
Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Sotos syndrome is a Mendelian disorder of the epigenetic machinery that is classically characterized by the triad of distinctive craniofacial features, overgrowth, and intellectual disability. NSD1 haploinsufficiency was determined to be the major cause of typical Sotos syndrome. Microdeletions of NSD1 were identified in Japanese Sotos patients whereas intragenic mutations were found in most non-Japanese patients. A Sotos-like presentation can also be associated with a number of other genes, most commonly variants of NFIX (Malan syndrome). The majority of Sotos syndrome cases are sporadic, but a few families with an autosomal dominant transmission have been reported. The clinical phenotypes of Sotos syndrome, Malan syndrome, and Sotos-like syndromes are discussed. In addition, the diagnostic and management guidelines are reviewed.
|
• Overgrowth, characteristic facial gestalt, and learning disability or behavioral abnormalities are considered the cardinal clinical criteria of Sotos syndrome. | |
|
• Cardiac anomalies, renal anomalies, hypodontia, seizures, or scoliosis are considered as major features. | |
|
• Genetic testing to confirm the diagnosis is highly recommended because of the remarkable clinical overlap with other overgrowth syndromes. | |
|
• NSD1 is the most commonly implicated gene in patients with Sotos-like presentation and should be included in the first step when testing any patient with Sotos syndrome. |
Sotos syndrome, historically referred to as cerebral gigantism, has been recognized for over 50 years, since the description of five children with macrocephaly, somatic overgrowth, characteristic facial appearance, and intellectual disability by Juan Sotos and his colleagues (49). Thirty years later, the major diagnostic criteria of this syndrome were established (10). In 2002, the genetic etiology of Sotos syndrome was unraveled in a patient carrying an apparently balanced de novo reciprocal translocation t(5; 8)(q35; q24). Since then, heterozygous inactivating variants of the gene NSD1 (nuclear receptor binding SET domain protein 1) have been identified as a major cause and are identified in about 90% of patients with Sotos-like presentation (22; 26). In 2010, pathogenic variants in NFIX were identified in patients with Sotos-like features and initially referred to as Sotos syndrome 2 (31; 42; 60). Subsequently, homozygous variants in the APC2 gene were detected in two siblings with Sotos-like features (03). This autosomal recessive type was originally known as Sotos 3. Further, DNMT3A, SETD2, and GPC3 gene variants or hypomethylation of KCNQ1OT1 may be responsible for clinical features in a few patients with Sotos-like presentation (05; 34).
Terminology. The Sotos syndrome 1/2/3 nomenclature is now nonpreferred. Instead, Sotos syndrome generally refers to affected individuals with NSD1 pathogenic variants, though this term is also used to describe individuals with a clinical diagnosis in the absence of confirmatory genetic testing. Malan syndrome is the current preferred nomenclature for individuals with Sotos-like phenotype and NFIX pathogenic variants. For optimal clarity, it is appropriate to use nomenclature that includes both the genetic etiology and the clinical phenotype in cases in which both are known, eg, NSD1-associated Sotos syndrome or APC2-associated Sotos-like syndrome.
