



Sleep Disorders
Sleep-related urologic dysfunction
Jul. 06, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Tuberous sclerosis complex is an autosomal dominant, multisystem genetic disorder with heterogeneous clinical manifestations. This update provides a succinct overview of the disorder and highlights advances in surveillance and treatment across organ systems. The authors summarize recent clinical trials and consensus recommendations, with particular emphasis on precision medicine approaches using mTOR inhibitors and on neurologic complications, including epilepsy, autism spectrum disorder, and tuberous sclerosis complex–associated neuropsychiatric disorders (TAND). Evolving evidence on early epilepsy management and its potential impact on neurodevelopmental outcomes is also reviewed.
|
• Tuberous sclerosis complex is a prototypical neurocutaneous disorder with substantial neurologic morbidity, particularly epilepsy, cognitive impairment, autism spectrum disorder, and tuberous sclerosis complex–associated neuropsychiatric disorders (TAND). Diagnostic and therapeutic capabilities have advanced markedly over the past decade. | |
|
• Epilepsy frequently presents in infancy, often as infantile spasms. Earlier onset correlates with higher risk of refractory epilepsy and developmental impairment. | |
|
• Autism spectrum disorder affects approximately 40% to 50% of individuals with tuberous sclerosis complex, far exceeding general population rates. TAND is highly prevalent and spans behavioral, psychiatric, intellectual, academic, neuropsychological, and psychosocial domains. | |
|
• mTOR inhibitors (eg, everolimus, sirolimus) have transformed management of several tuberous sclerosis complex manifestations, improving outcomes for subependymal giant cell astrocytomas (SEGAs), renal angiomyolipomas, lymphangioleiomyomatosis, and other hamartomas. | |
|
• Early epilepsy identification and treatment may favorably influence seizure burden and developmental outcomes, though some trial data are mixed. | |
|
• Prognosis has improved with contemporary surveillance and targeted therapies. Renal disease remains a major determinant of morbidity and mortality. |
Désiré‑Magloire Bourneville described “tuberous sclerosis of cerebral circumvolutions” in 1880 in a young girl with recurrent status epilepticus, noting sclerotic, firm cortical gyri and periventricular nodules with additional renal tumors (19). Nearly two decades earlier, von Recklinghausen had reported a neonate with cardiac “myomata” and multiple cerebral scleroses (139). Dermatologists subsequently described the characteristic facial hamartomas—formerly termed “adenoma sebaceum,” now “facial angiofibromas”—and their association with seizures and cognitive impairment (07; 108; 103; 105; 83). These observations culminated in the designation of tuberous sclerosis complex, reflecting the multiorgan nature and development of hamartomas. Early clinical recognition also relied on “epiloia” (Vogt’s triad: epilepsy, low IQ, and adenoma sebaceum) (138).
Key milestones include recognition of heredity (10), evolution of “phakomatoses” (134), radiologic identification of intracranial calcifications (84), and classic clinical syntheses (26). Molecular mapping and cloning of TSC1 (9q34.3) and TSC2 (16p13.3) were pivotal (52; 65). Over the last 2 decades, targeting mTOR pathway with rapamycin/sirolimus and everolimus demonstrated efficacy in epilepsy, SEGAs, renal angiomyolipomas, and lymphangioleiomyomatosis (50; 146; 16). The impact of preclinical management of epilepsy on epileptogenesis and neurodevelopment is being evaluated.
Neuroendocrine tumors (NETs) have been described in the last decade, with increasing identification of these during surveillance MRIs of the abdomen. Functional and nonfunctional pancreatic neuroendocrine tumors have been identified with insulinomas as the most common functional neuroendocrine tumors, with other neuroendocrine tumors including gastrinomas, glucagonomas, ACTHomas, and GHomas (05).
Tuberous sclerosis complex exhibits a broad phenotypic spectrum, occurring in approximately one in 6000 to one in 10,000 individuals (116; 102). Diagnosis is based on clinical features, imaging, pathology, and/or genetic testing. Consensus diagnostic criteria have been periodically refined to incorporate major and minor clinical features and to integrate genetic criteria (113; 96; 94).
Major features have high specificity for tuberous sclerosis complex; minor features are less specific. Although skin findings are most prevalent, the brain, kidneys, retina, heart, lungs, and large arteries are commonly involved. The 2013 revision added genetic diagnostic criteria and removed “probable” disease (96). The 2021 update replaced “cortical dysplasias” with “multiple cortical tubers and/or radial migration lines” and reinstated sclerotic bone lesions as a minor feature, while reaffirming genetic diagnostic criteria and noting the importance of mosaicism (94).
The identification of a heterozygous pathogenic variant in TSC1 or TSC2 from normal tissue is sufficient to make a definite diagnosis of tuberous sclerosis complex, recognizing that phenotypic features emerge over time. A pathogenic variant is defined as a variant that clearly inactivates the function of the TSC1 or TSC2 proteins (eg, frameshift or nonsense variant), prevents protein synthesis (eg, large genomic deletion), or is a missense variant whose effect on protein function has been established by functional assessment.
Other TSC1 or TSC2 variants whose effect on function is less certain do not meet these criteria and are not sufficient to make a definite diagnosis of tuberous sclerosis complex. Ten percent to 15% of patients with tuberous sclerosis complex meeting clinical criteria have no mutation identified by conventional genetic testing. Normal genetic results do not exclude tuberous sclerosis complex or have any effect on the application of clinical diagnostic criteria to diagnose tuberous sclerosis complex. High-read-depth approaches to next-generation sequencing yield evidence of mosaicism and intronic mutations in some of these patients.
Two major or one major and two minor criteria are required for definite diagnosis.
|
Major features | ||
|
1. Hypomelanotic macules (> /=3, at least 5 mm diameter) | ||
|
Minor features | ||
|
1. “Confetti” skin lesions | ||
|
Definite tuberous sclerosis complex | ||
|
1. Two major features or, | ||
|
Possible tuberous sclerosis complex | ||
|
1. One major feature or, | ||
|
If both renal angiomyolipoma and lymphangioleiomyomatosis are present, other features of tuberous sclerosis complex should be present for a definite diagnosis. | ||
The type, size, number, and sometimes the location of involved lesions and organ systems dictate clinical presentation.
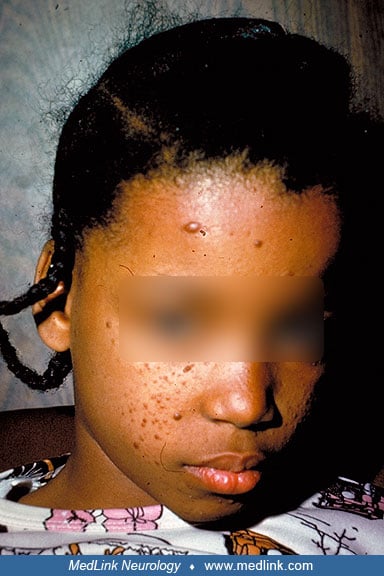
Skin. Cutaneous involvement occurs in 96% to 100% of individuals with tuberous sclerosis complex, often identified in the context of seizure evaluation. Common lesions include facial angiofibromas, fibrous cephalic plaques, shagreen patches, ungual fibromas, confetti skin lesions, and hypomelanotic macules (90; 115).
Of all patients with tuberous sclerosis complex, 87% have hypomelanotic macules, 75% have facial angiofibromas, 50% have shagreen patches, 21% have ungual fibromas, 3% to 58% have confetti skin lesions, and almost none are without skin lesions at all. The most common skin manifestation, the hypomelanotic macule, is best seen with a Wood’s lamp and is often noted over the buttocks and trunk. The facial angiofibromata are usually bilateral, in a butterfly distribution over the nasolabial folds and malar area. They have been recognized to manifest secondary to ultraviolet sunlight exposure as a form of a second hit (131). Shagreen patches occur over the lower trunk and flank. They may appear in the neck, upper trunk, buttocks, and thighs, too. Fibrous cephalic plaques may be the most specific dermatological finding in tuberous sclerosis complex. In addition, dental pits have been noted in 90% of patients with tuberous sclerosis complex, approximately 10 times more often than in the general population (45).
