







Epilepsy & Seizures
Driving and epilepsy
Jul. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Traumatic brain injury is one of the most common causes of morbidity and mortality, with posttraumatic epilepsy and functional disability being its major sequelae. Once posttraumatic epilepsy has developed, it remits less often than previously reported. Patients with penetrating head trauma have a very high incidence of posttraumatic epilepsy because of dural tear with intracerebral blood and metallic fragments. Hospital-acquired infections increase epilepsy risk after brain injury, highlighting infection prevention as crucial for improving patient outcomes. Several animal models have been used to explore the structural, chemical, and physiologic changes that are responsible for seizures in traumatic brain injury. Animal studies suggested two potential mechanisms of epileptogenesis in patients with head injury: occurrence of disinhibition and development of new functional excitatory connectivity. Possibly, reorganization of astrocytes may, along with dendritic sprouting and new synapse formation, form the structural basis for recurrent excitation in the epileptic brain. Structural imaging abnormalities on magnetic resonance imaging have been assessed as a biomarker to predict the risk of posttraumatic epilepsy following traumatic brain injury. Hippocampal volume deficit and inferior temporal cortex thinning predict early posttraumatic epilepsy. Genetic studies have suggested there is a significant genetic contribution to the development of posttraumatic epilepsy. Abnormalities in neuronal glutamate transporter genes were found to be associated with increased risk of epileptogenesis following severe traumatic brain injury. Epileptiform abnormalities in EEG performed in the acute period following traumatic brain injury predict first-year posttraumatic epilepsy. Traumatic brain injury increases epilepsy risk nearly seven-fold, and this effect is partly mediated by postconcussive symptoms, cognitive dysfunction, suicidal behavior, substance misuse, and stroke. No effective prophylaxis for posttraumatic epilepsy currently exists. Levetiracetam is now considered a preferred drug over phenytoin because use of phenytoin was associated with longer length of hospital stay and more dizziness. A metaanalysis found that surgical treatments, particularly resective surgeries like temporal lobectomy, significantly reduce seizures in posttraumatic epilepsy patients. Posttraumatic seizures add to the risk of death following traumatic brain injury. The risk of death is much higher in younger persons. In this article, the author summarizes the current data on epileptogenesis of posttraumatic epilepsy. The author also provides updated information on the epidemiology, clinical features, differential diagnosis, and management of posttraumatic epilepsy.
|
• Posttraumatic seizures may occur almost simultaneously with head injury or be delayed for several years. | |
|
• Once posttraumatic epilepsy has developed, it remits less often than previously reported. | |
|
• Patients with posttraumatic epilepsy appear to have a higher mortality rate than patients with traumatic brain injury without epilepsy. | |
|
• Posttraumatic epilepsy appears in many cases as temporal lobe epilepsy that possibly originates from the hippocampus. | |
|
• Neither phenytoin nor any other antiepileptic drugs have proven valuable for preventing the development of late posttraumatic epilepsy. | |
|
• The most effective way to prevent posttraumatic epilepsy is to prevent head trauma. |
Hippocrates (460 BC to 357 BC), in "Injuries of the Head," observed that a wound of one side of the head could cause convulsions on the other side of the body. By the time of the Renaissance, physicians were aware of the potential for head injuries to cause both acute convulsions and chronic epilepsy (98). In the 19th century, John Hughlings Jackson provided a detailed description of the association between the character of epileptic attacks and the location of causative head injury wounds (95). Penfield and colleagues extended Jackson's observations relating injury location to symptomatology and recognized meningocerebral scar formation in the pathophysiology of posttraumatic epilepsy (77; 80; 79; 78).
Military conflicts have been a major cause of head injuries throughout recorded history, and studies of posttraumatic epilepsy have been conducted following World War I (05), World War II (111; 104; 105; 106), the Korean War (110), and the Vietnam War (12; 89; 90). Studies of posttraumatic epilepsy in nonmilitary personnel have been fewer and reported most extensively by Jennett and colleagues (43; 39; 40; 41; 42) and later by several other groups (23; 24; 61; 53; 97).
Seizures occurring months or years after head injury are called "late seizures," and recurring "late seizures" make up the clinical syndrome of "posttraumatic epilepsy" (72). In several studies on posttraumatic seizures, head injury has been classified as mild, moderate, or severe. Mild injuries are defined by lack of skull fracture and a period of posttraumatic amnesia or loss of consciousness that is 30 minutes or less. Moderate injuries may or may not be associated with skull fractures, but there is a period of 30 minutes to 24 hours of posttraumatic amnesia or loss of consciousness. Severe injuries are characterized by brain contusion, intracranial hematoma, or 24 hours or more of either unconsciousness or posttraumatic amnesia (02; 32).
The frontal and temporal lobes are the most commonly affected regions, and the resulting epilepsy syndrome is typically localization related (38). Seizures remain focal in about one quarter of patients with posttraumatic epilepsy, partial-onset seizures with secondary generalization occur in about one half of patients, and generalized convulsions alone occur in the remaining one quarter of patients. Some posttraumatic seizures that occur early after head injury may result from diffuse brain insult, especially after closed head trauma, and, thus, may be generalized from onset. Two thirds to three fourths of patients at risk never develop posttraumatic seizures (12). Acute posttraumatic nonconvulsive seizures may also occur after traumatic brain injury and, in a selected subgroup, appear to be associated with disproportionate long-term hippocampal atrophy (103). A retrospective review of patients with moderate to severe traumatic brain injury with subsequent development of medically refractory epilepsy was performed. Information regarding details of brain injury, neuroimaging studies, seizures, video-electroencephalography, and surgery outcomes was analyzed. Out of 123 patients with posttraumatic epilepsy and the majority with localization-related epilepsy, 57% had temporal lobe epilepsy, 35% had frontal lobe epilepsy, and 3% each had parietal and occipital lobe epilepsy. Among patients with temporal lobe epilepsy, 44% had mesial temporal sclerosis, 26% had temporal neocortical lesions, and 30% were nonlesional. Epilepsy surgery was done in 22 patients. Findings suggested that careful evaluation with video-electroencephalography monitoring and high-resolution MRI can identify distinct syndromes among patients with posttraumatic epilepsy (35).
