Horner syndrome allows for definitive localization.
Lesions of the spinal cord caused by trauma (including surgery), demyelination, stroke, and tumors have been associated with Horner syndrome (46; 08). When Horner syndrome alternates from side to side, demyelinating, medullary, or cervical spinal cord lesions or systemic dysautonomias may be responsible (16; 67; 01).
Second-order (preganglionic) Horner syndrome. After exiting the spinal cord at C8-T1, the second-order neurons travel along the paraspinal sympathetic chain, over the pulmonary apex and into the neck. Synapse occurs in the superior cervical ganglion, which is located at the angle of the jaw.
Second-order (preganglionic) Horner syndrome often results from trauma or surgery to the paraspinal region, chest, and especially inferior jugular vein cannulation. Neuroblastoma, lymphoma, lung cancer, neurofibroma, and thyroid tumors are uncommon but important causes (44; 50).
Third-order (postganglionic) Horner syndrome. After synapsing in the superior cervical ganglion, the sudomotor and vasomotor innervation of the face travels with the external carotid artery. The fibers destined for the orbit travel in the posterior portion of the internal carotid artery sheath, entering the skull base, passing near the tympanic promontory and through the carotid canal to the cavernous sinus in close proximity to the carotid artery. In the superior orbital fissure, these third-order neurons jump onto the nasociliary branch of the first trigeminal division to enter the orbit. Two or three long ciliary nerves arise from the nasociliary nerve and travel with the lateral and medial suprachoroidal vascular bundles to reach the iris dilator. Other ciliary branches convey sympathetic innervation to Müller muscle and retractors of the lower lid.
Disorders of the third-order oculosympathetic pathway affect the high cervical neck region (internal carotid dissection, internal jugular venous cannulation, neck, dental, oropharyngeal, and parapharyngeal trauma and infections), middle ear (infections), carotid canal (aneurysms, trauma), and cavernous sinus (aneurysms, neoplasms, inflammations) (37). Horner syndrome is the most common—and often the only—sign of carotid dissection, occurring in 38.5% to 44% of patients (07; 33; 53; 42; 19; 34). Horner syndrome is reported in up to 10% of patients diagnosed with trigeminal autonomic cephalgias, including cluster headache. The mechanism of Horner syndrome in these trigeminal autonomic cephalgias is unknown. Horner syndrome should not, however, be attributed to a trigeminal autonomic cephalgia unless a structural cause has been ruled out.
|
First-order neuron |
Second-order neuron |
Third-order neuron |
|
Hypothalamus |
Paraspinal |
Internal carotid artery |
|
• stroke |
• trauma |
• dissection |
|
Brainstem |
Chest |
Skull base |
|
• dorsolateral medullary infarction |
• tumor |
• aneurysm |
|
Spinal cord |
Neck |
Cavernous sinus |
|
• demyelination |
• tumor |
• aneurysm |
|
Nonlocalizing | ||
|
• cluster headache |
Special consideration--infants. Although many of the causes of acquired Horner syndrome in children are similar to those in adults, most have no identifiable etiology (35).
Birth trauma causes Horner syndrome by injury to the lower trunk of the brachial plexus and to the carotid artery during forceps delivery (64). Agenesis of the carotid artery and orbital hemangioma are causes of congenital Horner syndrome (49; 14; 31; 62).
Isolated Horner syndrome is the presentation of 2% of neuroblastoma cases. The tumor compresses the sympathetic fibers in the neck and thorax (41). In suspected neuroblastoma, imaging should extend to the abdomen, where the more visible tumors may be identified (41; 17).
When Horner syndrome is discovered in hospitalized adult patients, the predominant causes will be neck, chest, and paraspinal tumors, trauma, and procedures, such as internal jugular venous cannulation (18; 50). Although dorsolateral medullary infarction is a common cause of Horner syndrome, it is never an isolated sign. In fact, it is often overlooked because it is a minor clinical component.
The site and cause of Horner syndrome in patients is likely to be obvious at the time of discovery of Horner syndrome, so that pharmacologic confirmation is not necessary (50). On the other hand, Horner syndrome diagnosed in outpatients is apt to be an isolated abnormality of uncertain site and cause. Pharmacologic confirmation is necessary. The cause often remains indeterminate, although carotid dissection, cluster headache, and neck and chest tumors must be considered (50).