Brain. The CNS manifestations of tuberous sclerosis complex, noted in about 90% of afflicted children, include seizures, cortical tubers, tumors, intellectual disability, autism spectrum disorder, ADHD, and sleep disorders. Tuberous sclerosis complex–associated neuropsychiatric disorders (TAND) is a term to describe the wide range of disorders including behavioral, psychiatric, intellectual, academic, neuropsychological, and psychosocial difficulties associated with tuberous sclerosis complex.
Epilepsy. Epilepsy occurs in approximately 84% overall and in over 95% of infants; infantile spasms affect approximately one third of children with tuberous sclerosis complex. Earlier seizure onset predicts greater risk of developmental delay and refractory focal epilepsy. Seizures may begin as subtle focal events and evolve into infantile spasms; controlling infantile spasms and focal seizures is associated with improved developmental outcomes.
Adults commonly have multiple seizure types; approximately 93% have focal‑onset seizures and approximately two thirds have refractory epilepsy. Risk factors for refractoriness include onset under 2 years of age, history of infantile spasms or Lennox–Gastaut syndrome, lower educational attainment, psychiatric comorbidity, and TSC2 variants (119).
Structural brain lesions. Cortical tubers and radial lines of migration occur in 90% of patients. Tubers are glioneuronal lesions, occurring in more than 90% of patients. Subependymal nodules are present in almost 80% of patients and can be identified prenatally or at birth. They are commonly low-grade dysplasias, which share histology with subependymal giant cell astrocytomas, whose presentation may vary from asymptomatic to obstructive hydrocephalus and death. CNS tumors are found in 5% to 15% of individuals afflicted with tuberous sclerosis complex.
These tumors differ in their location, radiological characteristics, and biological behavior (126). Subependymal giant cell astrocytomas are glioneuronal tumors located near the foramen of Munro, are greater than 0.5 cm in size, and grow and enhance with gadolinium on MRI. They occur in 10% to 15% of patients with tuberous sclerosis complex, almost exclusively in patients with the TSC2 mutation. Subependymal giant cell astrocytoma growth acceleration is related to young age, size greater than 2 cm, and TSC 2 genotype. They most commonly manifest between 8 and 14 years of age.
Less commonly, there are aggressive tumors like pineal giant cell astrocytomas, glioblastoma multiforme, or spongioblastoma. Ependymoma, neurinoma, acoustic neuroma, hemangioma, and neuroblastoma have also been reported. Hemimegalencephaly and scoliosis have been observed in a higher frequency of patients with tuberous sclerosis complex compared to normals.
Neuropsychiatric/behavioral. Tuberous sclerosis complex-associated neuropsychiatric disorders (TAND) is terminology that describes the interrelated functional and clinical manifestations of brain dysfunction seen in tuberous sclerosis complex, with seven natural TAND clusters identified in the literature: autism-like, dysregulated behavior, eat/sleep, mood/anxiety, neuropsychological, overactive/impulsive, and scholastic (37). More than 90% manifest one or more TAND concerns in their lifetime, yet under‑recognition and undertreatment are common (39).
Sleep. Night waking, early waking, seizure‑related sleep problems, and daytime sleepiness are frequent (57). A remarkably high association was found with TAND in a questionnaire-based study of EPISTOP, suggesting that attention to sleep may have a positive impact on attention and behavior (89).
Autism spectrum disorder and ADHD. Autism spectrum disorder and ADHD each affect about 40% to 50% of patients with tuberous sclerosis complex, in contrast to 2% and 4%, respectively, in the general population. Autism spectrum disorder phenotypes in tuberous sclerosis complex substantially overlap with nonsyndromic autism spectrum disorder (60). Autism spectrum disorder comorbidity confers a higher risk of global cognitive impairment (59).
Cognition and academic abilities. Approximately 36% to 58% of children with tuberous sclerosis complex have significant academic difficulties requiring intervention (36; 31; 68). A number of investigations have demonstrated that a history of infantile spasms or poor seizure control in general are associated with lower intellectual ability (23; 35). The PREVeNT and EPISTOP intervention studies on infants with seizures in tuberous sclerosis complex demonstrated small but notable gains in developmental outcomes likely associated with earlier diagnosis, monitoring epilepsy, early treatment, and referral to early intervention services (99).
Eye. Astrocytic retinal hamartomas are found in about 50% of patients and are histopathologically similar to subependymal nodules and subependymal giant cell astrocytomas. They may continue to grow past adolescence, but they are seldom symptomatic. Pigmentary changes and punched out lesions may also be found on retinoscopy. Retinal involvement correlates with higher rates of SEGA, renal angiomyolipoma, cognitive impairment, and epilepsy (114; 04).
Kidney. Renal lesions occur in more than half of children at the time of initial evaluation. Eighty percent of children are affected by 10 years of age (44). Patients with TSC2 are far more likely than those with TSC1 to have significant renal manifestations (86).
• Simple epithelial cysts occur in up to 50% of patients and may appear or disappear at any time; they commonly present with severe hypertension. | |
• Angiomyolipomas, which account for 75% of renal abnormalities, with a median age of detection of 8 to 13 years, increase in size with age and may become symptomatic, causing lumbar pain and hematuria, especially in women by the third decade. They predispose to aneurysms and massive bleeding (Wunderlich syndrome), especially when the angiomyolipomas are greater than 4 cm in diameter and the aneurysms are greater than 5 mm in size (149). Malignant transformation is rare (< 1%). | |
• Renal cell carcinoma occurs at an earlier age in tuberous sclerosis complex and is more frequent (2% to 5%) than in comparable cohorts (17; 25). Intriguingly, all major histological types of renal cell carcinoma have been reported in tuberous sclerosis complex. Age of diagnosis of angiomyolipomas in TSC2 is a decade earlier than TSC1, reiterating the severity of disease manifestation in TSC2 (86). | |
• TSC2/PKD1 contiguous gene deletion syndrome produces an autosomal dominant polycystic kidney disease phenotype with neonatal/infantile hypertension. Large kidney size may delay gross motor milestones, and reduced concentration can delay nocturnal continence. Patients are at risk for intracranial aneurysms and hepatic cysts (20; Bissler and Kingswood 2018). | |
• Premature decline of glomerular filtration rate occurs in 40% of patients with tuberous sclerosis complex. | |
• Hypertension develops in about 5% of children and 25% of adults (86). |
Heart. Cardiac involvement is common in tuberous sclerosis complex and is found in up to 80% of patients (109). Cardiac rhabdomyomas are common in infants (approximately 50% on echocardiography) and can be detected prenatally; most regress spontaneously (141; 130). Prenatal rhabdomyomas confer approximately 75% to 80% risk of postnatal tuberous sclerosis complex (06). Arrhythmias and mechanical effects can occur, especially perinatally. Wolff-Parkinson-White syndrome may occur with or without rhabdomyomas (133). Giant intracranial aneurysms of the internal carotid artery have also been reported (64).
Lung. Pulmonary issues include lymphangioleiomyomatosis, clear cell tumors of the lung, and multifocal micronodular pneumonocyte hyperplasia (107). Pulmonary lymphangioleiomyomatosis is the third leading cause of mortality after CNS and renal phenotypes (120) in tuberous sclerosis patients. The prevalence of lymphangioleiomyomatosis in tuberous sclerosis complex has been reported to be as high as 80% (27). It usually presents in women in their third or fourth decade with recurrent spontaneous pneumothorax, hemoptysis, chylothorax, and respiratory failure. An overwhelming majority of women have cystic lung disease by the age of 40. Women with tuberous sclerosis complex and lymphangioleiomyomatosis are also highly likely to manifest renal angiomyolipomas. Sporadic lymphangioleiomyomatosis occurs later, has a more severe course, and is also associated with renal angiomyolipomas about a third of the time. Thus, co‑presence of lymphangioleiomyomatosis and renal angiomyolipomas constitutes a single major criterion for tuberous sclerosis complex.