Patients with penetrating head injuries carry a high risk of posttraumatic epilepsy decades after their injury and, therefore, require long-term medical follow-up. Lesion location, lesion size, and lesion type are predictors of posttraumatic epilepsy in these patients (82; 88). Caveness summarized military experience from a number of reports: 40% to 50% of new cases of posttraumatic seizures occur within 6 months, 70% within one year, and 80% within 2 years after head injury (12). Posttraumatic seizures are most frequent within the first month after injury and more frequent in the first week than in the next several weeks. Seizures occurring within 5 minutes of traumatic injury to the head are called "immediate" posttraumatic seizures (60). Such seizures do not represent epilepsy, per se, and should be viewed as an acute response to the head injury. Almost all such seizures are generalized convulsions and probably are produced by the same mechanisms as experimental traumatic seizures in animal models. Seizures within the first 7 days after injury are called "early" posttraumatic seizures, whereas "late" posttraumatic seizures are those occurring more than 7 days after injury. Early posttraumatic seizures are less precisely related to the etiology or extent of injury than late posttraumatic epilepsy (111; 14). However, early seizures carry a definite risk for the occurrence of late seizures. One quarter to one half of patients with early seizures also have late seizures (42; 12). Early seizures are more common in children under the age of 5 years than in other groups, and their frequency tends to lessen with age.
Posttraumatic epilepsy does not persist in all patients. Within 5 to 10 years, one half of patients no longer have seizures (87; 106; 12). Patients with frequent seizures are less likely to undergo remission than patients who have fewer seizures (110).
Severe traumatic brain injury can lead to functional seizures as well. In a retrospective study, Asadi-Pooya and Farazdaghi evaluated association of traumatic brain injury with type of seizures (04). Among 1492 patients who were evaluated, 559 patients had idiopathic generalized epilepsies, 646 patients had temporal lobe epilepsy, and 287 patients had functional seizures. Overall, 77 (5.2%) patients had severe head injuries prior to the onset of their seizures. In this cohort, 12 persons (4.2%) with functional seizures reported prior severe head injuries. Compared to people with idiopathic generalized epilepsies, the odds ratio of having severe head injury in the functional seizures group was 2.67. Compared to patients with temporal lobe epilepsy, the odds ratio of having a prior severe head injury in the functional seizures group was 0.46. The authors concluded that all patients with functional seizures should be evaluated for history of severe head injury.
Once posttraumatic epilepsy has developed, it remits less often than previously reported. Moreover, patients with posttraumatic epilepsy appear to have a higher mortality rate than patients with traumatic brain injury without epilepsy (49).
Immediate (within the first 5 minutes), early (from 5 minutes to 1 week), and late (after the first week) posttraumatic seizures have different implications for prognosis. Immediate seizures are most frequently generalized convulsions and resemble those produced in acute experimental animal models (60). The pathological process is probably different from that operating in later posttraumatic seizures. Immediate posttraumatic seizures do not predict late posttraumatic epilepsy. Early posttraumatic seizures are usually focal in onset and are predictive of an increased risk for nonsurvival and for late epilepsy. Early seizures also predict an increased risk of convulsive status epilepticus, especially in children under 5 years (60).
Late posttraumatic epilepsy does not persist in all patients. Within 5 to 10 years, one half of patients cease having seizures (87; 106; 12). Patients with frequent seizures are less likely to undergo remission than patients who have few seizures (110). Patients with epilepsy showed a significantly higher incidence of personality disorders than patients without epilepsy. Uninhibited behavior, irritability, and agitated and aggressive behavior were significantly more frequent and severe in patients with posttraumatic epilepsy. The psychometric tests intended to explore memory, language, intelligence, attention, and spatial cognition did not show any significant difference between those with and without epilepsy (64). One study observed that patients who underwent surgical intervention and those who had presence of late-provoked seizures during the acute phase of traumatic brain injury experienced a poor outcome (107).
The risk of dementia associated with posttraumatic epilepsy was found to be 4.56 times higher than those without head injuries or seizures. A study investigated the association between posttraumatic epilepsy, a condition where seizures occur after a head injury, and the risk of developing dementia. The study analyzed data from over 12,500 participants in the Atherosclerosis Risk in Communities study, who were followed for a median of 25 years (92). The results showed that participants who developed posttraumatic epilepsy had a significantly higher risk of developing dementia compared to those who did not have head injuries or seizures.
Long-term mortality is higher in patients with posttraumatic epilepsy than in other patients with traumatic brain injury (101). Until one year, there was no difference in mortality. After that, the difference remained significant for 15 years from the injury. During this period, 65% of nonepileptic patients with traumatic brain injury were alive compared to only 45% of patients with posttraumatic epilepsy.
A very large Veteran Health Administration cohort of United States veterans (more than 210,000 persons with epilepsy diagnosed between 2005 and 2022) compared mortality in posttraumatic versus nontraumatic epilepsy. About 29,000 had posttraumatic epilepsy, and about 181,000 had nontraumatic epilepsy. Posttraumatic epilepsy was defined by a traumatic brain injury within 5 years before the epilepsy index date. Traumatic brain injury was subclassified by International Classification of Diseases codes into skull or facial fracture, diffuse cerebral injury, focal cerebral injury, extracerebral injury, or concussion. Average age at onset was younger in posttraumatic epilepsy. Overall mortality was significantly higher in posttraumatic epilepsy. The magnitude of excess mortality differed markedly by traumatic brain injury type: the highest excess mortality occurred after diffuse cerebral injury, focal cerebral injury, or skull or facial fracture. By contrast, concussion-related posttraumatic epilepsy had lower mortality than nontraumatic epilepsy. Age at onset also influenced risk: young adult onset posttraumatic epilepsy with extracerebral injury had more than two-fold higher mortality (69).