A robust claims database study of patients with Horner syndrome diagnosed between 2009 and 2018 in Korea disclosed 1331 cases in adults and 139 cases in children (21). The annual incidence in adults was 0.39 out of 100,000; in children, it was 0.20 out of 100,000. Peak age at diagnosis in adults was between ages 50 and 54; in children, it was before age 4. No cause was found in 43% of cases. Where a cause was found, it had been evident in 92% of cases by the time the Horner syndrome was recognized. Prominent among the causes identified after the diagnosis of Horner syndrome were neuroblastic tumors in children and thyroid tumors in adults. In an epidemiologic study of Horner syndrome in 20 children living in Olmsted County, Minnesota, the age-adjusted and sex-adjusted incidence was 1.42 per 100,000 (55). Eleven cases were caused by birth trauma, a birth prevalence of 1 in 6250 (95% CI 3333 to 10,000). There were no cases of neuroblastoma.
There is no primary prevention.
The clinical evaluation of suspected Horner syndrome depends on careful examination of ptosis and anisocoria, which are often mild and can have separate causes. Ptosis may be caused by myogenic disorders (previous trauma or inflammation, myotonic dystrophy, chronic progressive external ophthalmoplegia, senescent levator dehiscence), neuromuscular junction disorders (myasthenia gravis, botulism), and third nerve palsy. Exclusion of third nerve palsy is especially critical. It is based on finding pertinent ocular duction deficits, iris sphincter dysfunction, and diplopia. A common pitfall is to mistake ptosis for “pseudo-ptosis,” which can be caused by enophthalmos, dermatochalasis, contralateral lid retraction, hypertropia, blepharospasm, and postparetic facial contracture. Anisocoria may result from instillation of over-the-counter autonomically active agents, iris dysplasia, uveitis, or eye trauma, including intraocular surgery.
Anisocoria should be assessed in dim lighting, observing the size of the pupils by illuminating the patient’s eyes from below, in order to avoid stimulating the pupil response to a nearby stimulus. A bright light is then shined into each pupil from below. If both pupils constrict normally to light, the patient could have physiologic (“benign,” “essential”) anisocoria, which is far more common than Horner syndrome. It occurs in 20% of the population (29), and the smaller pupil often switches from one side to the other over time. As with Horner syndrome, physiologic anisocoria is minimal--never greater than 1.5 mm. It can be mimicked by exposure to topical medications with sympathomimetic or parasympathomimetic properties, whether deliberate or accidental. In sympathomimetic-induced anisocoria, pupil constriction to bright light is usually preserved. The presence of dilation lag (see “dilation lag,” above) distinguishes Horner syndrome from physiologic anisocoria but is difficult to elicit. A more reliable way to distinguish between physiologic anisocoria and Horner syndrome is with pharmacologic testing (see below).
If anisocoria is present in dim illumination and one of the pupils does not constrict briskly to light, you must reject a diagnosis of Horner syndrome. If the larger pupil does not constrict normally to direct light, you must first rule out a third nerve palsy by performing the cover test in search of (often subtle) ocular misalignment. If the anisocoria is the only pertinent feature, the differential diagnosis includes Adie tonic pupil, topical exposure to adrenergic or cholinergic substances, iris trauma including intraocular surgery, and congenital iris dysplasia. (See the MedLink article on pupillary abnormalities for further discussion.) If ptosis is the only pertinent feature, the differential diagnosis includes myasthenia gravis, chronic contact lens wear, an orbital roof lesion, old orbito-ocular trauma, and a congenital levator dystrophy. Bilateral ptosis should trigger the consideration of a mitochondrial disorder.
Examination of old photographs is helpful in determining the chronicity of ptosis and anisocoria.
Pharmacologic testing. Because the two common components of Horner syndrome—ptosis and anisocoria—can each have separate causes (see previous section), diagnosis should generally be confirmed with pharmacologic testing using apraclonidine 0.5% in patients older than 2 years and cocaine 10% in those under that age (60). Such pharmacologic confirmation can be omitted when the site and cause of Horner syndrome are obvious.
Apraclonidine. Apraclonidine is an alpha-2 receptor agonist with some weak alpha-1 activity. Disruption of the sympathetic pathway at any level causes upregulation (“denervation supersensitivity”) of the postsynaptic alpha-1 receptors on the iris dilator (38). This phenomenon leads to reversal of the anisocoria following instillation of apraclonidine. The Horner pupil becomes the larger pupil. The upper lid of the involved eye will also elevate, but that phenomenon also occurs in normal subjects.
Instill a single drop of apraclonidine 0.5% in each eye. After waiting 30 to 60 minutes, observe the pupils for reversal of the anisocoria, which is specific for Horner syndrome. This has been reported to occur as early as 24 hours after onset of lesion, but extensive documentation of this latency is still lacking (52). False negatives are rare (30). Apraclonidine testing should not be performed in children under 2 years of age, as it can precipitate an acute dysautonomia (63; 39).