Blood vessels. Vascular lesions of tuberous sclerosis complex reflect a defect in the arterial walls of large and medium arteries, predisposing to aneurysm formation.
Other findings. These include angiomyolipomas in various organs, thyroid and parathyroid adenomas, liver, colon and rectal polyps, gingival fibromas, chordomas, and bone cysts. Neuroendocrine tumors have been described with insulinomas as the most common functional neuroendocrine tumors, with other neuroendocrine tumors including gastrinomas, glucagonomas, ACTHomas, and GHomas. Standard MRI surveillance and treatment are recommended for nonfunctional pancreatic neuroendocrine tumors (05).
Rare manifestations and malignancies. Scoliosis and hemihypertrophy were more common in comparison to the general population, as were breast and thyroid cancers in a report of the TuberOus Sclerosis registry to increase disease Awareness (TOSCA) (118).
|
Fetal/neonatal |
Seizures, arrhythmias, Wolff-Parkinson-White, hydrops fetalis |
|
Infancy |
Infantile spasms, retinal hamartomas, hypomelanotic macules |
|
Early childhood |
Autism spectrum disorder, seizures, hypomelanotic macules |
|
Late childhood |
SEGA, facial angiofibromas, ungual fibromas |
|
Adolescence |
Shagreen patches |
|
Adulthood |
Lymphangioleiomyomatosis, renal angiomyolipomas (54) |
Overall morbidity and mortality reflect the extent and severity of multi-organ involvement. Renal disease remains a leading cause of death. TSC2 variants are generally associated with more severe phenotypes than TSC1, a finding supported by EPISTOP (98).
Major contributors to mortality include status epilepticus, SUDEP, severe respiratory infections (especially in those with profound cognitive impairment), and respiratory failure in biopsy‑proven lymphangioleiomyomatosis (approximately 40%) (120). Renal angiomyolipomas carry a 25% to 50% risk of hemorrhage and may present with hypovolemic shock. Cardiac dysrhythmias, including Wolff-Parkinson-White, can be problematic, though fatal outflow obstruction is rare (97). Thoracoabdominal aneurysms may occur in childhood and have substantial risk of rupture.
The natural history of the disease is changing with precision medicine, specifically with mTOR inhibitors in the management of angiolipomas and SEGAs, early use of antiseizure medications, and greater acceptance of epilepsy surgery. Early management of frequent interictal discharges has been shown to improve seizure control and may have a positive impact on cognitive functioning.
A 7-month-old girl presented with a history of spells manifesting for 1 week prior to presentation to the physician. These episodes were characterized by a brief flexion of her neck and abduction and extension of the arms. They occurred 15 to 20 times in a cluster, many times a day, predominantly while awakening. The clusters had been gradually lengthening in duration over time. She occasionally cried after a cluster. She had stopped smiling and tracking. She no longer cooed or played with her toys.
She was born vaginally at term following an uncomplicated pregnancy. Her birth was unremarkable except for an episode of tachycardia diagnosed to be Wolf-Parkinson-White syndrome. Her development had been age-appropriate, and the family history was characterized by a birthmark in her father over his back, which was consistent with a shagreen patch on examination.
On exam she was non-dysmorphic and alert. She did not smile, coo, or track consistently. On Wood’s light exam, she had a few hypopigmented macules on her trunk, in addition to café-au-lait spots. The cranial nerve exam was normal for her age. There was neck lag on pulling to sit and appendicular hypotonia.
Brain MRI revealed cortical tubers and subependymal nodules on both T1 and T2 images. The cardiac echo revealed a rhabdomyoma. EEG revealed multifocal interictal discharges and electrodecrement correlating with clinical events of infantile spasms but did not reveal hypsarrhythmia. The child was initiated on vigabatrin at an escalating dose to a maximum of 150 mg/kg/day and did well without clinical seizures for the next 10 days, after which seizures recurred. She was initiated on adrenocorticotropic hormone with good benefit.
This child met the criteria for definite tuberous sclerosis complex, having four major criteria for diagnosis. The high likelihood of seizures in the first year of life is exemplified in this case, as well as the occurrence of infantile spasms without hypsarrhythmia. The case reiterates the difficulty in the management of seizures despite the use of the currently recommended first-line therapy, vigabatrin.
|
• mTOR pathway dysregulation underlies most manifestations of the disease complex and provides a therapeutic target for precision medicine. | |
|
• Biallelic loss of either TSC1 or TSC2 within a lesion (via a germline hit plus a somatic “second hit”) drives hamartoma formation. | |
|
• Neurodevelopmental and epileptogenic mechanisms include altered neuronal migration/lamination, dendritic architecture, synaptic function, ion channel expression, glial glutamate uptake, inflammation, and myelination. |
Genetics. Tuberous sclerosis complex is an autosomal-dominant disorder caused by a genetic mutation in one of two different genes. Chromosomal bands 9q34.3 and 16p13.3 are the loci for the two genes; they are respectively called TSC1 (tuberous sclerosis complex 1) and TSC2 (tuberous sclerosis complex 2) (52; 65). The 16p13.3 TSC2 gene was identified first, and its 5.5 kilo base TSC2 transcript encodes a 200 KDa protein called tuberin. The 9q34.3 TSC1 gene was identified from a 900-kilobase region containing over 30 genes, and its 8.6 kilo base TSC1 transcript encodes a 140 KDa protein called hamartin.
As of March 1, 2026, 1455 unique allelic DNA variants of TSC1 and 5017 unique DNA allelic variants of TSC2 have been reported. More information is available at LOVD tuberous sclerosis database.
Pathogenic variants in TSC2 and TSC1 occur in a 2:1 ratio among tuberous sclerosis complex patients. Only 5% to 10% of patients have no mutation identified or a variant of unknown significance after assessment with ultra-deep next-generation sequencing. Most patients previously assessed to have no mutation identified have low level mosaic pathogenic variants or pathogenic variants in introns that affect splicing.
There are differences in the phenotypic expressions of TSC1 and TSC2 gene mutations. TSC2 mutations express a more severe phenotype – more severe renal involvement, intellectual disability, more cerebral and facial lesions, less reproductive fitness (61), and more pulmonary involvement. Complex partial seizures, focal seizures, and infantile spasms are more likely in TSC2. About 80% of cases are caused by de novo mutation (113), with an estimated 15% or more of the individuals with tuberous sclerosis complex exhibiting somatic mosaicism and another 1% or so having germline mosaicism. Tuberous sclerosis complex could appear as sporadic disease in the general population.
Response of tumors including subependymal giant cell astrocytomas and angiomyolipomas to mTOR inhibitors is independent of mutation type and also occurs in patients with no mutation identified (104).
Cell biology and pathophysiology. The pathology of tuberous sclerosis complex reflects abnormalities in cell size, number, morphology, and location, implying multiple roles of the genes. Wild TSC2 and TSC1 function as tumor suppressor genes. With the mutation of either one, their defective product is unable to inactivate the tumor growth caused by a second random somatic cell mutation (loss of heterozygosity). The multifocal nature of tuberous sclerosis complex is best explained by the Knudson 2-hit hypothesis, where the second hit is a somatic mutation that completely abrogates TSC1-TSC2 function by accelerating the effect of the first systemic hit/mutation. Both alleles of either TSC1 or TSC2 need to be inactivated for the development of tumors, ie, loss of heterozygosity.