Posttraumatic epilepsy may occur as the result of closed head injury or of penetrating wounds to the brain.
Late seizure occurrence is highly correlated with the extent of focal brain destruction (05; 104; 42; 12). For both early and late posttraumatic seizures, prolonged impairment of consciousness increases the likelihood of posttraumatic seizures. Although seizures may result from injury to any portion of the cerebral cortex, injury to regions adjacent to the central sulcus are significantly more likely to cause posttraumatic epilepsy than injury to either pole (05; 87; 106).
In a study, 783 patients at high risk of developing posttraumatic seizures were followed for 2 years. Indicators for a significantly elevated risk of posttraumatic seizures included:
|
• Surgery for removal of a subdural hematoma |
In another prospective observational study, risks for the development of late posttraumatic seizures were assessed within 24 hours of injury. A total of 647 young individuals (16 years of age or less) who fulfilled any of the following criteria, were included:
|
• Abnormal computed tomography scan findings | ||
|
- Midline shift or cisternal compression | ||
|
• Best Glasgow Coma Scale score of 10 or less during the first 24 hours following traumatic brain injury. | ||
Sixty-six individuals had late posttraumatic seizures during full 24-month follow-up. The highest probability for late posttraumatic seizures was associated with biparietal contusions, dural penetration with bone and metal fragments, multiple intracranial operations, multiple subcortical contusions, subdural hematoma with evacuation, midline shift greater than 5 mm, and multiple or bilateral cortical contusions.
A low Glasgow Coma Scale score was associated with the higher probabilities for development of late posttraumatic seizures at 24 months (29).
A metaanalysis identified potential risk factors for posttraumatic epilepsy. Men had a higher risk of developing posttraumatic epilepsy than women. A history of alcohol abuse, posttraumatic amnesia, focal neurologic signs, and loss of consciousness at initial traumatic brain injury were associated with an increased risk of posttraumatic epilepsy. Traumatic brain injury-related abnormal neuroimaging findings, including skull fracture, midline shift, brain contusion, subdural hemorrhage, and intracranial hemorrhage were strong risk factors for posttraumatic epilepsy. The risk of developing posttraumatic epilepsy after skull fracture, mild brain injury, and severe brain injury peaked within the first year after traumatic brain injury, and then gradually decreased. However, a high risk of posttraumatic epilepsy was sustained for greater than 10 years (114). Posttraumatic epileptic epilepsy is a serious complication in patients with subdural hematoma (113).
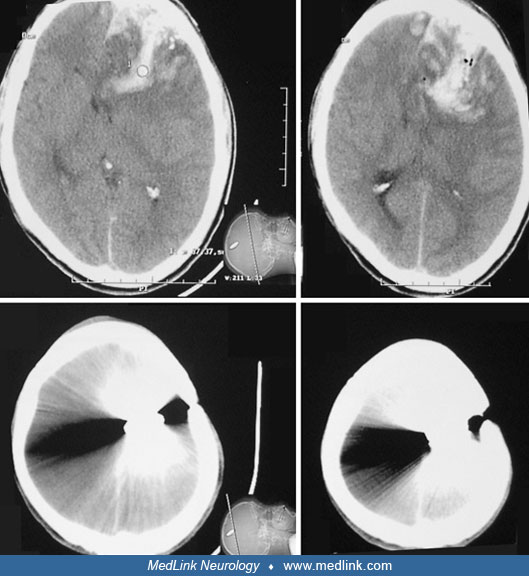
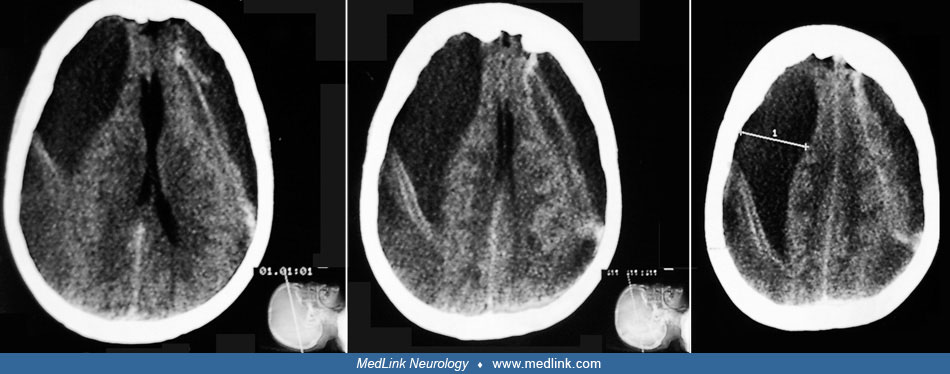
The neuropathology of penetrating and nonpenetrating cerebral injuries is distinct (07). Penetrating injuries cause loss of neuropil from the direct trauma and invariably develop into a dense gliotic and fibrotic scar, which injures blood vessels and may cause even more extensive secondary brain loss. Closed head injuries from blunt head trauma subject the brain to acceleration, deceleration, rotation, compression, and shearing of fiber tracts and vessels. Focal brain damage as the result of angular acceleration of the head occurs "characteristically at the frontal and temporal poles and on the inferior surfaces of the frontal and temporal lobes where brain tissue comes in contact with bone protuberances in the base of the skull" (19). Diffuse brain damage also may result from closed head trauma. An NINDS report described four categories of diffuse brain damage: (1) axonal injury, (2) hypoxia, (3) vasogenic brain edema (which may be exacerbated by early posttraumatic seizures or status epilepticus), and (4) multiple petechial hemorrhages (01).