Sotos syndrome is characterized by the following:
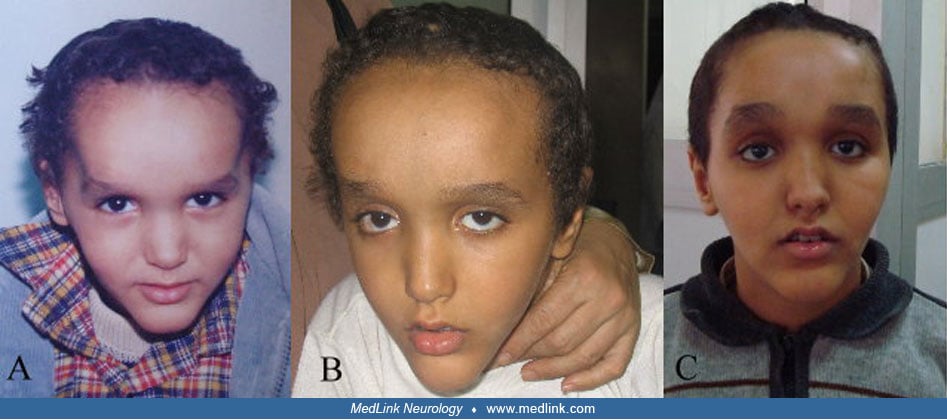
Craniofacial features. Characteristic facial features are present in essentially all Sotos patients with NSD1 variants. This most typically includes a high, prominent forehead and a small, pointed chin, giving the appearance of an inverted pear. Further, individuals frequently have macrodolichocephaly, a receding anterior hairline, hypertelorism, malar flushing, downslanting palpebral fissures, anteverted nostrils, relatively large ears, and highly arched palate (Allanson and Cole 1996; 53; 54). These characteristic facial features change with time; in youth, the face is round with disproportionate prominence of the forehead, whereas in adolescence the face lengthens, with less prominence of the square or pointed chin, and macrocephaly is no longer pronounced but the downslanting palpebral fissures and the high hairline remain distinctive (10; 41; 17). In comparison, patients with Malan syndrome have a long or triangular face, prominent forehead, depressed nasal bridge, deeply set eyes, downslanting palpebral fissures, short nose with anteverted nares and upturned tip, long philtrum, small mouth that is often held open with a thin upper vermillion in a cupid bow shape, an everted lower lip, and a prominent chin. These facial features became more prominent among adults with Malan syndrome, showing more elongated face, prominent chin, deeper skin folds, and more open mouth (43). Mild dysmorphic facial features with normal head circumference were reported in Sotos-like patients (07).
Overgrowth. Height or head circumference more than two standard deviations above the mean have been reported in 90% of children with Sotos syndrome (51). Overgrowth usually starts prenatally and increases rapidly in early childhood before stabilizing above the 97th percentile. However, adult height often ends up in a high-normal range, especially in females (54). This is likely explained by the presence of advanced bone age, which results in earlier closure of epiphyses. However, 20% of patients were described to have normal growth and bone age (29; 07).
Developmental delay and learning disability. Sotos patients typically present with neonatal hypotonia that improves with age. Therefore, walking is usually delayed.
In general, the majority of patients with Sotos syndrome have some degree of learning disability, which may vary from mild to profound learning difficulties requiring lifelong care (18). Although approximately 20% of patients harboring NSD1 variants are reported to have normal learning, patients harboring NFIX variants usually display severe to profound disability (43; 30). Males with Sotos syndrome may be more likely to have a greater degree of intellectual disability than females.
Patients with missense mutations had milder phenotype than those with truncating mutations (45). Severe cognitive impairment was observed in patients harboring large NSD1 deletions (58; 51; 39).
The Sotos syndrome cognitive profile is characterized by relative strength in verbal ability and visuospatial memory but relative weakness in nonverbal reasoning ability and quantitative reasoning. Patients with Sotos syndrome may display greater disability in expressive language when compared to receptive language (28). Furthermore, speech articulation difficulties, including delayed or no speech development, were observed.
Behavioral disturbances with aggressiveness. Although no characteristic behavioral profile has been specified among patients with Sotos and Malan syndrome, neurobehavioral disorders, such as autism-like features, ADHD, and anxious behavior, are common. However, it is not clear that these are more common in individuals with Sotos and Sotos-like syndromes than in other individuals with similar levels of intellectual disability (28). Aggression and self-injurious behavior were also noted. One study suggested that individuals with inactivating variants of NSD1 show fewer behavioral problems, an easier temperament, and fewer internalizing behaviors than those with a clinical diagnosis of Sotos syndrome without identified variants of NSD1, but the study was limited to molecular analysis for large deletions of NSD1 or small variants in a single NSD1 domain (12).