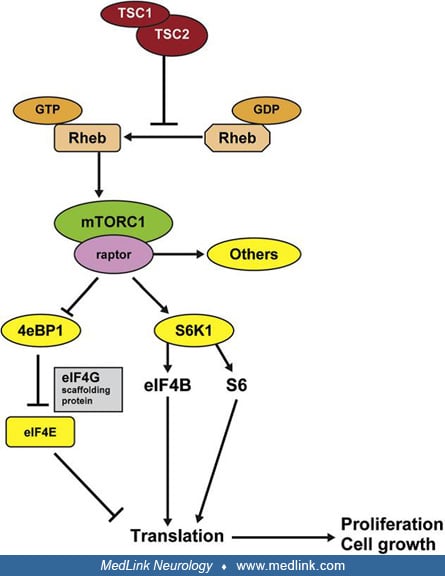
The protein products tuberin and hamartin function together with the protein product of the TBC1D7 gene in cellular signaling pathways (135; 136), forming the TSC protein complex. The TSC protein complex is purported to participate in protein translation, cell growth, proliferation, adhesion, migration, and intracellular trafficking. The TSC complex is the principal cellular inhibitor of the mechanistic target of Rapamycin (mTOR, previously called mammalian target of rapamycin) (100), while also being the sensor of cellular growth conditions. Thus, mutation of hamartin or tuberin in tuberous sclerosis complex leads to hyperactivation of the downstream mTOR pathway and the associated kinase signaling cascades and translational factors, resulting in increased cell growth and proliferation. Tuberin and hamartin are co-expressed in several cells, including kidney, brain, lung, and pancreas, and mutations in either hamartin or tuberin lead to a single disorder.
By sharing homology with a GTPase activating protein for Rap 1 and GTPase Rheb (Ras homologue enhanced in brain), the TSC complex inactivates GTP-bound Rheb (150). Active Rheb is an upstream positive modulator of mTOR. mTORC1 complex includes mTOR protein kinase and the subunits Deptor, Raptor, PRAS40, and mLST8. Activated mTORC1 stimulates synthesis of proteins, nucleotides, and lipids as well as biogenesis of ribosomes and inhibits autophagy and apoptosis (24). Therefore, a loss of function mutation in TSC1-TSC2 will enhance Rheb, activate mTORC1 constitutively, and critically upregulate cell growth and proliferation through p70S6kinase (78), ribosomal S6 proteins, and eukaryotic initiation factor 4E binding protein 1 (4E-BP1).
Mechanistic target of rapamycin, mTOR, exists as two complexes with differing functions. mTOR complex 1 (mTORC 1) with its cofactor, Raptor (regulatory associated protein of mTOR), activates mTOR’s protein kinase domain and is sensitive to rapamycin. This activation results in increased mRNA transcription and protein synthesis. TSC mutation leads to loss of inhibition of mTORC1. This causes constitutive activation of mTOR and, in turn, abnormal cellular proliferation and differentiation, producing the hamartomatous lesions of tuberous sclerosis complex. mTOR complex 2 (mTORC 2), with its cofactor Rictor (rapamycin insensitive component of mTOR), regulates protein synthesis in a manner distinct from mTORC1 and is unaffected by Rheb or rapamycin.
Loss of TSC1 or TSC2 in mature postmitotic hippocampal neurons in vitro causes enlarged somas, abnormal dendritic spines, and enhancement of glutamatergic neurotransmission. Elevated extracellular glutamate levels are assumed to contribute to excitotoxic neuronal death, abnormal glutamatergic synaptic physiology, and impaired behavioral conditioning and learning (92). mTOR dysregulation is also involved in tuberous sclerosis–related epileptogenesis through a range of potential mechanisms, including alteration of neuroblast migration, cortical lamination, cell body size, and dendritic arborization, synaptic plasticity, and by altering neuronal excitability through modulation of the expression of voltage-gated potassium channels.
Risk of epilepsy and encephalopathy are associated with TSC2 genotype, TSC2 mutation, abnormal neuronal morphology, disruption of GABAergic interneuron development, abnormal astrocyte glutamate uptake, synaptic abnormalities, inflammatory changes, impaired long-term potentiation, high tuber brain proportion, and white matter abnormalities (147; 28). mTOR dysregulation is thought to affect the maturation of the GABAergic system, leading to a ‘‘GABAergic immaturity,’’ which may contribute to the development of epilepsy. Widespread hypo-/ dysmyelination has emerged as an important pathological component underlying the complex clinical phenotypes in patients with tuberous sclerosis complex.
Tuberous sclerosis complex occurs in one of 6000 to 10,000 individuals, with two thirds of cases being sporadic (102). A study in Sweden showed a peak prevalence of one in 6800 for individuals between 11 and 15 years of age, and one in 12,900 for all individuals younger than 20 years of age. The TSC2 gene is associated with the vast majority of cases, both familial and sporadic. Boys are typically more severely affected.
|
• Inheritance is autosomal‑dominant with high penetrance. An affected parent has a 50% risk of having an affected child; however, most cases are sporadic due to high de novo mutation rates. | |
|
• There is marked intrafamilial variability. A minimally symptomatic or unrecognized affected parent can have a child with severe epilepsy and intellectual disability/cognitive impairment. | |
|
• Mosaicism complicates risk assessment. Somatic and germline mosaicism and variable expression can obscure true inheritance patterns and recurrence risk. | |
|
• Reproductive options are available. Genetic counseling, preimplantation genetic testing, and prenatal diagnostic testing (when a familial pathogenic variant is known) should be offered. | |
|
• Prenatal imaging can raise suspicion. Fetal echocardiography/ultrasonography may detect cardiac rhabdomyomas or multicystic kidneys. Fetal brain MRI (after approximately 20 weeks) may reveal large cortical tubers. |
Tuberous sclerosis complex is inherited in an autosomal‑dominant pattern with high penetrance; thus, the risk of transmission from an affected parent to offspring is 50%. Nevertheless, the majority of cases are sporadic, reflecting a high de novo mutation rate. Intrafamilial heterogeneity is substantial: a mildly affected or asymptomatic parent (including those with mosaicism) may have a child with severe epilepsy and significant cognitive impairment. Accurately estimating mutation rates is challenging because of reduced/variable expression, non‑expression in some tissues, and somatic or germline mosaicism.
Genetic counseling is essential. When a familial TSC1 or TSC2 pathogenic variant is known, preimplantation and prenatal genetic testing are available. In families without a known variant, linkage analysis may be considered if multiple informative relatives are available. Contemporary mutational analysis methods (including next‑generation sequencing and multiplex ligation‑dependent probe amplification) support genotype–phenotype correlation and reproductive planning.
Prenatal detection of features suggestive of tuberous sclerosis complex may be possible. Fetal echocardiography or ultrasonography can identify cardiac rhabdomyomas or multi-cystic kidneys. Fetal brain MRI after approximately 20 weeks can reveal large cortical tubers when present.
Tuberous sclerosis complex is a multisystem disorder affecting the brain, skin, heart, lungs, kidneys, and other organs. Consequently, the differential diagnosis depends on the presenting problem and is broad. The following should be considered in the evaluation of tuberous sclerosis complex:
|
• Autism spectrum disorder and attention‑deficit/hyperactivity disorder (ADHD) | |
|
• Focal neurologic deficits (eg, hemiplegia) | |
|
• Intra‑abdominal hemorrhage | |
|
• Respiratory failure (particularly in women with cystic lung disease) | |
|
• Seizures (including infantile spasms) and developmental delay/cognitive impairment | |
|
• Uremia or renal masses/cysts |
Tuberous sclerosis complex should also be in the differential when rare tumors (eg, angiomyolipomas, SEGAs, astrocytic retinal hamartomas) are detected, as many are uncommon in the general population.