Posttraumatic epilepsy often develops following traumatic brain injuries, with the likelihood of developing epilepsy increasing with the severity of the injury. Repeated seizures can severely hinder neurologic recovery and lead to poor outcomes, making prompt and effective treatment crucial. Much research into posttraumatic epilepsy aims to understand the complex process of epileptogenesis, where previously normal brain tissue becomes prone to recurrent seizures. Researchers are working to develop various diagnostic tools, including clinical assessments, imaging tests, EEG monitoring, and fluid biomarkers, to identify patients at high risk of developing posttraumatic epilepsy. Studies using animal models have identified potential therapeutic strategies to prevent epilepsy, which may also apply to other types of epilepsy. By exploring the underlying mechanisms and developing innovative treatments, researchers hope to improve outcomes for individuals with posttraumatic epilepsy (75).
In an in vivo model of posttraumatic epilepsy in the rat, chronic spontaneous recurrent seizures following a single episode of fluid percussion injury have been described. Posttraumatic epilepsy, studied during the first 2 months postinjury, was focal; seizures originated predominantly from the frontal-parietal neocortex at or around the injury site. However, rarer bilateral seizures originating from a different and undefined focus were also observed. Posttraumatic epilepsy is thought to result from a progressive process that occurs between brain injury and spontaneous recurrent seizures (21; 33; 20). Animal studies suggested two potential mechanisms of epileptogenesis in patients with head injury. Occurrence of disinhibition and development of new functional excitatory connectivity are primarily responsible for epileptogenesis. Experiments have shown that tetrodotoxin applied to injured cortex during a critical period can prevent epileptogenesis in the undercut model of posttraumatic epilepsy. It has also been shown that such treatment markedly attenuates histologic indices of axonal and terminal sprouting and presumably associated aberrant excitatory connectivity. A second finding in the undercut model is a decrease in spontaneous inhibitory events (81).
Kharatishvili studied the electrophysiologic, behavioral, and structural features of posttraumatic epilepsy induced by severe, nonpenetrating lateral fluid-percussion brain injury in rats. Data from two independent experiments indicated that 40% to 50% of injured animals developed epilepsy, with a latency period between 7 weeks and 1 year. Mossy fiber sprouting was increased in the ipsilateral hippocampus of animals with posttraumatic epilepsy. Stereologic cell counts indicated a loss of dentate hilar neurons ipsilaterally (48). Formation of new recurrent excitatory circuits after brain injuries has also been hypothesized as an important factor contributing to epileptogenesis (44). Possibly, reorganization of astrocytes may, in concert with dendritic sprouting and new synapse formation, form the structural basis for recurrent excitation in the epileptic brain (71).
Posttraumatic epilepsy appears in many cases as temporal-lobe epilepsy that possibly originates from the hippocampus (34). A single episode of experimental closed-head trauma may produce long-lasting alterations in the hippocampus in rats. One study suggests that significant head trauma in human adults can result in hippocampal cell loss, particularly in hilar and CA3 neurons, similar to that observed in animal models of traumatic brain injury. In this study, 21 tissue specimens (out of 200 sequential temporal lobectomies, where trauma was the only risk factor for epilepsy) were analyzed. Hippocampal neuronal loss was found in 94% of specimens and all of these had cell loss in the hilar region of the dentate gyrus. Hilar cell loss ranged from mild, when cell loss was confined to the hilus, to severe, when cell loss extended into CA3 and CA1 (94). Authors in an experimental study used the model of closed head injury to analyze the electrophysiological changes in hippocampus CA1 and CA3 pyramidal cells and in interneurons of the CA1 field, which is extremely sensitive to ischemia. These authors observed that closed head injury causes hyperexcitability in rat hippocampal CA1 but not in CA3 pyramidal cells (34).
Another important observation regarding the development of posttraumatic epilepsy is the occurrence of a “latent period” between the acute insult and the development of epilepsy. In animal models, this latent period is characterized by neuronal necrosis, apoptosis, synaptogenesis, changes in gene expression, and axonal sprouting. Therapeutic intervention during this latent period may provide an opportunity to prevent the development of posttraumatic epilepsy (37).
Several experimental studies also highlight the concept of the “critical period,” during which a therapy could potentially be administered to prevent epileptogenesis following traumatic brain injury. In the rat undercut model of neocortical posttraumatic hyperexcitability, suppression of neuronal activity by exposing the injured cortex to tetrodotoxin in vivo for approximately 2 weeks prevents the expression of abnormal hypersynchronous discharges in neocortical slices. In this model, it was concluded that a critical period for development of hyperexcitability exists, and the critical period depends on cortical activity (21; 33; 20). Epileptiform abnormalities in EEG performed in acute period following traumatic brain injury predict possibility of first year posttraumatic epilepsy (50). Chen and colleagues evaluated the role of quantitative electroencephalograms to predict first-year possibility of posttraumatic epilepsy (17). The authors noted that greater epileptiform burden, suppression burden, and beta variability were associated with 4.6 times higher risk of posttraumatic epilepsy.
Neuroinflammation is crucial in the process of epileptogenesis. Traumatic brain injury leads to an intense neuroinflammatory change in the brain that is considered responsible for posttraumatic epilepsy. Intense inflammation causes substantial tissue damage and epileptogenesis following brain injury. The inflammatory changes in the brain are mediated via complement system (16).
Reactive oxygen species and reactive nitrogen species have been demonstrated to be involved in epileptogenesis induced by iron ions in the rat brain, an experimental animal model for posttraumatic epilepsy. Head injury or hemorrhagic cortical infarction results in extravasation of blood and breakdown of red blood cells and hemoglobin. Hemoglobin and iron liberated from hemoglobin are associated with the generation of reactive oxygen species and reactive nitrogen species. Reactive oxygen species are responsible for the induction for peroxidation of neural lipids, ie, an injury of neuronal membranes, and also could induce disorders in the excitatory and inhibitory neurotransmitters (67). Other potential pathogenic mechanisms include release of excitotoxins such as glutamate, alteration of energy metabolism, and free radical damage. Although early seizures predict late seizures, whether early seizures actually contribute to the development of late posttraumatic epilepsy is still unclear (37).