Neuroradiologic anomalies. In general, disturbed development of the midline structures of the brain, such as ventricular dilatation, hypoplastic corpus callosum, heterotopias, periventricular leukomalacia, macrocerebellum, open operculum, and cavum septum pellucidum, are the main neuroradiologic anomalies described in Sotos and Malan syndromes (46). Mild ventriculomegaly may be a relatively common prenatal finding; it was noted in three of eight fetuses with Sotos syndrome in a small retrospective cohort (61). In comparison, Chiari malformation type I, nodular heterotopias, and cortical dysplasia were reported in few patients with Malan syndrome (48; 43). In one study, hypoperfusion of the frontal lobe was noticed in two of the three patients with Sotos syndrome, suggesting some associations with the abnormal behavior noticed in Sotos syndrome (21).
Seizures. Seizures have been described in 50% of patients with Sotos syndrome and 18% of patients with Malan syndrome (43). Of individuals with Sotos syndrome and seizures, 10% to 50% had febrile seizures only (51; 37; 16). In a cohort of individuals with a clinical diagnosis of Sotos syndrome and seizures, two thirds had multiple seizure types, with staring spells, tonic-clonic seizures, and febrile seizures being the most common (16). Temporal lobe epilepsy may include olfactory, gustatory, or auditory hallucinations, automatisms, fear, auras (eg, abdominal aura) with or without behavioral arrest that could be confused with behavioral disorders. Infantile spasms, absence, and myoclonic seizures were also reported. In the majority of cases, seizures were easy to control with common antiepileptic drugs (37; 16).
Dental and oral findings. These include premature or ectopic tooth eruption, hypodontia, enamel hypoplasia, excessive tooth wear, maxillary and mandibular recession, talon cusps, fused teeth, expanded pulp cavity of deciduous teeth, and tooth agenesis (found in 69%) involving the premolars (especially the second) are noted (25). High arched palate and dental crowding were reported in few patients with Malan syndrome (43).
Cardiac findings. Congenital heart defects reported in Sotos syndrome range from single, self-limiting anomalies to complex anomalies requiring interventional surgery. Patent ductus arteriosus and atrial septal defect are among the most frequent congenital heart defects reported (35; 29). In one cohort of patients with Sotos and Malan syndrome, there was a significant association between the presence of neonatal hypotonia and congenital heart defects (30). Noncongenital heart disease, including aortic enlargement and recurrent pericarditis requiring chronic immunosuppressive therapy, have also been reported in rare individuals (20; 30).
Urogenital abnormalities. The most common renal abnormalities are vesicoureteric reflux, hydronephrosis, and hydroureter, but anatomical abnormalities such as duplex kidney, urethral stenosis, pelviureteric junction obstruction, and absent kidney are also recognized (50). They are commonly associated with NSD1 gene microdeletions (35).
Ocular features. Cataracts, megalophthalmos, exotropia, megalocornea, hyperopia, nystagmus, esotropia, optic disk pallor, and retinal dystrophy were reported in Sotos syndrome (23), whereas strabismus, optic nerve atrophy, or hypoplasia were described in 30% of Malan patients (13; 43).
Skeletal anomalies. Scoliosis has been reported in about up to 52% of individuals with NSD1 variants (15; 17). It develops at an early age and can range from a mild to severe with rapidly progressive curving. In comparison, individuals with Malan syndrome often have a marfanoid body habitus, with tall and slender build, long narrow face, and long slender fingers, though these features may only become apparent with age (32; 43).
Tumor risk. The risk of tumors was estimated to be about 2% to 3% (15; 17). Most tumors (45%) developed after the age of five years and were of hematopoietic and neuroectodermal origin (29).
