Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Porencephaly, an encephaloclastic brain lesion, is one of the most frequent causes of infantile cerebral palsy. This lesion results mainly from ischemic insults occurring in late pregnancy or in early postnatal life or from intrauterine exposure to infections and toxins. The identification of mutations in the COL4A1 and COL4A2 genes, which encode the type IV alpha1 and alpha2 collagen chains, has demonstrated that a subset of porencephaly may be genetically determined. In this article, the author reviews the clinical features and the pathogenetic aspects of porencephaly and provides an update on the pathophysiology of this disorder. Moreover, the author summarizes findings on the role of mutations in COL4A genes in determining small vessel disease and brain malformations. The phenotypic spectrum of mutations of the COL4A genes, which besides cerebrovascular damage may include systemic involvement with ocular, renal, muscular, and cardiac features, is progressively widening. COL4A1/2 gene mutations, originally reported to be associated with familial porencephaly, are in fact increasingly recognized as causative genes of an array of brain malformations, including schizencephaly, polymicrogyria, and subependymal heterotopia, as well as of minor, nonspecific brain abnormalities.
|
• Porencephalies are focal cavitary lesions frequently communicating with the lateral ventricles; they result from brain destruction during late fetal or early postnatal developmental stages. | |
|
• The more common etiologic factors are ischemic and hemorrhagic insults, but toxic agents, infections, trauma, and drugs have also been reported. | |
|
• Porencephaly may also be due to dominant mutations (either inherited and de novo) of genes COL4A1 and COL4A2 encoding for type IV collagen alpha-1 and type IV collagen alpha-2, respectively. The type IV collagen, which is a component of nonfibrillary collagen, is a main constituent of the basement membrane of many tissues, among them vascular endothelia. | |
|
• The clinical expression of the porencephaly depends on its site, its size, and the timing of the causative insult. Clinical symptoms include motor deficits, intellectual impairment, and seizures. | |
|
• The management of porencephaly is that of the associated clinical manifestations: cerebral palsy, intellectual impairment, and seizures. Management includes rehabilitation and antiepileptic treatment. Patients with drug-resistant epilepsy may benefit from resective surgery, which may consist of functional hemispherectomy in patients with large, hemispheric cysts. | |
|
• Genetic counseling is appropriate in familial and genetically determined porencephaly. Special attention in genetic counseling must be paid to families carrying COL4A mutations, given the variable phenotypic expression and penetrance. |
The term "porencephaly" (from the Latin word porus, meaning "communication") was first introduced by Heschl in 1859 to describe clefts or cavities in the cerebral mantle (20). He attributed these lesions to the destruction of the developing brain. Since Heschl's first description, the term "porencephaly" has been widely used to describe etiologically different brain cavities communicating with either the subarachnoid space or the lateral ventricles.
Most authors recognize two major subgroups: (1) developmental or true porencephaly, and (2) congenital or encephaloclastic porencephaly. The former refers to cases characterized by full-thickness defects of the cortical mantle associated with polymicrogyria or heterotopic gray matter. For these cases, the term "schizencephaly" is preferred. The second subgroup refers to lesions arising from cortical destruction in the developing brain. Reports of COL4A1 and COL4A2 mutations, both in porencephaly and schizencephaly, suggest that a common genetic mechanism may be responsible for brain damage at least in a subset of patients (50; 51; 06). Nevertheless, from a clinical perspective, schizencephaly and porencephaly must be differentiated because the MRI pictures, the clinical and epileptic phenotypes, and the outcome have distinctive features.
The clinical expression of the encephaloclastic porencephaly depends on its site, its size, and the timing of the causative insult. Clinical findings are intellectual impairment, seizures, and various forms of cerebral palsy, including spastic hemiparesis, diplegia, and quadriplegia.