Inheritance of the apolipoprotein E (APO-E) epsilon-4 allele (like in Alzheimer disease, progression to disability in multiple sclerosis, and poor outcome after traumatic brain injury) is associated with increased risk of late posttraumatic seizures. This risk appears to be independent of an effect of epsilon-4 allele on functional outcome after traumatic brain injury (28). The proposed mechanisms by which APO-E affects the clinicopathological consequences of traumatic brain injury include amyloid deposition, disruption of cytoskeletal stability, cholinergic dysfunction, oxidative stress, neuroprotection and central nervous system plasticity in response to injury (45). Some reports suggest that interleukin-1β gene variability is a risk factor for posttraumatic epilepsy in patients with traumatic brain injury (25; 26). Higher cerebrospinal fluid/serum interleukin-1β ratios were associated with increased risk for posttraumatic epilepsy. Possibly, genetically mediated signaling mechanisms contribute to interleukin-1β cerebrospinal/serum associations with posttraumatic epilepsy. Abnormalities in neuronal glutamate transporter genes were found to be associated with increased risk of epileptogenesis following severe traumatic brain injury (83). A systematic review investigated the association of single-nucleotide polymorphisms (SNPs) with posttraumatic epilepsy and poststroke epilepsy. Out of 420 articles, 16 studies were reviewed, revealing 127 single-nucleotide polymorphisms from 32 genes. Eleven single-nucleotide polymorphisms significantly increased posttraumatic epilepsy risk, whereas five influenced poststroke epilepsy risk. However, the association of APOE ɛ4 with posttraumatic epilepsy was nonsignificant (66).
Tubi and colleagues noted that the temporal lobe traumatic lesion is related to both a high incidence of early seizures and longitudinal development of posttraumatic epilepsy (100). In addition, patients with early seizures following injury are more likely to develop posttraumatic epilepsy. Patients who develop posttraumatic epilepsy have greater chronic temporal lobe atrophy.
Posttraumatic epilepsy is common in pediatric patients with traumatic brain injury admitted into an intensive care unit. Older age, moderate injury severity, obliterated suprasellar cisterns, seizures during intensive care unit stay, and surgical treatment predicted an increased risk of posttraumatic epilepsy (68).
The incidence of posttraumatic seizures varies with the time period after injury and population age range under study. In addition, the risk of developing epilepsy is directly proportional to the severity of the brain injury. The incidence of posttraumatic seizures has been reported to be anywhere from 4% to 53%. Penetrating brain injury produces fibrosis in the cerebral cortex and is associated with a risk of posttraumatic epilepsy of approximately 50%, whereas nonpenetrating head injury may produce focal contusions and intracranial hemorrhages and is associated with a risk of posttraumatic epilepsy up to 30%. Closed head injury often produces diffuse concussive injury, with shearing of axons and selective damage to vulnerable brain regions, such as the hippocampus (27).
As many as 86% of patients with a seizure after traumatic brain injury will have a second in the next 2 years (31). Significant risk factors for the development of seizures in the first week after injury include acute intracerebral hematoma (especially subdural hematoma), young age, increased injury severity, and chronic alcoholism. Risk factors for the development of seizures within a week after traumatic brain injury include seizures within the first week, acute intracerebral hematoma (especially subdural hematoma), brain contusion, increased injury severity, and age older than 65 years at the time of injury (31). Overall, traumatic brain injury is responsible for 4% of all cases of epilepsy. Children with traumatic brain injury are less likely to develop late epilepsy compared to adults (37). Posttraumatic epilepsy in the pediatric age group is mostly seen within the first week.
A study of 1785 pediatric patients, younger than 6 years of age, was performed to define the risk factors for posttraumatic early epilepsy and the indications for prophylactic therapy. Only 149 (8.4%) of the patients had posttraumatic early epilepsy. The data showed that approximately 12% of the patients were 3 years of age or younger. Thirty-one percent of the patients with severe head injury (Glasgow Coma Scale=3 to 8; Children's Coma Scale = 3 to 8), 19.3% of the patients with depressed skull fractures, 13.7% of the patients with intraparenchymal hemorrhage, and 21.6% of the patients with cerebral edema had posttraumatic early epilepsy. Patients 3 years of age or younger with severe head injury, cerebral edema, intraparenchymal hemorrhage, or depressed skull fracture had a higher incidence of posttraumatic early epilepsy (06). In another study of pediatric patients, predictors of early posttraumatic seizure activity during the first 7 days were determined. Of the 275 patients included in the study, 34 (12%) had early posttraumatic seizures. Risk factors identified included prehospital hypoxia, young age, nonaccidental trauma, severe traumatic brain injury, impact seizure, and subdural hemorrhage. Independent risk factors identified by multivariable analysis included age younger than 2 years, Glasgow Coma Scale score 8 or lower, and nonaccidental trauma as a mechanism of injury (55).
Karlander and colleagues performed a register-based cohort study with the objective of assessing the risk of epilepsy in adult patients hospitalized for traumatic brain injury (46). The authors analyzed the data for 111,947 patients admitted between 2000 and 2010; 325,881 sex- and age-matched controls were also included. Karlander and colleagues observed that the 10-year risk of epilepsy was 12.9% for focal cerebral injuries, 8.1% for diffuse cerebral injuries, 7.3% for extracerebral injuries, 2.8% for skull fractures, and 2.6% for mild traumatic brain injury. The overall risk for the traumatic brain injury group was 4%, whereas the risk of seizures was only 0.9% in controls. A study in Finland examined the long-term risk of epilepsy in children with traumatic brain injury (52). Analyzing data from 1998 to 2018, it compared 71,969 children with traumatic brain injury to 64,856 with extremity fractures. Results showed higher epilepsy rates in the pediatric traumatic brain injury group, especially in the first 4 years and among those requiring neurosurgery.