Other features. These include neonatal jaundice, neonatal cutis laxa, feeding problems, hypothyroidism, laryngomalacia, inguinal hernia, craniosynostosis, conductive or sensorineural hearing loss, pneumothorax associated with multiple subpleural blebs, arthrogryposis, hyperpigmentation, hypopigmentation, hypoplastic nails, and recurrent onychocryptosis requiring surgical correction (10; Allanson and Cole 1996; 54; 04; 36; 30).
Sotos syndrome is not a progressive disorder. Thus, long-term survival is typical. Adults with Sotos syndrome who have mild to moderate intellectual disability usually display social isolation, depression, and anxiety. The most common medical problems are scoliosis or contractures, visual difficulties (as a result of strabismus, myopia, glaucoma, nuclear cataract, and retinal atrophy), and increased risk of tumorigenesis (15). Less common medical problems are lymphedema, contracture deformities, dilatation of the aortic arch, and tremors (17). The severity of these medical problems could affect life expectancy (54; 29).
Patient 1. A three-year-old boy presented with macrodolichocephaly and developmental delay. He was the fourth child of a healthy nonconsanguineous 39-year-old mother and 45-year-old father. The pregnancy was unremarkable. There was no history of toxemia, preeclampsia, or exposure to any teratogenic agent during the pregnancy. The boy was delivered via normal spontaneous vaginal birth at 38 weeks of gestation. Birth weight, length, and occipitofrontal head circumference measurements were unknown. He had neonatal jaundice, hypotonia, and feeding difficulties, for which he was kept in an incubator for two weeks. Examination showed weight, height, and head circumference of 18.5 kg (+2.5 SD), 101.2 cm (+1.8 SD), and 54 cm (+2.2 SD), respectively. A triangular face, dolichocephaly, hypertelorism with downslanting palpebral fissures, anteverted nostrils, and a pointed chin were noticed.
He had hypotonia, but the deep tendon reflexes were slightly brisk. His psychomotor development showed mild delay in terms of head control at the age of seven months, sitting at the age of nine months, and walking alone at 21 months.
The patient’s Portage development test results were abnormal. His most notable deviations were in language and cognitive behavior. His self-help and gross motor skills were mildly delayed at a 3-year level. His EEG showed nonspecific abnormalities. Electromyography and nerve conduction showed axonal neuropathy. An MRI of the brain showed dysplastic corpus callosum, especially in the posterior part, and mild enlargement of the lateral ventricles.
Bone age at chronological age six years was six years. Examination at the age of seven years showed weight, height, and head circumference of 29.5 kg (+2 SD), 125 cm (+1 SD), and 56.3 cm (+3 SD), respectively. Craniotomy was performed at the age of nine years for cosmetic purpose to improve the shape of the skull. Follow up cranial MRI did not reveal further changes. Ophthalmologic examination, echocardiogram, abdominal ultrasound, and karyotype of cultured peripheral blood lymphocytes were normal. His mother reported that he was inattentive, hyperactive, and aggressive toward the other children. At the age of 11 years, weight, height, and head circumference were 56 kg (+3.2 SD), 147.3 cm (+1 SD), and 58.4 cm (+3.2 SD), respectively. Dental examination at this age showed high-arched palate and malposed teeth with missing left upper second premolar. Molecular genetic analysis of NSD1 was not done.
Patient 2. This 15-month-old male was the third child born to nonconsanguineous healthy parents. His mother’s age at the time of conception was 34 years, and his father’s age was 45 years. The patient had an older brother with epilepsy and a sister with hydroureter and learning disability. Further, the familial history showed maternal and paternal cousins with learning disability. The patient was born at term by normal vaginal delivery, and the pregnancy had been uneventful. His birth weight was 4.5 kg. His birth length and his head circumference were not recorded. From the 3rd day after birth, he developed physiological jaundice that lasted for 15 days. He raised his head at five months, sat alone at eight months, stood with support at nine months, and walked at 13 months. He presented to us at 15 months of age. Weight, length, and head circumference were 16.5 kg (+4.2 SD), 85.5 cm (+2.5 SD), and 50 cm (+2.2 SD), respectively. His face was long with prominent forehead, antimongoloid slanting, large ears, long philtrum, high-arched palate, and pointed chin. The patient demonstrated slight generalized hypotonia and umbilical hernia.