Tuberous sclerosis complex can underlie tumors across organ systems. Although most tuberous sclerosis complex–associated tumors are benign hamartomas, renal cell carcinoma occurs earlier and with greater frequency in tuberous sclerosis complex than in comparable cohorts (17; 25). Given tuberous sclerosis complex’s systemic nature, organ‑specific evaluations are often required to distinguish tuberous sclerosis complex–related pathology from mimics and to identify clinically silent comorbidities.
|
• A three‑generation pedigree is essential to identify at‑risk relatives and inform genetic testing and counseling. | |
|
• Genetic testing is recommended for counseling and when clinical criteria are insufficient for a definite diagnosis. A negative result does not exclude tuberous sclerosis complex. | |
|
• Organ‑system evaluation at diagnosis and for surveillance includes skin, eyes, brain (MRI/EEG), kidneys (MRI and renal function), lungs (CT/pulmonary function tests in appropriate adults), heart (echo/ECG), and dental evaluation. |
Obtain a three‑generation family history for all patients to identify additional at‑risk relatives. Genetic testing is recommended for reproductive counseling and when tuberous sclerosis complex is suspected but clinical criteria are not fully met. Clinical diagnostic criteria confer the diagnosis regardless of genetic results.
Initial and longitudinal evaluation should include the following:
|
Brain |
MRI and EEG |
|
Dental |
Comprehensive dental examination |
|
Eyes |
Dilated indirect ophthalmoscopy to assess for astrocytic hamartomas and retinal achromic patches |
|
Heart |
Fetal echocardiogram if prenatal ultrasound suggests rhabdomyomas; in children under 3 years, echocardiography; ECG across the lifespan (53). |
|
Kidneys |
Abdominal MRI, glomerular filtration rate assessment (eg, creatinine or cystatin C), urinalysis/proteinuria, and blood pressure |
|
Lungs |
In adults—particularly women and symptomatic men—low‑dose or high‑resolution chest CT and pulmonary function testing |
|
Skin |
Detailed exam in natural and Wood’s lamp light for facial angiofibromas, fibrous cephalic plaques, shagreen patches, periungual/ungual fibromas, and hypomelanotic macules |
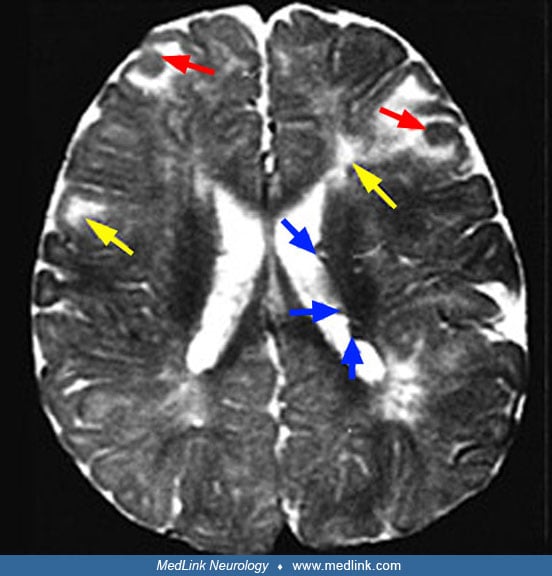
Neuroimaging. On brain MRI, T1‑weighted sequences best identify subependymal nodules, whereas FLAIR is more sensitive than spin‑echo for cortical/subcortical tubers (66). T2‑weighted images may show subcortical white matter changes; subependymal nodules can indent the ventricular wall. To minimize cumulative exposure, avoid contrast unless there is clinical concern for SEGA or for a lesion demonstrating growth, given lifelong surveillance needs. MR imaging demonstrates cyst-like structures that are commonly seen immediately adjacent to cortical tubers or within dysplastic lesions (137).
Advanced neuroimaging. Magnetization transfer imaging can reveal additional tubers and white‑matter anomalies in older children and adults. Dual inversion recovery MRI may depict tubers with high conspicuity compared to high‑resolution T2/FLAIR. Computational morphometry quantifies lesion burden and distribution. Proton MR spectroscopy typically shows decreased N‑acetylaspartate/creatine and increased myoinositol/creatine in tubers, consistent with neuronal loss and gliosis. Diffusion tensor imaging demonstrates perilesional abnormalities and may assist in mapping epileptogenic networks; increased ADC has been associated with epileptogenic tubers. FDG‑PET may show broader hypometabolism than FLAIR suggests, and AMT‑PET (alpha[11C] methyl-L-tryptophan positron emission tomography) can assist in localizing epileptogenic cortex (82). HASTE (Half Fourier Acquisition with Single shot Turbo spin Echo) and MPRAGE (Magnetization Prepared Rapid Gradient Echo) sequences can increase sensitivity for subtle tubers and subependymal nodules (74). Arterial spin labeling may help identify early childhood tubers correlating with EEG slow waves, but further validation is required.
Genetic testing. The preferred first‑line test is a concurrent TSC1/TSC2 next‑generation sequencing panel with deletion/duplication analysis. A broader multigene epilepsy/neurocutaneous panel (including TSC1/TSC2) is reasonable when phenotypic certainty is lower, ideally with deletion/duplication analysis.
Testing is indicated when (1) clinical features do not permit a definite diagnosis, and/or (2) mutation identification will guide family screening or prenatal/preimplantation genetic testing options. Conventional Sanger plus deletion/duplication analysis identifies approximately 75% to 90% of cases. Next-generation sequencing increases yield by detecting mosaicism and intronic splice variants. The LOVD database can assist in variant interpretation. Low‑level variants may be detectable only in saliva or skin; confirm very low‑level mosaic variants across multiple tissues when feasible.
• Multidisciplinary care and guideline‑based surveillance (neurology, nephrology, pulmonology, cardiology, dermatology, ophthalmology, dentistry, genetics, psychiatry/psychology, developmental pediatrics, and rehabilitation) are essential (94; 121). | |
• Early epilepsy detection and treatment are critical for developmental outcomes; evidence from EPISTOP and PREVeNT informs EEG‑guided surveillance and timing of therapy. | |
• mTOR inhibitors (everolimus, sirolimus) have transformed care for SEGA, renal angiomyolipomas, lymphangioleiomyomatosis, and refractory focal epilepsy in tuberous sclerosis complex. | |
• Surgical and device‑based therapies (resective surgery, LITT, vagus nerve stimulation, responsive neurostimulation) are important options for drug‑resistant epilepsy. | |
• Comprehensive TAND screening and intervention across the management and lifespan improve functional outcomes and quality of life. |
Multidisciplinary care and resources. Care is optimized in regional tuberous sclerosis complex clinics with coordinated specialty services. The Tuberous Sclerosis Alliance provides comprehensive resources for clinicians and families. Consensus clinical management and surveillance recommendations and large registries (eg, TOSCA) guide practice and characterize natural history (68; 94; 121).
Transition to adult care. Transition from pediatric to adult services is high‑risk for care gaps; child neurologists play a central role in structured transition planning (21).
Organ system or | Recommendations for surveillance |
Genetics | 1. Obtain three-generation family history to assess for additional family members at risk of tuberous sclerosis complex. |
Brain | 1. Obtain MRI of the brain at diagnosis to assess for the presence of tubers, subependymal nodules, migrational defects, and SEGA. Continue with MRIs every 1 to 3 years to monitor SEGA growth. |
TAND | 1. Perform comprehensive assessment for all levels of potential TAND manifestations. Repeat annually between ages 1 and 16, then again between ages 28 and 35. |
Kidney | 1. Obtain MRI of the abdomen at diagnosis and every 1 to 3 years to assess for the presence of angiomyolipomas and renal cysts. |
Lung | 1. Inquire about tobacco exposure, connective tissue disease manifestations, signs of chyle leak, and pulmonary manifestations of dyspnea, cough, and spontaneous pneumothorax in all adult patients with tuberous sclerosis complex. |
Heart | 1. Consider fetal echocardiography to detect individuals with a high risk of heart failure after delivery when rhabdomyomas are identified via prenatal ultrasound. |
Eye | Perform a complete ophthalmologic evaluation, including dilated fundoscopy, to assess for retinal findings (astrocytic hamartoma and achromic patch) and visual field deficits. Repeat annually if symptomatic. |
Skin | Detailed skin examination annually. |
Teeth | Detailed dental exam every 6 months. Panoramic dental radiograph at the age of 7. |
(94) | |
Surveillance for epilepsy. Seizures in infants with tuberous sclerosis complex are a medical emergency given the strong association between refractory epilepsy and adverse developmental outcomes.