Pregnancy is usually uneventful, although in severe cases, decrease in fetal movements may be reported by the mother or detected by ultrasonography. In few cases, the delivery may be complicated, leading to the wrong diagnosis of perinatal hypoxic-ischemic damage. The porencephalic cyst that follows intraventricular hemorrhage in the preterm infant manifests at birth with nonspecific symptoms, such as decreased alertness, irritability, irregular respiratory rhythm, hypotonia, and seizures. The long-term clinical symptoms are hemiplegia or asymmetric diplegia with greater involvement of the leg, and cognitive difficulties (45). Term infants may exhibit no symptoms or signs at birth. When symptoms manifest, they are represented in most cases by focal motor seizures, or slight hemiparesis, contralateral to the involved side. The long-term clinical outcome again depends on the site and size of porencephaly. Children with mild lesions may have improvement or even disappearance of motor deficit by the age of 2 years. Children with more extended lesions may develop hemiplegic or tetraplegic cerebral palsy. Intellectual impairment is present in about one third of unilaterally affected children and in most patients with bilateral involvement (45; 41).
Epilepsy may manifest as West syndrome in the first year of life or as partial epilepsy later in childhood (43; 52; 42). Infantile spasms are usually controlled by steroid therapy, but partial seizures can subsequently appear, particularly in infants with frontal lobe involvement. In childhood, epilepsy occurs in about one third of term infants with focal brain damage, whereas it is uncommon in preterm children with intraventricular hemorrhage (45). In most patients, epilepsy is characterized by partial seizures whose semeiology is consistent with the epileptogenic area located close to porencephaly. Epilepsy usually starts during childhood; only a few patients with seizure onset during adulthood have been reported (34). A minority of patients with circumscribed MRI lesion may show a more diffuse epileptogenicity (related to widespread brain damage or resulting from secondary bilateral synchrony of frontal foci) with tonic and atonic seizures.
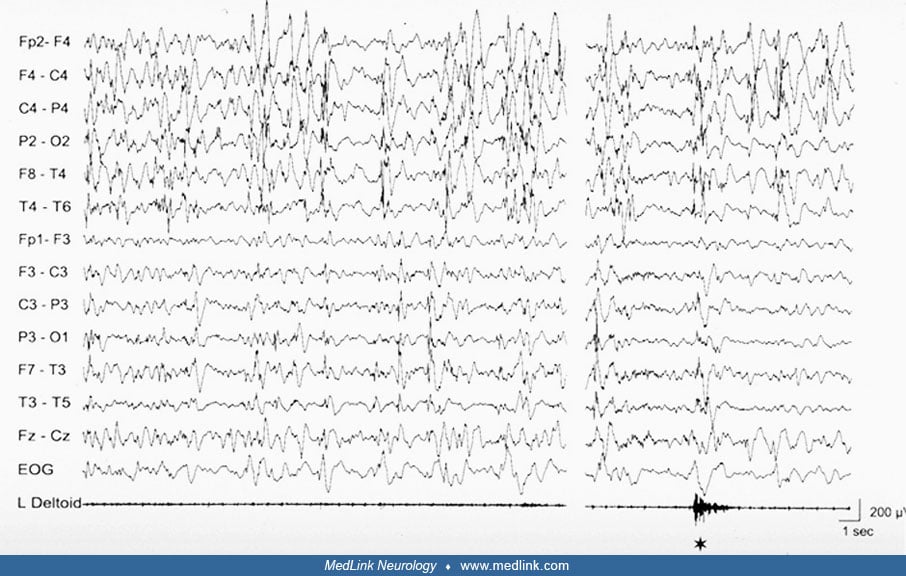
A peculiar epileptic syndrome frequently present in patients with porencephaly, likewise in patients with hemiplegic cerebral palsy resulting from congenital (or acquired early in life) unilateral brain damage is startle epilepsy (05). In startle epilepsy, sudden tonic contraction of the paretic limbs are induced by auditory, or less frequently, somatosensory stimuli. The reflex seizures are brief, lasting less than 30 seconds, but are usually frequent and may cause falls to the ground, thus impairing everyday life. The ictal EEG of reflex seizures is characterized by a vertex high voltage discharge followed by diffuse flattening or low amplitude 6 to 11 Hz activity. In most cases, startle epilepsy is refractory to antiepileptic drugs. Sporadic unprovoked seizures, mostly of focal type, are associated and usually precede the onset of startle epilepsy.