Electromyography and nerve conduction showed axonal neuropathy. Abdominal ultrasonography revealed dilatation of left renal pelvis at the pelviureteric junction. His EEG showed bilateral focal epileptogenic dysfunction. Routine blood examination, thyroid hormone levels, electrocardiogram, and ophthalmologic examination were within normal ranges. His bone age at chronological age 15 months was 32 months. His developmental quotient test was 73. Brian CT revealed brain atrophy. Unfortunately, molecular genetic analysis of NSD1 for the family was not performed.
Truncating mutations, missense mutations in functional domains, partial gene deletions, or 5q35 microdeletions encompassing the entire NSD1 gene are identifiable in the majority (≥ 90%) of individuals with a classic Sotos phenotype. Protein truncation mutations are found throughout the NSD1 gene, whereas missense mutations cluster in the latter half of the gene where functional domains are located (51). The mechanism of generation and size of 5q35 microdeletions differ depending on the ethnic origin of the affected individual. Outside Japan, 5q35 microdeletions are variable in size and predominantly arise through interchromosomal rearrangements (51). In contrast, a uniform 1.9Mb microdeletion, arising through intrachromosomal rearrangements, has been identified in the majority of individuals of Japanese descent with Sotos syndrome (51; 59; 39). Low-copy repeats flanking the Sotos common deletion showed that the deletion arises through nonhomologous recombination utilizing the low-copy repeats (59). An inversion polymorphism that predisposes to the microdeletion is common in Japan and may explain the high recurrence of the 1.9Mb 5q35 microdeletion in the Japanese population, although the frequency of this inversion polymorphism outside Japan is currently not known (59). Generally, the paternal allele is preferentially deleted in the majority of cases with NSD1 microdeletions. This could be partly attributed to the greatly increased recombination rate in men compared with women at the 5q telomere. Mutational hotspots do not exist (51).
NSD1 encodes the protein nuclear receptor-binding SET domain-containing protein 1 (NSD1), also known as histone-lysine N-methyltransferase, H3 lysine-36 specific. NSD1 falls in the class of epigenetic writers, catalyzing the transfer of methyl groups to lysine residues of histone tails. More specifically, it mediates mono- and dimethylation of lysine residue 36 of histone H3 (H3K36) and, at least in vivo, of lysine residue 20 of histone H4 (H4K20) (44). NSD1 regulates transcription via interactions with H3K36 methylation and RNA polymerase II (09).
As a global epigenetic regulator, NSD1 impacts expression of a large number of genes in a manner that is likely both cell type–specific and developmental stage–specific. The precise genetic pathways by which NSD1 haploinsufficiency results in the features of Sotos syndrome are still under investigation. Several possible mechanisms have been proposed and supported by initial studies. The role of NSD1 in the pathogenesis of Sotos syndrome could be mediated through its action as a corepressor of genes that promote growth (26). It is expressed in several tissues including fetal/adult brain, kidney, skeletal muscle, spleen, and thymus, and the NSD1 protein is probably involved in the transcriptional silencing of developmentally regulated genes during embryogenesis. NSD1-mediated H3K36 methylation may also protect a subset of genes from the repressive activity of polycomb repressive complex 2 (PRC2). In Sotos syndrome, reduced NSD1 levels may, therefore, result in excessive PRC2 activity and, thus, to decreased expression of PRC2-regulated genes, a pattern that has been seen in samples from patients with Sotos syndrome (06). It has also been proposed that deregulation of the MAPK/ERK pathway in Sotos syndrome results in altered hypertrophic differentiation of NSD1-expressing chondrocytes and may be a determining factor in statural overgrowth and accelerated skeletal maturation in Sotos syndrome (57). Further, haploinsufficiency of NSD1 results in dysregulated insulin expression (19).