TOSCA registry (5‑year, multinational). Epilepsy prevalence is approximately 85%; TSC1 variants associate with relatively milder phenotypes. Vigabatrin improved infantile spasms and is recommended as first line; focal seizures often show drug resistance (91).
EPISTOP. EPISTOP was the first prospective study of epileptogenesis in human infants with tuberous sclerosis complex and revealed that infants with tuberous sclerosis complex presenting with multifocal interictal discharges had a higher risk of drug-resistant epilepsy and benefited more from preventive vigabatrin treatment in delaying seizure onset (119; 35). The study revealed that epileptiform discharges on EEG typically appear before the age of 3 months and often with a multifocal distribution. Early onset of epileptiform discharges as well as focal slowing were associated with earlier seizure onset. Preventive vigabatrin delayed seizure onset approximately 4‑fold (median 614 vs. 124 days), reduced risk of clinical seizures by two thirds and drug‑resistant epilepsy by more than 2-fold, and prevented infantile spasms in pooled analyses (73).
PREVeNT (multicenter randomized phase IIb, double‑blind, placebo‑controlled study). Early vigabatrin at first abnormal EEG delayed onset and reduced incidence of infantile spasms but did not improve Bayley‑III or Vineland‑II developmental outcomes at 24 months, nor reduced focal seizures or incidence of drug‑resistant epilepsy at 24 months (09).
Eligibility criteria, EEG monitoring, and follow-up procedures were similar between the two studies. The PREVeNT study was more statistically robust, with the double-blind, placebo-controlled design. In contrast, EPISTOP treatment trial was open label, with randomization or study site determining whether vigabatrin therapy was started at time of EEG abnormalities versus waiting until onset of clinical seizures. A centralized EEG review process determined randomization in PREVeNT versus local EEG review and determined randomization in EPISTOP. Findings from both the randomized control and open label trials were combined to reach conclusions in the EPISTOP trial. Both studies demonstrated small gains in developmental outcomes likely due to earlier diagnosis, regular monitoring of epilepsy, early initiation of treatment, and referral to early intervention services (99).
EEG monitoring schedule. At diagnosis of tuberous sclerosis complex, baseline EEG is recommended for all patients, irrespective of seizure history. Follow with 8- to 24‑hour video‑EEG if routine EEG is abnormal or if clinical concern exists.
For infants without clinical seizures, (1) EEG every 4 weeks to 6 to 8 months, then every 4 to 8 weeks to 12 months, and every 3 months to 24 months, or (2) every 6 weeks to 12 months, then every 12 weeks to 24 months (95).
Both regimens aim to detect preclinical EEG change and initiate timely therapy.
First‑line therapy in infancy. Vigabatrin is recommended as first‑line for infantile spasms and often for focal seizures associated with tuberous sclerosis complex (01; 94). A practical approach is 25 to 50 mg/kg/day, rapidly titrating approximately every 3 days to 100 to 150 mg/kg/day as needed. FDA approvals include infantile spasms and adjunctive therapy for refractory focal seizures (125); generics have been available since 2019. In Europe, presymptomatic vigabatrin on EEG abnormality is common practice (121), though PREVeNT introduces nuance (03).
Safety and monitoring of vigabatrin. Approximately one third may develop visual field defects with cumulative exposure, with reported thresholds including approximately 720 g cumulative dose and approximately 24-month duration (111). Monitoring methods include electroretinography (ERG), electro‑oculography, field‑specific visual evoked potentials, and optical coherence tomography; some require sedation in infants. Programs vary; many centers emphasize regular ophthalmologic evaluation. Recommended schedules include baseline every 3 months for 18 months, then every 6 months thereafter. Long‑term U.S. registry data (over 9000 patients, up to 16 years) reported no clinical vision loss attributed to vigabatrin and very few new vision findings over time (46). Other adverse events include insomnia, agitation, constipation, and transaminase suppression (which can mask hepatotoxicity from concomitant drugs). Vigabatrin‑associated brain MRI abnormalities (brainstem/cerebellum/basal ganglia T2 signal) occur in approximately 35%. These have no proven clinical significance and resolve after discontinuation; higher doses and possible mitigation with concurrent mTOR inhibitors have been reported.
Second‑line therapy for infantile spasms. If spasms (and hypsarrhythmia, when present) do not resolve within approximately 2 weeks, add adrenocorticotropic hormone (natural or synthetic) or prednisolone.
New or changing seizures in older patients. Evaluate patients for new structural etiologies (eg, SEGA growth, stroke, hemorrhage, glioma) with appropriate imaging.
mTOR inhibitors for refractory focal epilepsy. In the EXIST‑3 trial of everolimus, both low and high exposure in 366 participants with refractory focal seizures achieved 50% or greater seizure reduction versus placebo (p< 0.008 and p< 0.001, respectively), with the largest effect in children under 6 years of age (51). Open‑label extension spanning 48 weeks to 2 years or more showed increasing response over time, 50% or greater reduction in over 40% by 2 years, and 19% seizure freedom in some cohorts (47; 77). A small study reported sustained interictal discharge reduction and improved seizures over 27 months; cognitive improvement aligned with elimination of interictal discharges (145).
In therapeutic drug monitoring, typical trough is 5 to 9 ng/mL (consider up to approximately 15 ng/mL if inadequate response) (49).
Sirolimus is also used for epilepsy in tuberous sclerosis complex.
Cannabidiol. Cannabidiol was approved in 2021 for tuberous sclerosis complex–related seizures in patients aged 12 months and older (GWPCARE6). Efficacy of 25 mg/kg/day was similar to 50 mg/kg/day, with fewer adverse events (123). In a long‑term extension, median seizure reduction was approximately 54% at 12 weeks and sustained at 48 weeks, with quality‑of‑life gains over 80%; cannabidiol with clobazam was more efficacious than cannabidiol alone (124).
Administration and interactions. Take consistently with food (systemic bioavailability is increased approximately 4‑fold with high‑fat meals; approximately 3‑fold with low‑fat/whole milk). With clobazam, reduce clobazam if sedation emerges. With valproate, monitor liver function tests closely—transaminitis is common early and typically reversible. With mTOR inhibitors, monitor trough levels and adjust doses to avoid toxicity (144).
Other antiseizure medications and diets. GABAergic agents (topiramate, carbamazepine, oxcarbazepine) are commonly used (30). Ketogenic diet and carbohydrate‑restricted variants (modified Atkins, low glycemic index) are effective options for refractory epilepsy, even alongside vigabatrin (32). In the TRUST‑TSC phase 3 randomized controlled trial, ganaxolone did not demonstrate superiority over placebo (11).
New studies. TSC-STEPS evaluates the benefit of early treatment with sirolimus before 6 months of age in infants with tuberous sclerosis complex, with expected completion in June 2026.
ViRap is testing the benefits of sirolimus versus vigabatrin on occurrence of clinical seizures in infants and the volume of tuberous sclerosis complex-associated tumors at 2 years of age, with expected completion in March 2026.
PROTECT assesses the long-term neuropsychologic outcome of preemptive mTOR inhibitor treatment in children with tuberous sclerosis complex under 4 months of age (41).