Prognosis depends on the severity of the associated static encephalopathy or seizure disorder. The general prognostic criteria for motor and mental functions are those of cerebral palsy (45). Severe and long-lasting symptoms in the first days of life and difficult-to-treat neonatal seizures are unfavorable prognostic factors. EEG recordings, CT, and cranial ultrasonographies may contribute as early prognostic factors. Occurrence and prognosis of epilepsy is related to the site of clastic damage. In preterm infants with porencephaly following periventricular hemorrhage, and in term infants with circumscribed lesions involving the rolandic or parieto-occipital regions, spasms and later focal seizures are easily controlled by treatment. A less favorable course is common in children with extensive porencephaly and cerebral atrophy, particularly when the frontal lobes are involved.
In patients with COL4A1 or COL4A2, the possibility of spontaneous intracerebral hemorrhages and strokes in childhood and adulthood must be considered (19).
The boy was born by Cesarean section at 30 weeks' gestation for acute fetal distress due to abruptio placentae. Weight at birth was 1215 g. The Apgar scores were 2 and 5 after 1 and 5 minutes, respectively. The infant had anemia, transient renal failure, hemorrhagic colitis, and respiratory distress requiring assisted ventilation. Transcranial ultrasonography at birth showed a right periventricular hyperechogenic area. Serial examinations in the following weeks demonstrated posterior enlargement of the lateral ventricles and cystic cavitation communicating with the right lateral ventricle. The presence of porencephaly was later confirmed by MRI.
Neonatal EEG recordings were consistently characterized by focal slow and sharp waves over the right hemisphere. The newborn was hyperexcitable, and a mild left hemiparesis was evident.
General condition progressively improved, and the infant was discharged from neonatal care at 2 months of age. Spastic tetraparesis, more severe on the left side, then became evident, associated with motor and cognitive delay. At the corrected age of 6 months, infantile spasms occurred. The EEG recordings showed right hemihypsarrhythmia.
Steroid treatment led to disappearance of spasms and hypsarrhythmia. No seizures occurred in the follow-up period. By the age of 4 years, spastic tetraparesis was still evident, but motor and cognitive skills were slowly improving.
Porencephalies are due to encephaloclastic brain lesions occurring from the late second trimester of gestation to the first postnatal days or weeks (39). Porencephaly is not due to a single etiology, and various factors, both genetic and acquired, contribute to its occurrence. Porencephalies have been reported in association with maternal exposure to toxins, infections, and drugs.
Intraventricular hemorrhage in the preterm infant may obstruct the terminal vein in the subependymal region, leading to hemorrhagic infarction and formation of porencephalic cysts. In the twin embolization syndrome, embolic phenomena or thromboplastin release from the deceased to the surviving twin through the monochorionic placenta may result in porencephaly (35). Porencephaly has also been rarely reported in association with developmental failure of cerebral venous sinuses and linear sebaceous nevi, pyknodysostosis, neonatal intravascular transfusion for Rhesus alloimmunization, alloimmune thrombocytopenic purpura, deficiencies in the protein C anticoagulant pathway, and factor V Leiden mutation. The role of coagulation abnormalities is supported by the high prevalence of prothrombotic risk factors, either isolated or combined, observed in patients with porencephaly. A case-control study has demonstrated a significantly higher prevalence of FV G1691A mutation (factor V Leiden mutation) and of multiple prothrombotic defects in a large series of patients with porencephaly compared to controls (08). The presence of at least one prothrombotic abnormality has also been reported in 60% of children with perinatal stroke (26).