Lolk and colleagues performed a population-based cohort study on 2,476,905 individuals born in Denmark between 1977 and 2016 (58). Traumatic brain injury was sustained by 167,051 subjects (71,162 females and 95,889 males), and 37,200 individuals developed epilepsy. There were 10 age- and sex-matched controls for each subject with traumatic brain injury. The relative risk of epilepsy increased after a first traumatic brain injury (hazard ratio 2.04) and even more after a second traumatic brain injury (hazard ratio 4.45). The risk of epilepsy was higher after severe traumatic brain injuries. Females were more likely to develop epilepsy after mild traumatic brain injury; in contrast, males were more likely to develop epilepsy after severe traumatic brain injury.
In a multicenter, prospective case-control cohort study, Burke and colleagues followed 1885 participants including 1493 patients with traumatic brain injury, 182 orthopedic controls, and 210 healthy controls, and all were followed up for one year (11). The authors noted that during follow-up period, 41 (2.8%) patients with traumatic brain injury and no controls had evidence of posttraumatic epilepsy. Poor Glasgow Coma Scale score and abnormal neuroimaging were significant predictors of posttraumatic epilepsy. Pease and colleagues assessed the incidence among 392 survivors of severe traumatic brain injury (74). After hospital discharge, cumulative incidence of posttraumatic epilepsy was 25% at 5 years and 32% at 15 years. Among patients with one late seizure (> 7 days post-trauma), the risk of seizure recurrence was 62% after 1 year and 82% at 10 years. Age, decompressive hemicraniectomy, and intracranial infection were independent predictors of posttraumatic epilepsy. The authors noted that patients with severe brain injury and a single late posttraumatic seizure required long-term antiepileptic treatment (74).
Sødal and colleagues reviewed the data of early posttraumatic seizures among 1827 patients of traumatic brain injury. Approximately, 6% of patients had at least one seizure within 7 days of head injury. The multivariate logistic regression analysis revealed that alcohol abuse, moderate and severe brain injury, brain contusion, and subdural hematoma were the significant predictors of early posttraumatic seizures (93).
The most effective way to prevent posttraumatic epilepsy is to prevent head trauma. Head injuries are inevitable as long as armed combat occurs. However, missile injuries in the civilian population could be greatly reduced by the enactment of strict gun control laws and the elimination of private ownership of handguns (91). Head injuries from vehicular accidents can also be reduced by the universal use of protective devices such as seat belts and air bags, enforcement of speed laws, and severe penalties for driving under the influence of alcohol (10).
When head trauma does occur, the incidence of posttraumatic epilepsy can be reduced by preventing or minimizing the development of the meningocerebral cicatrix and reactive gliosis by definitive debridement of the damaged brain tissue and prevention of complications from brain edema, hemorrhage, or infection (12).
Willmore has reviewed prophylactic and preventive use of antiepileptic drugs (112). Prophylactic antiepileptic drug therapy has not been shown to reduce the incidence of posttraumatic epilepsy. An American Academy of Neurology Practice Parameter concluded, “prophylaxis with phenytoin in patients with severe traumatic brain injury is established as effective in decreasing the risk of early posttraumatic seizures (those occurring within 7 days).” Consequently, the AAN recommends that patients with severe traumatic brain injury receive prophylactic phenytoin for the first 7 days after injury. It is not recommended for prophylaxis of late posttraumatic epilepsy (15).
The efficacy of prophylactic phenytoin for the prevention of early posttraumatic seizures in children with moderate to severe blunt head injury was also investigated (117). A total of 102 children younger than 16 years of age with moderate to severe blunt head injury were randomized to receive phenytoin or placebo within 60 minutes of presentation. The primary endpoint was posttraumatic seizures within 48 hours; secondary endpoints were survival and neurologic outcome 30 days after injury. During the 48-hour observation period, three (7%) of 46 patients given phenytoin and three (5%) of 56 patients given placebo experienced a posttraumatic seizure. There were no significant differences between the treatment groups in survival or neurologic outcome after 30 days. Phenytoin did not substantially reduce early posttraumatic seizures in children (117).
Perhaps excitotoxicity proximate to the brain injury also leads to the neurologic deficits seen after severe trauma, initiating and promoting epileptogenesis. It has been suggested that disrupting this process may prevent epilepsy (09). Development of an epileptic focus in posttraumatic epilepsy, as in iron-induced epilepsy, seems to be due to a cascade of events beginning with hemorrhage, hemolysis, iron or heme compound liberation, free radical formation, peroxidation, and cell death (72). From time to time, prophylactic efficacy of several other drugs, like lipid peroxidation inhibitors, neuroprotectors (antioxidants), glutamic receptor blockers, N-methyl-D-aspartate receptor blockers, and drugs that modulate apoptosis via caspase inhibition have been considered. However, these newer ways of therapeutic intervention, as of 2022, have only an experimental basis.
In a Cochrane review, Thompson and co-workers noted that early treatment with an antiepileptic drug (phenytoin or carbamazepine) may reduce the risk of early posttraumatic seizures (99). However, the available evidence was of low quality. The authors also noted that there was no evidence to support a reduction in the risk of late seizures or mortality following early antiepileptic treatment. There was inconclusive data to draw any conclusions about the superiority of other antiepileptic drugs over phenytoin.
Seizures immediately following severe head trauma are most likely to be caused by the head trauma, but toxic effects from substance abuse or seizures secondary to electrolyte imbalance or other metabolic derangements must be considered. Sometimes head trauma may occur as the result of a seizure in a patient with known epilepsy or as the result of a first seizure in new-onset epilepsy. In late posttraumatic epilepsy, the longer the period between head trauma and the development of epilepsy, the greater the possibility the seizures may be due to some other cause. Depending on age, other causes that must be considered include CNS infection, cerebral infarction or mass lesion, systemic metabolic disorders, and substance abuse or other toxins. Late seizures (more than 7 days after head trauma) are virtually always partial-onset seizures. Primarily generalized seizures are rare in posttraumatic epilepsy, and absence seizures, with generalized 3-second spike-wave discharges on EEG, were not observed in studies of Korean War and Vietnam War head injury victims (12) nor in a civilian population in Poland (60). Even with a history of severe head trauma, the diagnosis of posttraumatic epilepsy should not be made until other causes of new-onset epilepsy have been eliminated.