Malan syndrome is caused by heterozygous variants of NFIX clustered mostly in exon 2. NFIX encodes the protein nuclear factor 1 X-type (NFIX). NFIX variants associated with Malan syndrome are thought to cause haploinsufficiency, either by nonsense variants predicted to result in nonsense-mediated decay and, thus, reduced protein translation, or by missense and in-frame deletions predicted to reduce function of the DNA-binding/dimerization domain (32). Functional studies showed the role of NFIX in chondrocyte differentiation and bone formation and its specific involvement in the endochondral ossification process. The overgrowth in patients with dominant NFIX mutations supports a dysregulation of the switch between proliferation and differentiation stages and also suggests that NFIX could act as a negative regulator of the endochondral ossification process (31; 34; 42; 60). There is no genotype-phenotype correlation in Malan syndrome except for an increased risk for epilepsy with 19p13.2 microdeletions (43).
In two Egyptian siblings with Sotos-like features, homozygous mutation of the APC2 gene was identified. Expression of APC2 was revealed to be under the control of NSD1. The knockdown of NSD1 in neuronal cells suppressed the expression of the APC2 gene, which led to developmental defects in the brain. These results strongly indicated that APC2 is a crucial target of NSD1 (03). DNMT3A, SETD2, and GPC3 gene variants or hypomethylation of KCNQ1OT1 may be responsible for clinical features in a few patients with Sotos-like presentation (05; 34).
The prevalence of Sotos syndrome is estimated at about 1 per 15,000 (54). Pathogenic variants of NSD1 were described in approximately 90% of patients with classic Sotos phenotype (24). Microdeletions encompassing the NSD1 gene have been reported in approximately 50% of Japanese patients and in less than 10% of non-Japanese patients (27; 51). The differences in NSD1 microdeletion frequency are due to differences in genomic architecture rather than case ascertainment bias (27). There have not been any reports of unaffected individuals who carry known pathogenic NSD1 variants. Thus, the penetrance of Sotos syndrome is 100%. The vast majority of NSD1 variants in individuals with Sotos syndrome are de novo, but autosomal dominant transmission has been reported in a few families. Each child of an individual with Sotos syndrome has a 50% risk of inheriting the NSD1 pathogenic variant and having Sotos syndrome. The majority of individuals with Sotos-like features in the absence of NSD1 variants have Malan syndrome associated with specific variants in NFIX. Like Sotos syndrome, most individuals with Malan syndrome have de novo NFIX variants, but one case of transmission from a "possibly affected” mother has been reported (60). Thus, the penetrance of Malan syndrome is high but may be less than 100%.
Genetic counseling is crucial and depends on the family history and genetic testing (54).
Prenatal diagnosis of Sotos syndrome may be considered in cases with a normal fetal karyotype where there is increased risk for Down syndrome demonstrated by maternal serum screening, especially in the presence of supportive ultrasound findings such as macrocephaly, polyhydramnios, and decreased fetal movements (56). When Sotos syndrome is suspected prenatally, genetic testing for NSD1, NFIX, or both should be performed, as it provides a relatively simple, safe, and sensitive confirmatory test in the great majority of cases (54).
Phenotypic overlap with other overgrowth syndromes exists, in particular with the following:
Weaver syndrome. The facial features of Sotos syndrome and Weaver syndrome are similar, particularly in infancy. However, as children get older, patients with Sotos usually have the characteristic prominent chin. Further, advanced dental maturation is remarkably observed in Sotos syndrome but rarely commented on in Weaver syndrome. Moreover, Sotos syndrome may carry an increased risk for cancers, whereas Weaver syndrome does not. Mutations in the histone methyltransferase EZH2 were shown to cause Weaver syndrome (52).