Genetics. A possible genetic origin of a subset of porencephaly had been suggested by the report of a few familial cases and then confirmed by the identification of a locus for familial porencephaly on chromosome 13qter. In 2005, Gould and coworkers, using a mouse model generated by random mutagenesis, characterized a novel mutant mouse with severe perinatal cerebral hemorrhage resulting in porencephaly (15). The genetic abnormality was found to reside in a defect in collagen IVA1 (COL4A1), a major component of the vascular basement membrane (15). COL4A1 forms a sheet-like network beneath the endothelium and surrounding smooth muscle cells with a role in the formation of basement membranes during embryogenesis, in the cohesiveness of basement membrane, and in the preservation of vascular tone and endothelial cell function (48). The mutant mice bearing COL4A1 mutations (namely, in-frame deletion of exon 40) had severe cerebral hemorrhage, were smaller than controls, and could associate ocular and renal abnormalities. Homozygous mutant mice were not viable after midembryogenesis; half of the heterozygous died in the first day of life, and about 18% of adult heterozygous had porencephalic lesions. Structural integrity of the vascular basal membrane was demonstrated to be impaired in this animal model, resulting in vessel susceptibility to disruption, and supporting a possible predisposition of this animal model toward cerebral hemorrhage. Because not all mutant mice developed porencephaly, it has been suggested that environmental factors, such as birth trauma, could interact with the genetic background in determining the outcome (15); this hypothesis was supported by the fact that perinatal hemorrhages were dramatically reduced by surgical delivery of mutant pups (16). The concurrent role of environmental factors, including delivery-induced stress, is in line with the observation that in patients harboring COL4A1 mutations, the hemorrhage may develop during the first few days of a complicated neonatal course. It is also supported by the report of a sporadic case of a boy with a history of infantile hemiparesis who experienced recurrent intracerebral hemorrhage during sport activities (47).
However, parenchymal hemorrhage may also occur during fetal life, and the mutation itself may be responsible for the brain damage as initially demonstrated by the antenatal onset of parenchymal hemorrhage in two preterm siblings (09).
Following the initial description of COL4A1 mutations in the animal model and in familial porencephaly, the crucial role of type IV collagen alpha 1 in preserving the integrity of the vascular basement membrane has been increasingly investigated, and different mutations of the COL4A1 gene have been identified. Correspondingly, the clinical phenotype of COL4A1 mutations has also widened in different members of a given affected family.
Dominant, inherited, or apparently de novo mutations in the COL4A2 gene have also been identified in patients with porencephaly and other cerebrovascular disorders, such as periventricular leukoencephalopathy, cerebral aneurysm, intracerebral hemorrhage, and small vessel disease, isolated or associated with ocular abnormalities (19). COL4A2, which encodes for the type IV alpha2 collagen chain, is structurally and functionally associated with COL4A1. Both genes are ubiquitously expressed, they are regulated by the same promoter, and the encoded procollagen alpha1 and alpha2 chains form a heterotrimeric helix with a constant 2:1 ratio (alpha1 alpha1 alpha2). The putative role of COLA42 mutations in determining vascular accident is supported by experimental data from animal models; mutant Col4α2 mice feature defects in the eye, brain, and vascular stability with focal brain hemorrhagic lesions that are similar to those observed in mutant Col4α1mice (10).
The disorders related to COL4A1/A2 mutations are now considered systemic diseases, which involves the brain, kidney, eyes, heart, and skeletal muscles (30; 28).
A report based on clinical and genetic data of a personal series and on a thorough literature review has provided an overview of the phenotypic and genotypic spectrum of disorders related to COL4A1 and COL4A2 mutations (30). The authors report their own series of 21 COL4A1 and three COL4A2 mutations, with a high percentage of de novo mutations. A high rate of de novo mutations is also reported in the literature: among 67 families (60 with COL4A1 and seven with COL4A2 mutations), 25% of cases carried a de novo mutation and 50% an inherited (no parental data were available in the remaining 25% of cases).
The study confirms the wide phenotypic spectrum of COL4A-related disorders that are noted in the following table:
|
1. Prenatal and neonatal intracerebral hemorrhage. Porencephaly is the most frequent brain damage observed, isolated, or associated with intracranial calcification, hemolytic anemia, elevated creatine kinase, myopathy, ophthalmologic features, and hematuria. | |
|
2. Extensive bilateral porencephaly resembling hydranencephaly. | |
|
3. Periventricular leukomalacia with intracranial calcifications in periventricular areas, basal ganglia, or deep white matter, also in the absence of porencephaly. | |
|
4. Ocular manifestations consisting of retinal small vessel tortuosity, cataract, and anterior segment dysgenesis are always associated with leukoencephalopathy and small vessel disease. | |
|
5. HANAC syndrome (hereditary angiopathy with nephropathy, aneurysms, and cramps), which is due to mutations in COL4A1 affecting glycine residues in close proximity to exons 24 and 25, and which constitutes a distinctive phenotype dominated by the systemic symptoms, in the absence of porencephaly. In this syndrome, the angiopathy manifests with retinal vessel arterial tortuosity and cerebral vessel disease with aneurysm of the carotid siphon; the kidney manifestations include persistent hematuria or proteinuria with or without renal cysts. An additional characteristic symptom is the presence of painful muscle cramps with elevated creatine kinase. | |
|
6. Stroke in childhood and young adulthood. | |
|
7. Sporadic late-onset hemorrhagic stroke. |
In the last few years, several reports further widened the phenotypic spectrum of COL4A1- and COL4A2-related disorders. Studies have clarified that mutations in these genes may cause an array of brain malformations, including schizencephaly, polymicrogyria (either isolated or associated to porencephaly), and subependymal heterotopia (50; 51; 06; 32).