Hospital-acquired infections significantly increase the risk of developing epilepsy after a brain injury. The study of adults with moderate to severe brain injuries in Australia analyzed data from a large database and found that nearly a quarter of patients developed infections, with pneumonia and urinary tract infections being the most common (18). Two years after the injury, 11% of patients had developed epilepsy, and those who had infections were more likely to develop epilepsy, even after accounting for other risk factors. Infections were also linked to poor outcomes overall. The study suggests that preventing hospital-acquired infections could be a crucial step in improving outcomes for patients with brain injuries (18).
In the immediate posttrauma period, the diagnostic evaluation should be focused on determination of the nature and extent of the brain damage caused by the trauma. This usually involves CT or MRI of the head, depending on the patient's clinical state. Serum chemistry determinations and complete blood count, as well as a toxicological screening panel, should be obtained. If the first posttraumatic seizure occurs after recovery from the acute head injury, then that seizure should be treated in the same manner as any new-onset seizure. Again, toxic and metabolic causes of seizures must be evaluated with appropriate blood and urine samples. An EEG, recorded both awake and asleep, is essential, as is a cerebral imaging study, preferably an MRI.
In many patients with moderate or severe brain injury, posttraumatic seizures are nonconvulsive and, therefore, continuous electroencephalography monitoring is needed (118). In one study, authors reported EEG patterns observed in patients with traumatic brain injury. Continuous digital EEG was recorded in 70 patients with traumatic brain injury requiring intensive care. Twenty-three of these patients (33%) developed seizures. Eighteen patients (26%) displayed focal high-frequency activity that proceeded to seizures in eight cases. Twelve patients (17%) developed recurrent paroxysmal delta activity. The patients in the seizure group were significantly older and more often exposed to low-energy trauma compared to the paroxysmal delta pattern group (85). Continuous video-EEG monitoring significantly improves detection of subclinical seizures or status epilepticus in children with traumatic brain injury. Significant risk factors for subclinical seizures or status epilepticus are younger age, abusive head trauma, and parenchymal cerebral hematoma (03).
A study evaluated risk factors for posttraumatic epilepsy by using brain MRI after traumatic brain injury. Brain MRI hyperintense (gliosis) or hypointense (hemosiderin) areas or both were assessed in the images of 135 adult traumatic brain-injured patients who completed a 2-year clinical, electroencephalography, and magnetic resonance imaging study protocol. Overall clinical follow-up for the development of posttraumatic epilepsy was 5 to 10 years. In 20 patients, posttraumatic epilepsy developed. Kaplan-Meier curves showed that gliomesenchymal sequelae of focal brain lesions (subdural hematomas or contusions) that required surgical treatment were a posttraumatic epilepsy risk factor, as were sequelae of nonsurgical hemorrhagic contusions with gliosis wall incompletely surrounding hemosiderin remains (65). T1-weighted magnetization transfer imaging may be of value in predicting the intractability of the seizure in delayed posttraumatic epilepsy. Patients with a magnetization transfer abnormality beyond a T2 abnormality had a significantly higher intractability of seizures compared with those with a magnetization transfer abnormality within a T2 abnormality. The authors suggested that the mere presence of hemosiderin deposit was not associated with seizure intractability; however, gliosis around the hemosiderin as seen on T1-weighted magnetization transfer images was associated with seizure intractability (51).
Lutkenhoff and colleagues assessed structural imaging abnormalities as a biomarker to predict the risk of posttraumatic epilepsy in patients with traumatic brain injury (59). In a cohort of 96 patients with moderate to severe traumatic brain injury, the authors performed magnetic resonance imaging-based shape analysis of local volume deficits in subcortical areas and cortical ribbon thinning as potential biomarkers for posttraumatic epilepsy. Right hippocampal volume deficit and inferior temporal cortex thinning were significantly associated with early posttraumatic epilepsy.
A 20-year matched cohort of almost three lakh United States veterans evaluated whether the link between traumatic brain injury and later epilepsy is mediated by specific postinjury conditions. Overall, traumatic brain injury was associated with nearly seven-fold higher epilepsy risk. Median interval between traumatic brain injury and epilepsy diagnosis was about 3 years. Mediation analyses showed that post-concussive symptoms, cognitive dysfunction, suicidal ideation or attempts, overdose or drug abuse, and stroke explained a significant portion of this association. These intermediaries may be useful targets for future prevention strategies (102).
Management of posttraumatic seizures is determined by the timing of their occurrence in relation to the head injury. Because of the potential for convulsions to cause a dramatic increase in intracranial pressure, intense efforts should be made to prevent seizures during the recovery phase of the acute head injury. Phenytoin (or fosphenytoin) is usually the drug of choice in this situation because a loading dose can be administered intravenously to rapidly achieve therapeutic concentrations and because phenytoin does not impair consciousness. Diazepam can be used for the acute treatment of posttraumatic seizures, but it will produce at least transient impairment of consciousness. Other benzodiazepines (eg, lorazepam) have a longer effective duration of action against seizures but also a longer period of impaired consciousness. Neither phenytoin nor any of the benzodiazepines or any other antiepileptic drugs have proven effective for preventing the development of late posttraumatic epilepsy. Consequently, prophylactic treatment with antiepileptic drugs is not indicated after 7 days (21).
After recovery from the acute head injury, posttraumatic epilepsy should be managed in the same manner as symptomatic partial-onset epilepsy from any etiology. Available data suggest carbamazepine, phenytoin, and valproate are drugs of choice for the initial management of secondarily generalized convulsions (62; 63). Carbamazepine and phenytoin are drugs of choice for complex partial seizures (62), but valproate is also effective (63). Newer agents, such as gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide, are also effective in the management of partial-onset seizures and generalized convulsions. Among newer drugs, levetiracetam looks the most promising in children with posttraumatic seizures (73). Levetiracetam is now considered a preferred drug over phenytoin for early seizure prophylaxis in traumatic brain injury. In a study, use of phenytoin was associated with longer length of hospital stay and more dizziness, compared to levetiracetam (36).