Prompt referral for epilepsy surgery evaluation is recommended when two to three or more appropriately chosen medications have failed (08). Even with multiple tubers or diffuse EEG abnormalities, a single epileptogenic zone can often be localized and resected. Corpus callosotomy may benefit selected patients with unlocalizable patterns (70). Early surgical intervention correlates with better outcomes (148).
Outcomes. Seizure freedom is achieved in approximately 50% to 60%; approximately another 15% have a 50% or more reduction, even with multiple tubers (80).
Large multicenter studies demonstrate improvements in seizure control, quality of life, and IQ (81). Seizure freedom is associated with complete resection of epileptogenic tubers, presence of large/calcified tubers, and tuberectomy plus lobectomy.
Real‑world registries show variable outcomes (eg, ILAE 1–2 in approximately 41% of 229 surgical patients) reflecting heterogeneity in presurgical testing and center expertise (55). Tuberectomy/sublobar resections confer greater medication reduction than callosotomy.
A meta‑analysis of 44 studies (n=2058) reported approximately 66% good surgical outcome (56).
A prospective multicenter cohort (China; 21 centers; n=200) found resective surgery provided superior seizure control, higher likelihood of IQ/QOL improvement, and greater medication reduction at 1 to 4 years versus continued medical therapy. Seizure‑free rates were approximately 77% at 1 year and approximately 66% at 4 years (143).
Minimally invasive and neuromodulatory options. MRI‑guided laser interstitial thermal therapy (LITT) can achieve over 50% seizure reduction in approximately 80% and over 90% reduction in approximately 63% at 2‑year follow‑up, with 58% freedom from the targeted seizure type; there are perceived developmental gains in approximately 63% (128; 101). Vagus nerve stimulation and responsive neurostimulation may benefit selected patients.
Surveillance. Brain MRI is recommended every 1 to 3 years through at least the age of 25, even if asymptomatic. Brain MRI is recommended every 6 months for SEGAs 1 cm or larger, growing lesions, or in patients unable to reliably report symptoms. They should continue beyond the age of 25 if growth occurs in adulthood or if new lesions are suspected.
Clinical vigilance. Urgent neuroimaging is warranted for headache (positional or progressive), irritability, signs of raised intracranial pressure, or seizure exacerbation.
Surgery. Gross total resection has been the treatment of choice for acute deterioration and is often curative (63), but mTOR inhibitors are replacing it. Consider surgery for asymptomatic enlarging lesions, ventriculomegaly, or failure/intolerance of medical therapy. Minimally invasive/endoscopic approaches and LITT are increasingly utilized.
mTOR inhibitors. Everolimus (FDA 2010) is recommended for asymptomatic growing, large, or multiple SEGAs; for mild–moderate symptoms in nonsurgical candidates; for regrowth after incomplete resection; or to shrink tumors pre‑operatively (reducing vascularity and improving planes). Tumor regrowth can occur after discontinuation (75). Surgery is now generally reserved for mTOR‑refractory cases or medication intolerance (112).
EXIST‑1 and extensions. Everolimus was effective in children as young as 12 months, reducing SEGA volume and often improving seizure control. Sustained efficacy was noted over approximately 4 years with 30% or more primary SEGA reduction in approximately 60% (72; 47).
EMINENTS and 60‑month follow‑up. After 1 year or more of standard dosing (trough 5 to 15 ng/mL), reduced/fractionated maintenance dosing of everolimus to maintain a serum level of 5 to 15 ng/ml can maintain tumor control in selected patients (129; 18).
Early psycho-educational or neuropsychological assessment is important to identify problems in cognitive development and to develop appropriate teaching strategies. Problems with inattention, hyperactivity, aggression, or autistic features may necessitate psychological or psychiatric consultation. Periodic assessment is advised at each developmental age in childhood to tailor appropriate support services through the school. The transition from special education resources in the classroom to vocational rehabilitation opportunities in young adulthood should be carefully monitored (38). An excellent review on TAND is available (40), as are the consensus recommendations for identification and management of TAND (37). Tools like the TAND-L or TAND-SQ checklists are well suited for regular screening by a professional supporting an individual with tuberous sclerosis complex (TAND-L) or by families themselves (TAND-SQ). If concerns are identified on screening, referral to relevant professionals is indicated.
The core principles for the identification and treatment of tuberous sclerosis complex–associated neuropsychiatric disorders were noted as follows (37):
1. Everyone with tuberous sclerosis complex is at risk. | |
2. Screen at least annually and act. | |
3. Aim for early identification and early intervention. | |
4. TAND features cluster. | |
5. Consider effects of physical comorbidities and medications. | |
6. Partner with families/caregivers | |
7. Generate a “bio-psycho-social” “whole-system” plan for intervention. | |
8. Be evidence-based and evidence-informed. | |
9. Optimize function and quality of life. |
EPISTOP cohort. The Bayley Scales of Infant Development (BSID) and Autism Diagnostic Observation Schedule (ADOS) at 12 months helped identify infants at higher risk of autism spectrum disorder (88). The normal cognitive quotient at 12 months had 100% negative predictive value for later autism spectrum disorder. Abnormalities in socio‑communication preceded stereotypies.
PREVeNT follow‑up (36 months). There is no cognitive or developmental benefit from pre‑seizure vigabatrin (99). Epilepsy correlated with lower Bayley‑III/Vineland‑II. Autism spectrum disorder diagnosis was approximately 31% overall; none without epilepsy developed autism spectrum disorder. Minimal treatment‑timing differences, close monitoring, and universal access to early intervention likely attenuated between‑group cognitive differences.
Monitoring. Annual blood pressure, urinalysis/proteinuria, and eGFR (creatinine or cystatin C) are recommended. Consider 24‑hour ambulatory blood pressure when office readings are high. Low muscle mass can inflate creatinine‑based eGFR.
Imaging. Renal MRI is recommended at diagnosis and every 1 to 3 years lifelong (angiomyolipomas can enlarge rapidly in children and recur) (44). MRI is preferred over ultrasound because approximately 25% of angiomyolipomas are fat‑poor; use contrast CT if MRI is unavailable. Increase frequency to annual imaging when tumors approach approximately 3 cm or are growing.
Pathology. HMB‑45 (angiomyolipoma) and cytokeratin (RCC) immunostains help distinguish fat‑poor angiomyolipomas from renal cell carcinoma.
Hypertension. Treat aggressively, especially in TSC2/PKD1 contiguous gene syndrome (lower blood pressure targets recommended) (86). RAAS inhibitors are first‑line; evidence does not support avoiding ACE inhibitors in patients on mTOR inhibitors. Monitor eGFR and proteinuria every 3 to 12 months when receiving mTORC1 inhibitors. Note that zonisamide and topiramate increase nephrolithiasis risk. Use caution when adrenocorticotropic hormone is used.
mTOR inhibitors for angiomyolipoma. Consensus guidelines recommend mTORC1 inhibition for asymptomatic, rapidly growing angiomyolipomas over 3 cm and for lesions over 4 cm (94). Proteinuria may occur but rarely mandates discontinuation. Continue therapy for 12 or more months and as long as tolerated.
Everolimus (FDA 2012). EXIST‑2 (double‑blind, placebo‑controlled) demonstrated significant volume reductions in angiomyolipomas and benefit in sporadic lymphangioleiomyomatosis (14), with durable response and safety at 4 years (15). In the EXIST‑1 extension, approximately 73% achieved angiomyolipoma response; over 75% had over 50% volume reduction over approximately 4 years (47; 12).
Interventional management. For large or hemorrhagic lesions, transarterial embolization is preferred—even for lesions 4 cm or more—and is favored over nephrectomy whenever possible (43; 71). Acute hemorrhage should be managed with embolization and corticosteroids.
Avoid routine biopsy of fat‑poor lesions. Biopsy should be considered if growth is over 5 mm/year or nonresponsive to mTORC1 therapy (86). Avoid nephrectomy when feasible.