Moreover, the growing use of next-generation sequencing techniques has led to the identification of patients harboring COL4A1 and COL4A2 mutations with epilepsy as their main clinical problem. In these patients, MRI did not detect brain malformations nor porencephaly, but only nonspecific findings such as asymmetric ventricular enlargement, diffuse periventricular leukoencephalopathy, and white matter thinning (53).
A further possible cause of genetic porencephaly has been reported by Sato and colleagues (38). In this paper, the authors reported on an infant carrying a novel missense mutation in the TUBA1A gene. Her MRI showed a complex and diffuse brain malformation characterized by a simplified gyral pattern with areas of focal polymicrogyria and porencephaly. Tubulin-related genes regulate neuronal migration and axonal guidance; therefore, brain malformations due to mutations in these genes are typically complex and may involve multiple cortical and subcortical structures (01). However, porencephaly has never been associated with tubulinopathies. Should this association be confirmed by further observations, including tubulin-related genes in the genetic testing for patients with porencephaly, will be considered (38).
Finally, homozygous variants in COLGALT1 gene have been reported in a patient with a severe form of congenital porencephaly and leukoencephalopathy. The clinical picture was characterized by severe spastic tetraparesis, developmental delay, and refractory epilepsy (44).
COLGALT1 encodes for CoLGalT1, a galactosyltransferase that initiates glycosylation of collagen. The reduced ColGalT activity might decrease COL4A1 production or glycosylation and cause the fragility of the vascular basement membrane (31).
Pathology. A porencephalic cyst is the result of focal necrosis occurring in the developing brain. The pathogenetic mechanisms leading to porencephaly are related to ischemic or hemorrhagic lesions in most cases, and it should be emphasized that the timing of the insult is more important than the type of the insult in determining brain cavitation in the developing brain (36). Several factors are important in facilitating cyst formation from the late second trimester of gestation to the first postnatal stages. The higher water content, the larger vascular territories with insufficient collateral circulation, and the limited capacity to establish adequate gliotic scarring for the smaller number of mature astrocytes are all typical features of the immature brain in this developmental phase, contributing to its higher propensity to cavitation (36).
Porencephaly communicates with the lateral ventricle. Imaging studies often give the impression that the cavity also communicates with the subarachnoid space overlying the cerebral hemisphere, but pathologically the pial membrane, and often some of the arachnoid, is nearly always preserved and prevents the free communication of fluid between the two spaces, though this membrane is so thin that it is below the resolution of CT or MRI. Loculated cysts within the deep white matter of the cerebrum that do not communicate with the ventricle are not porencephalic by definition. The walls of the cyst are smooth and not covered by reactive gliosis. The basal ganglia, cerebellum, and brainstem are usually not involved, but the thalamus may be small for the hypoplasia of the thalamic nuclei projecting to the affected cortex. Secondary degeneration of descending fiber systems, particularly the corticospinal tracts, depends on the location and extent of the lesion (11). Porencephalic cysts are often surrounded by a zone of polymicrogyria. These gyri are atrophic secondary to ischemia during development and, therefore, do not represent a primary disturbance of neuroblast migration or other primary malformations as does pachygyria. This polymicrogyria may or may not be evident with MR imaging. The gray matter surrounding porencephaly undergoes structural and functional changes for a prolonged period of time after the original injury (27). A further type of cortical malformation that may be observed in surviving cerebral cortical tissue adjacent to a porencephalic cyst is focal cortical dysplasia. In this case, the dysplasia results from a "maturational arrest" caused by the ischemic cerebral insult that was not severe enough to include this zone in the frank infarction but that is severe enough to prevent maturation of the cortical lamination or transition from micro-columnar to laminar architecture shortly after midgestation (37). This type of malformation, together with the other dysplasia that occurs adjacent to lesions acquired during early life, is termed focal cortical dysplasia IIId (04).