Antiepileptic therapy should be initiated with a single agent. For most antiepileptic drugs, treatment should be initiated with a low dose, which is slowly increased until serum concentrations are in midtherapeutic range or the patient experiences dose-related side effects. If seizures remain controlled and there are no side effects, the dose should be maintained. If seizures recur, the dose should be pushed upward until seizures are controlled or unacceptable side effects develop. If seizures persist at maximum tolerated doses, a second drug should be slowly added. If seizure control is achieved with the addition of a second drug, the first drug can be slowly tapered to see if the second drug can be used as monotherapy. This crossover procedure from one to two drugs to one to two drugs can be repeated until control is achieved or all potentially effective drugs have been tried. Many patients with extratemporal posttraumatic epilepsy can achieve good to excellent seizure control with epilepsy surgery. Vagus nerve stimulation should be considered in patients with medically refractory posttraumatic epilepsy who are not good candidates for resection (30).
Authors of a study evaluated the role of glucocorticoid administration after traumatic brain injury. Patients who were given glucocorticoids within one day of their traumatic brain injury were more likely to develop first late seizures than were those without. In addition, glucocorticoid treatment was not associated with decreased late posttraumatic seizures (109).
Results from studies of experimental models and human epilepsy suggest that alterations in GABAergic interneurons and formation of excessive new excitatory synaptic connectivity are prominent potential targets for prophylactic therapies. In the future it will be possible to experimentally modify these aberrant processes and interfere with epileptogenesis (54).
Karlander and colleagues reviewed mortality data of 111,947 individuals with traumatic brain injury and noted that presence of posttraumatic epilepsy enhanced the risk of death with a crude hazard ratio of 2.3 (95% CI: 2.2-2.4) (47). The authors further noted that there was a greater risk of death in younger persons with a crude hazard ratio of 7.8 (95% CI: 6.5-9.4). In a retrospective analysis of severe traumatic brain injury patients from 2002 to 2018, it was found that posttraumatic epilepsy is linked to worse functional outcomes (76). Of 392 patients, 25% developed posttraumatic epilepsy. The study revealed that posttraumatic epilepsy patients had significantly lower rates of favorable outcomes (based on the Glasgow Outcome Scale) at 6, 12, and 24 months post-injury. Although mortality rates were similar, posttraumatic epilepsy patients were more likely to have severe disability or be in a vegetative state. Multivariate analysis confirmed posttraumatic epilepsy as a predictor of poorer recovery (76).
A metaanalysis assessed surgical interventions' effectiveness for posttraumatic epilepsy. Analyzing 14 studies with 430 posttraumatic epilepsy patients, it found a 77.1% overall seizure reduction rate for surgical treatments (both resective surgery and vagus nerve stimulation). Resective surgery was more effective than vagus nerve stimulation, particularly temporal lobectomy. However, results indicate surgical interventions, especially resective, are beneficial for posttraumatic epilepsy management (108).
The risk of posttraumatic epilepsy to the pregnant woman and fetus is the same as the risk of epilepsy of any etiology. All commonly used antiepileptic drugs have risks of causing major fetal malformation, but seizures, especially generalized convulsions, also pose a major risk to the developing fetus (116). Valproate (84; 57) and carbamazepine (86; 56) have been reported to increase the risk of spina bifida, and these and other drugs may produce the fetal anticonvulsant syndrome. As a generalization, the risk of major fetal malformation for a child born to a mother with epilepsy using antiepileptic drugs is 4% to 6%, whereas the risk of major fetal malformation for a child born to a normal mother is 2% to 3% (115). Most of the risk of antiepileptic drug-induced fetal malformations is in the first trimester of gestation. The risk of fetal malformation increases with the antiepileptic drug serum concentration (22) and with the number of antiepileptic drugs used (70). Because of these considerations, most epileptologists recommend treating a pregnant woman with epilepsy with the antiepileptic drug most likely to prevent that patient's seizures and to use the fewest possible drugs at the lowest possible doses to keep the patient seizure free and especially to prevent all generalized convulsions. Adjustments of drugs and doses must be made before conception, and the patient should be started on folic acid (0.4 to 1.0 mg/day) before conception.
If anesthesia is necessary in the acute management of a recent head injury, it is reasonable to assume that intracranial hypertension is present and that cerebral blood flow is being compromised (08). If anesthesia is necessary, agents least likely to increase intracranial pressure and compromise cerebral perfusion should be used. If it is necessary to use neuromuscular blockade, it is essential to monitor the EEG to ensure the absence of electrical seizure activity if coma persists after discontinuation of general anesthesia. After recovery from the acute head injury, anesthetic management, if necessary, is the same as for epilepsy of any etiology. If a patient requires general anesthesia it is important that adequate serum concentrations of antiepileptic drugs be maintained by temporarily switching to parenteral administration of the drug if a parenteral form is available. If not, it may be necessary to temporarily switch to an antiepileptic drug, such as phenytoin, for which a loading dose of the parenteral form can be administered safely so that adequate serum concentrations can be achieved rapidly.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Ravindra Kumar Garg DM
Dr. Garg of King George's Medical University in Lucknow, India, has no relevant financial relationships to disclose.
See Profile
Solomon L Moshé MD
Dr. Moshé of Albert Einstein College of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Epilepsy & Seizures
Jul. 08, 2026
Epilepsy & Seizures
Jun. 02, 2026
Movement Disorders
May. 18, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Epilepsy & Seizures
May. 08, 2026
Epilepsy & Seizures
May. 01, 2026
Epilepsy & Seizures
Apr. 30, 2026
Epilepsy & Seizures
Apr. 28, 2026