Marshall-Smith syndrome. Like Malan syndrome, Marshall-Smith syndrome is associated with genetic variants in NFIX but presents with a distinct phenotype. It is characterized by increased birth length with subsequent failure to thrive, prominent forehead, prominent eyes, underdeveloped midface and prominent premaxilla micrognathia, anteverted nares, broad proximal and middle phalanges, disharmonic bone maturation, and respiratory compromise secondary to upper airway obstruction, (31; 42; 60).
Variants associated with Marshall-Smith syndrome have exclusively been found in regions outside of those coding for the DNA binding and dimerization domain, especially in exons 6-8 and sometimes in exons 9 and 10. For several Marshall-Smith-associated variants it has been demonstrated that mutant mRNA does not undergo nonsense mediated decay (31; 47), whereas variants associated with Malan syndrome are instead restricted to the 5’ part of the gene encoding for the DNA binding and dimerization domain (43).
Tatton-Brown-Rahman syndrome. Tatton-Brown-Rahman syndrome is a newer overgrowth syndrome characterized by a tall stature, large head circumference, intellectual disability, and a facial gestalt that is characterized by a round face, heavy, horizontal eyebrows, and narrow palpebral fissures. It is caused by DNMT3A (DNA cytosine 5 methyltransferase 3A) mutations or 2p23 microdeletion (55; 40).
Bannayan-Riley-Ruvalcaba syndrome. Bannayan-Riley-Ruvalcaba syndrome is differentiated by the characteristic lipomatosis and hemangiomatosis and penile freckling in boys, which has not been reported in Sotos syndrome. It is due to PTEN mutations in about 60% of cases.
Beckwith-Wiedemann syndrome. Macroglossia is the hallmark in Beckwith-Wiedemann syndrome but has never been reported in Sotos syndrome. Further, visceromegaly, hemihypertrophy, and embryonal tumors are usually associated with Beckwith-Wiedemann syndrome (05). Hypomethylation of KCNQ1OT1 is the most common cause of Beckwith-Wiedemann syndrome, although it was reported in three patients with Sotos-like features (34).
Benign familial macrocephaly. Benign familial macrocephaly lacks the distinctive facial features of Sotos syndrome.
Simpson-Golabi-Behmel syndrome. Coarse facies, vertebral segmentation defects, supernumerary nipples, dental malocclusion, organomegaly, and diaphragmatic hernia make Simpson-Golabi-Behmel syndrome quite distinct from Sotos syndrome. Besides, it has an X-linked mode of inheritance. It is caused by mutations and deletions in GPC3 (05).
In the majority of individuals with Sotos-like presentation, molecular genetic testing is able to confirm a diagnosis of Sotos syndrome, Malan syndrome, or another overgrowth syndrome. Molecular testing is frequently performed using a multigene panel for overgrowth syndromes that includes NSD1 and NFIX as well as genes for other potentially overlapping syndromes. However, narrower testing, ie, single-gene sequencing of NSD1 or NFIX, and broader testing, eg, exome sequencing, may also be used. Given the relatively high prevalence of 5q35 microdeletions in Sotos syndrome, it is important to select a test that includes deletion/duplication analysis.
In cases in which genetic testing results in a variant of uncertain significance (VUS), genome-wide DNA methylation (DNAme) profiling can be used for variant disambiguation. Disorder-specific DNAme signatures have been defined for a number of genetic conditions caused by disruption of epigenetic regulators, and Sotos syndrome was among the first. For individuals in whom genetic testing was not definitive but did reveal an NSD1 VUS, presence of a characteristic DNAme signature can confirm the diagnosis of Sotos syndrome (14). In the United States, clinical DNAme profiling, including for NSD1-associated Sotos syndrome, is available.