Neuropathological studies in fetuses carrying COL4A1–A2 mutations demonstrate the presence of hemorrhagic encephalopathy, which was constantly associated with post-ischemic damage in grey and white matter that can worsen throughout the pregnancy. At pathological examination, the multifocal hemorrhagic lesions of different ages were found close to capillaries and small vessels with discontinuous walls, in agreement with the known small vessel fragility due to COL4A diseases.
Porencephaly is relatively rare, and little epidemiologic information is available. A descriptive epidemiologic study, based on the Texas Birth Defect Registry and including 2,042,554 live births over the period 1996 to 2002, analyzed the prevalence of birth defects thought to be related to vascular disruption (23). The prevalence of isolated porencephaly was 0.05 for every 10,000 live births, whereas the prevalence of porencephaly associated with other defects with vascular component etiology (eg, gastroschisis, intestinal atresia, renal agenesis) was 0.35 per 10,000 live births. A similar estimated incidence rate (5.2 per 100,000 live births) has been reported in a population-based survey conducted in Japan (Miyagi prefecture) on 96,136 babies born between 2007 and 2011 (22). Both the Texan and Japanese studies report a higher prevalence in male newborns and in multiple pregnancies. In the Texan study, multivariate analysis showed a higher prevalence of porencephaly in male newborns, particularly when delivered by mothers younger than 20 years of age and in multiple gestation pregnancies.
Studies aimed to evaluate the prevalence of mutations in COL4A1 and COL4A2 genes in cohorts of patients with hemorrhagic or ischemic cerebral lesions. Pathogenic or likely pathogenic mutations in COL4 genes were identified in four of 19 patients with hemolytic anemia and congenital brain malformations (33) and in nine of 18 fetuses in whom prenatal MRI had detected multifocal hemorrhagic or ischemic lesions (44). These studies confirm the relevant role of mutations in COL4 genes in determining not only porencephaly and schizencephaly but also diffuse encephaloclastic lesions.
Prenatal and perinatal care may reduce the risk of porencephaly. Prevention of intrauterine asphyxia and prematurity, avoidance of toxins and infection during pregnancy, and close monitoring of the high-risk fetus during labor and delivery are of major importance. Recognition of maternal risk factors must lead to appropriate monitoring and, when available, treatment. Pregnancy at risk for fetal and neonatal alloimmune thrombocytopenia (ie, women with a positive family history of the disorder, or who had a previous affected neonate) should be referred to a specialized center where information can be given about the risk of fetal hemorrhage, along with the benefits and risks of management strategies. With this regard, a characteristic antenatal and early postnatal neuroimaging pattern (ie, large hemispheric porencephaly associated with lateral ventriculomegaly) has been described in neonatal alloimmune thrombocytopenia (07). Recognition of this pattern could be useful in prompting the diagnosis and treatment to prevent the catastrophic sequelae of neonatal alloimmune thrombocytopenia in both the index case and subsequent offspring. Prenatal ultrasonography or MRI in a large cohort of patients harboring COL4A1/A2 variants detected the following characteristics: fetal ventriculomegaly with increased echogenicity and irregularity of the ventricular walls, deformed frontal horn, cerebellar anomaly, and fetal growth restriction (18; 14). In pregnant women carrying COL4A1 mutations, cesarean section is recommended to reduce the risk of vascular injury to the newborn as a result of birth trauma (47).
The possibility of familial occurrence from either maternal or genetic causes and the possible association with cerebral and extracerebral malformation must be considered.