Cocaine. Cocaine blocks the reuptake of norepinephrine in the presynaptic sympathetic nerve terminal, leading to a relative increase in norepinephrine available to the postsynaptic receptors located on the iris dilator. Topical instillation leads to mydriasis if the oculosympathetic pathway is intact. If the sympathetic pathway is disrupted, there is a decrease in the amount of norepinephrine released into the synapse, blunting its mydriatic effect. Thus, the affected pupil in Horner syndrome will not dilate to the same degree as the contralateral normal pupil.
Cocaine testing for Horner syndrome is reserved for children under 2 years of age. For others, testing is inaccurate and impractical, as cocaine drops are hard to obtain, expensive, and must be secured in a locked cabinet (27).
Instill a single drop of 10% cocaine solution into each eye. After 1 to 5 minutes, instill a second drop into each eye. If neither pupil dilates after 40 minutes, repeat the procedure or consider that the cocaine drops may have expired. The cocaine test is positive for Horner syndrome if the miotic pupil remains smaller than the normal pupil by at least 1 mm (28). Patients should be counseled that a urine drug screen may be positive for at least 2 days after cocaine testing (24), but that there are no psychoactive effects.
Hydroxyamphetamine. Instillation of this agent was formerly used to distinguish postganglionic from preganglionic and central causes of Horner syndrome. Its reliability was always poor, and it is no longer available.
Imaging evaluation. Imaging can be omitted if the location and cause are obvious. Otherwise, imaging should be aimed at sites identified by localizing features. If there are no localizing features, imaging must cover a span from the angle of the jaw to the upper thoracic spine (03; 61; 10; 50).
Targeted imaging. Use the following constellation of clinical features, as a guide to directed imaging:
|
• Somnolence, weakness, sensory loss, or autonomic dysregulation: hypothalamus | |
|
• Ataxia, nystagmus, intractable singultus, or swallowing difficulty: medulla and cerebellum | |
|
• Myelopathy: cervical spine | |
|
• Arm pain, weakness, or numbness: brachial plexus | |
|
• Acute unilateral headache, face or neck pain: cervical region | |
|
• Hearing loss or ear pain: temporal bone (57) | |
|
• Sixth nerve palsy, third nerve palsy, or trigeminal dysfunction: cavernous sinus |
Nontargeted imaging. When Horner syndrome is isolated (the only pertinent neurologic abnormality), the lesion is unlikely to be in the brain, cavernous sinus, or orbit. It could lie within the cervical spinal cord, paraspinal region, chest, or neck. Therefore, imaging must cover the region between the skull base and the T1 spinal level. Either MRI/MRA and CT/CTA will provide adequate resolution of the soft tissues in the neck and excellent vascular imaging. In carotid dissection, CTA may show luminal narrowing and MRI may show mural hemorrhage. However, MRI is easily degraded by motion and may be difficult to obtain urgently (61).
A history of birth trauma or the finding of heterochromia does not negate the need for imaging in children. Isolated Horner syndrome in a child, even if noted at birth, should be evaluated with imaging (66; 61; 04) because it may be caused by neuroblastoma (41; 17). Normal urine catecholamines are useful in ruling out neuroblastoma, but a low tumor burden may account for the mere 60% to 70% sensitivity of this test (32; 59; 56).
The principal identifiable causes of isolated Horner syndrome are: (1) neck and chest tumors, trauma, or surgery; (2) internal jugular vein cannulation; (3) acute carotid dissection; or (4) the cluster variant of trigeminal autonomic cephalalgia. Those causes will often be known at the time of discovery of Horner syndrome (50).
Imaging is urgent if ipsilateral acute face or neck pain is present because carotid dissection is a stroke-prone state within the first few weeks of onset (06; 02). One-third of strokes occur in the first 24 hours (11) and 88% within the first 7 days of symptom onset (06). In chronic isolated Horner syndrome in adults, imaging is often negative (05; 04) but must be performed to rule out tumors (50). In Horner syndrome of uncertain duration, imaging is dedicated to ruling out carotid dissection and neck and chest lesions. Even after proper imaging, over 50% of isolated Horner syndrome cases will remain of idiopathic origin (36; 51; 50).
Diagnosis of Horner syndrome is out acute carotid dissection, where aspirin is believed to be protective against stroke. Early diagnosis of neuroblastoma and other neck, chest, and paraspinal tumors is also beneficial. For patients bothered by the appearance of ptosis, apraclonidine 0.5% or surgery is helpful in palliation of ptosis.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026