After diagnosis of Sotos syndrome, or if clinical concern is high, thorough evaluation for known features should be performed, if not already done. This may include the following:
|
• In the neonatal period, glucose and insulin levels in blood should be evaluated, especially for those displaying irritability and poor feeding (33). | |
|
• Echocardiography to detect cardiac anomalies. | |
|
• X-rays of hands and spine for bone age and scoliosis assessment, respectively. | |
|
• Dental examination must be checked to detect premature eruption or ectopic tooth. | |
|
• Audiometric assessment. | |
|
• Ophthalmologic evaluation. | |
|
• Neuropsychological evaluation, including IQ testing. | |
|
• Echocardiography to detect cardiac anomalies. | |
|
• Abdominal ultrasound is to identify renal anomalies such as vesicoureteric reflux, hydronephrosis, and hydroureter and solid tumors. | |
|
• Brain MRI should be considered, although anomalies of the midline structures are common, but nonspecific. However, the presence of Chiari malformation type 1 is commonly associated with Malan syndrome. | |
|
• EEG should be performed, especially in case of seizures or behavioral disorders. | |
|
• Finally, genetic testing for NSD1, NFIX, or both genes should be performed to confirm the clinical diagnosis (42; 60). |
Rehabilitation. No specific treatment is available, but symptomatic treatment for the associated complications depends on age and severity.
|
• Treat hypoglycemia with glucose infusion; in persistent hypoglycemia treatment with diazoxide (6 mg/kg/day) should be added (33). | |
|
• Nasal tube feeding should be used in case of poor feeding. | |
|
• For severe congenital abnormalities, cardiac surgery may be recommended. | |
|
• In patients with scoliosis, brace wearing can provide control of the curve progression and delay surgical intervention (11). | |
|
• In patients with tremors propranolol and clonidine may be recommended. | |
|
• Monotherapy with valproic acid is the most frequently used in epilepsy. Less frequently, polytherapy with antiepileptic drugs is used. | |
|
• Methylphenidate (successively up to 30 mg/day) was described to significantly reduce aggression and impulsivity (38). | |
|
• It is noteworthy to mention that management guidelines do not include tumor screening, as the types of tumors detected in Sotos syndrome are diverse and do not have well-defined screening protocols (54). | |
|
• Compression stockings and a lymphedema pump three hours per day should be practiced in case of lymphedema. |
|
• Most of the neonates with hypoglycemia displayed normal glucose level within three weeks, although few cases required continuation of the treatments for 3 months. | |
|
• Seizure control was obtained in the majority of individuals with seizures, most often without treatment (eg, in those with febrile seizures only; 19%) or with monotherapy (48%) (16). | |
|
• Aggression and impulsivity improved significantly after two months of therapy with methylphenidate (38). | |
|
• Surgical correction may be required for severe scoliosis not responsive to more conservative management. |
Transmission in families is uncommon. When pregnancy occurs, it is described to have normal prenatal history. However, toxemia or preeclampsia have been reported in several cases (54; 29). After birth, neonates with Sotos syndrome may develop neonatal hyperinsulinemic hypoglycemia, neonatal jaundice, or feeding difficulty.
Regional techniques of anesthesia are the reasonable choice. They should be performed after induction given the high prevalence of intellectual disability potentially leading to decreased ability and willingness to cooperate and, to a lesser extent, aggressive behavior in children with Sotos syndrome (08). The use of muscle relaxants may not be necessary in patients with marked hypotonia. Further, it can be suggested to avoid opioids, drugs that decrease seizure threshold, and antiepileptic drugs (01).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Christina Grant MD PhD
Dr. Grant of Children’s National Hospital received a consulting fee from Amicus Therapeutics and honorariums from Sanofi Genzyme for serving on an advisory committee.
See ProfileAndrea Cohen MD
Dr. Cohen of the National Human Genome Research Institute has no relevant financial relationships to disclose.
See Profile
Ganeshwaran H Mochida MD PhD
Dr. Mochida of Boston Children's Hospital and Harvard Medical School has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026