An accurate family history and examination must also be performed in apparently isolated cases to identify possible carriers of cerebral small vessel disease (46). In families carrying COL4A1 or COL4A2 mutations, genetic counseling is warranted. Genetic counseling must consider the high intrafamilial variability of COL4A-related disorders, with a wide range of phenotypes and the apparent incomplete penetrance, as well as the possibility of de novo mutations (29).
Couples who already have an affected child must be investigated for the mutation, and if one of the two parents carries the mutation, the couple should be informed of the risk for any following pregnancies.
Few authors reported on prenatal diagnosis of COL4A1 gene mutations detected on fetal DNA from amniotic fluid (12). As underscored by Garel and colleagues, given that the prenatal identification of this autosomal dominant gene raises ethical and medical questions concerning both the fetus (given the wide range of clinical presentation) and the parents, parental consent for the genetic analysis requires complete and clear information (12). Identifying the asymptomatic carrier implies obtaining the correct information about the risk for stroke in the carrier and his/her family. At-risk pregnancy requires accurate follow-up with ultrasound (and, when available, MRI) evaluation of the fetus. Cesarean delivery is recommended to decrease the risk of perinatal hemorrhagic events in the baby and in the mother when she carries the mutation.
Porencephaly should be differentiated from schizencephaly, characterized by full-thickness clefts of cortical mantle surrounded or lined by polymicrogyria or heterotopic gray matter (17). Encephaloclastic porencephalies are lined by white matter (02). MRI examination should differentiate between the two brain lesions.
Multicystic encephalomalacia is characterized by multiple cavities of various sizes, separated by glial septae, usually bilateral, determined by multiple foci of necrosis due to prolonged severe ischemia. Hydranencephaly is a massive bilateral lesion wherein most of the cerebral hemispheres, with relative sparing of occipital lobes, are destroyed in utero and replaced by fluid. The term "basket brain" has been used to describe a condition that falls between porencephaly and hydranencephaly. In this condition, extensive bilateral hemispheric defects leave a thin central arch of tissue, with residual cingulate and adjacent gyri that connect with frontal and occipital parts of the brain (11). An arachnoid cyst is a fluid-containing sac covered by a transparent membrane found on the surface of the brain and derived from the pia-arachnoid.
The diagnosis of porencephaly is made through imaging techniques. Prenatal ultrasonography can detect intracranial hemorrhage and cystic lesions in the fetus’s brain. Porencephaly should also be suspected when isolated cerebral ventriculomegaly is consistently detected in the late stages of pregnancy (03).
Porencephaly, along with other brain abnormalities, may be detected by prenatal MRI. Fetal MRI should be considered a valuable diagnostic tool when ultrasonography is doubtful (for example, when it only shows ventriculomegaly) and in selected at-risk pregnancies because it can give a more precise characterization of the lesions in the cerebral parenchyma and detect associated malformations (03; 49).
In preterm infants, serial cranial ultrasonographies demonstrate increased periventricular echogenicity and, some weeks later, cystic formation, periventricular leukomalacia, and ventriculomegaly. In term newborns, a focal area of increased echogenicity is then replaced by cystic cavitation. In older infants, CT scans reveal porencephaly as a circumscribed area of hypodensity, often associated with cerebral atrophy of varying degrees. On MRI, porencephalies appear as smooth-walled cavities with no internal structure, lined by white matter. MRI demonstrates the convolutional pattern of the surrounding cortex, as well as the presence of gliosis and white matter injury; it distinguishes porencephaly from other clastic encephalopathies (multicystic encephalomalacia, hydranencephaly, "basket brain") and true brain malformations (schizencephaly). The prenatal and early postnatal imaging findings in infants with serologically proved alloimmune thrombocytopenia are characterized by large hemispheric porencephaly associated with lateral ventriculomegaly (07).
In patients harboring COL4A1 mutations, with or without porencephaly, MR images may demonstrate changes in the periventricular and deep white matter with sparing of U fibers, and irregular dilatation of the lateral ventricles. MRI and CT scans may also show spot and linear calcification in the subependymal region and around areas of porencephaly in the deep cerebral white matter and the basal ganglia (25).
Mutational analysis of the COL4A1 and COL4A2 genes should ideally be performed in all infants with porencephaly of unknown cause, especially if retinal vascular tortuosity, congenital cataract, or renal cysts are associated or if the family history is positive for the symptoms included in the COL4A1 – COL4A2 phenotypes (46). In a case suspected or proven to have COL4A1 or COL4A2 mutations, a comprehensive evaluation aimed at detecting the possible involvement of the eyes, kidneys, heart, skin, and muscles must be performed (13).
The management of porencephaly is that of the associated clinical manifestations: cerebral palsy, intellectual impairment, and seizures.
Based on evidence linking COL4A1 and COL4A2 mutations to a broad spectrum of multiorgan disorders (often with variable expressivity even within families), a clinical management protocol has been proposed to guide diagnosis, monitoring, and treatment.
In a systematic review of the literature and data from a cohort of 43 individuals from 23 families (including asymptomatic or mildly symptomatic individuals), the authors found that the most frequent and severe manifestations are neurologic, including stroke (from the prenatal period to adulthood), epilepsy, intellectual disability, motor deficits, and structural brain abnormalities, such as porencephaly and schizencephaly (13). Other systems commonly affected include the eyes, kidneys, cardiovascular system, and muscles. The authors propose a multidisciplinary clinical management protocol, which includes an initial comprehensive multi-organ assessment following genetic diagnosis (encompassing neurologic, ophthalmological, cardiac, and renal evaluations) with follow-up tailored by each specialist according to individual patient needs. Prenatal monitoring is advised for women who are carriers of pathogenic variants, including fetal MRI after 20 weeks of gestation to assess for brain malformations, vascular events, and other multiorgan anomalies in the fetus. Genetic counseling is also recommended to explain the inheritance risks and potential complications during pregnancy. Finally, the authors emphasize that anticoagulants and thrombolysis should be used with caution, blood pressure must be tightly controlled, and patients should avoid head trauma and smoking.
Most patients have static neurologic deficits and seizures requiring rehabilitation and antiepileptic treatment. In patients with drug-resistant seizures, particularly when startle epilepsy occurs, surgery must be considered. The surgical exclusion of the affected hemisphere by hemispherectomy must be considered in children with drug-resistant seizures and hemiparesis due to a large porencephalic cavity; this surgical procedure may indeed lead to excellent seizure control (40; 24). Successful functional hemispherectomy has been reported in a patient carrying the COL4A1 mutation. However, as underscored by the authors, the presurgical risk-benefit assessment must also include the potential risk of hemorrhagic complication during surgery, a risk that is increased in patients harboring a COL4A mutation (21).
In a small number of cases, the progressive enlargement of the cyst and the ensuing increased intracranial pressure will cause hydrocephalus and worsening neurologic deficits. In these cases, a ventriculoperitoneal shunt may be necessary. The enlarging size of some porencephalic cysts is not due to intrinsic progressive intracranial hypertension but is believed to be due to the pressure effects of the pulsations of the choroid plexus on the enlarged surface with less resistance to stretching.
Given the possibility of spontaneous intracerebral hemorrhage in patients bearing COL4A1 or COL4A2 mutations, particular attention should be given to avoiding precipitating factors such as oral anticoagulants and incidental trauma.
Genetic counseling to families harboring COL4A mutations must consider the wide variability of phenotype, also in patients carrying the same mutation. Along with genetic and environmental modifying factors, reduced penetrance is probably crucial in determining the phenotype. A screening protocol in families with a COL4A mutation has been proposed (30). The workup includes a medical history, neurologic examination, brain MRI (if abnormalities are found), ophthalmologic examination, renal ultrasound, and functional tests (including urine analysis for the presence of hematuria), serum creatine kinase, and EKC. Prenatal testing can be offered in high-risk pregnancies, always taking into account the medical and ethical issues related to the phenotype variability (13).
In women carrying COL4A1 or COL4A2 mutations, cesarean section should be recommended to reduce the risk of maternal vascular injury during expulsive effort and to reduce the risk of vascular injury in the newborn as a result of birth trauma (16; 12). However, it must be remembered that porencephaly, or other brain lesions, may have been established during fetal life.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Tiziana Granata MD
Dr. Granata of the Fondazione Istituto Nazionale Neurologico Carlo Besta in Milan, Italy, has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026