Screening and imaging. In women with tuberous sclerosis complex (and in symptomatic men), non‑contrast chest CT is recommended once at approximately 18 years of age. If there are no cysts, repeat every 5 to 7 years to menopause, or sooner if symptoms evolve (76). Use ultra‑low‑dose protocols to limit radiation. High‑resolution CT helps with diagnosis but is not required to diagnose typical lymphangioleiomyomatosis cysts.
Pulmonary function tests. Baseline is at approximately 18 years of age in women; annual pulmonary function tests and 6‑minute walk are recommended when lymphangioleiomyomatosis/cystic disease is present. Consider spirometry every 3 to 6 months when newly diagnosed, symptomatic, for advanced disease, or monitoring mTOR response. Post‑bronchodilator values are preferred. When pulmonary function tests are not feasible, serial CT can track progression.
Biomarkers and counseling. A VEGF‑D level can serve as a baseline for potential lymphangioleiomyomatosis development and progression but is not yet a routine screening tool. Counsel patients regarding smoking cessation, estrogen exposure (oral contraceptives), and pneumothorax risk. Talc pleurodesis and pleurectomy complicate future lung transplantation; avoid if transplant is likely. Doxycycline is not recommended. Ensure influenza and pneumococcal vaccination (48).
Treatment. Sirolimus is first‑line for qualifying lymphangioleiomyomatosis and stabilizes or improves lung function; everolimus is effective in sporadic lymphangioleiomyomatosis and should be continued if already used for other tuberous sclerosis complex indications (14; 85). Chylous effusions often decrease or resolve with sirolimus. Pregnancy in lymphangioleiomyomatosis is associated with accelerated decline in lung function and higher pneumothorax risk. Lung transplantation is an option in end‑stage disease; talc pleurodesis is not a contraindication.
Screening. In patients under 3 years of age, 12‑lead ECG and echocardiogram assess for arrhythmias and rhabdomyomas. Baseline ECG is recommended for adults (new diagnosis); echo if symptomatic or as clinically indicated. For asymptomatic rhabdomyomas, echo every 1 to 3 years until regression is documented. ECG is recommended every 3 to 5 years thereafter to screen for conduction disease. Vascular aneurysm screening in childhood and adolescence has been suggested with abdominal ultrasound and chest X‑ray at intervals (62).
Management. Cardiac symptoms (heart failure, arrhythmias) may require medications, ablation, pacemakers, resection for obstructive tumors, and, rarely, transplant (34). The ORACLE trial (everolimus for symptomatic cardiac rhabdomyomas) was planned; updated results are not yet available (122). A systematic review suggests that everolimus/sirolimus may be beneficial in neonates with hemodynamic compromise (58). Sirolimus dosing guidance in neonates and infants has been proposed: initial dose 0.25 mg/day (premature newborns), 0.5 mg/day (term newborns), 0.5 to 1 mg/day (2 to 6 months), 1 to 1.5 mg/day (6 to 12 months), with a 10% to 15% dose increase based on sirolimus serum level until steady state is achieved (93).
Screening and surveillance. Baseline dilated fundus exam detects astrocytic hamartomas and retinal achromic patches. Annual examinations are recommended in asymptomatic patients and more frequent examinations if lesions involve the macula or threaten vision. Periodic imaging to track lesion growth is reasonable, though standardized surveillance intervals are not firmly established.
Interventions. For vitreous hemorrhage from retinal hamartomas, pars plana vitrectomy is an option (87). Other modalities include laser photocoagulation, photodynamic therapy, intravitreal anti‑VEGF, and steroids.
Surveillance and general care. Skin examination in children should be annual; frequency in adults should match severity. Maintain serial photographs for objective comparison. Use Wood’s lamp for hypomelanotic macules. Sun protection is recommended for hypomelanotic macules and especially when using topical sirolimus.
Topical mTOR therapy. The TREATMENT trial (ages 3–61 years) found rapamycin 1% and 0.1% both improved angiofibromas, with greatest benefit within 6 months (69). Other studies support topical sirolimus, with better responses in children (smaller, less fibrotic lesions) (79; 140). Systemic everolimus for other tuberous sclerosis complex indications also reduces facial angiofibroma size/density/erythema (142), but the risk–benefit is inadequate to recommend systemic mTOR inhibitors solely for cutaneous lesions. Topical sirolimus 0.1% to 1%, once or twice daily, is a reasonable first‑line option; emphasize sun protection.
Establish dental care with eruption of the first tooth and no later than 12 months. Schedule dental assessments every 3 to 6 months and obtain a panoramic radiograph by approximately 7 years, if not previously done. Manage enamel pits preventively (eg, topical fluoride, sealants).
Pancreatic neuroendocrine tumors (functional and nonfunctional) are increasingly recognized on annual abdominal MRI (with fine pancreatic cuts) and were a preventable cause of death in one mortality study (02). Manage functional neuroendocrine tumors per standard of care, similar to patients without tuberous sclerosis complex.
Agents and dosing. Everolimus and sirolimus are widely used in tuberous sclerosis complex. Everolimus is the only oral rapalog and is typically initiated at approximately 4.5 mg/m²/day, titrated to a trough of 5 to 15 ng/mL. Sirolimus is available parenterally and topically; dosing varies by indication and age.
Baseline and follow‑up labs. Obtain fasting lipids, comprehensive metabolic panel, cystatin C (when needed), urinalysis, and CBC with differential before initiation. Repeat after starting therapy, alongside trough levels, and check for drug–drug interactions (particularly potent CYP3A4 inhibitors/inducers). Practical adverse event management strategies are available (33; 117).
Adverse effects. Common adverse events include stomatitis, upper respiratory infections, rash, gastrointestinal upset, dyslipidemia, leukopenia, and urinary tract infections. Long‑term adverse events include increased creatinine, glucose, triglycerides, osteopenia/osteoporosis, and, rarely, pneumonia and death from immunosuppression. Alcohol and peroxide wipes may worsen stomatitis.
Drug interactions. Levels increase with CYP3A4 inhibitors (clarithromycin, erythromycin, ketoconazole, verapamil, grapefruit). Higher doses may be needed with CYP3A4 inducers (phenobarbital, phenytoin, St. John’s wort, glucocorticoids). PGP inhibitors (amiodarone, quinidine, erythromycin) can also raise levels (U.S. Food and Drug Administration 2023). Cannabidiol can increase mTOR inhibitor levels: monitor and adjust.
Perioperative and vaccination considerations. Because of impaired wound healing and immunosuppression, temporarily hold mTOR inhibitors 1 to 2 weeks before planned procedures or live vaccines and during significant infections. Resume approximately 1 to 2 weeks after vaccination and approximately 2 to 4 weeks after surgery once recovered.
Life expectancy in tuberous sclerosis complex is reduced by approximately 1 to 2 decades in some cohorts; renal disease remains a leading cause of death. Other important causes include brain tumors, epilepsy/SUDEP, and pulmonary complications. Renal cysts can be associated with renal failure. Angiomyolipomas may cause massive intra‑abdominal hemorrhage.
Epilepsy prognosis. Two‑thirds of patients experience drug‑resistant epilepsy. Even after surgery, approximately 40% may have persistent seizures (22). Adverse prognostic markers include infantile onset, multiple seizure types, multifocal EEG discharges with sleep generalization or secondary bilateral synchrony and evolving new EEG foci (29).
Neurocognitive prognosis. Risk factors for intellectual disability include frontal/temporal cortical tubers (110), cerebellar tubers, TSC2 variants, and refractory epilepsy. Infantile spasms and early epilepsy onset predict poorer cognition; TACERN longitudinal data support these associations (23).
SEGA complications. Before modern imaging and neurosurgery, obstructive hydrocephalus was a major cause of death. Intraventricular hemorrhage within a SEGA is rare but can be fatal.
Pregnancy risks primarily relate to renal and pulmonary disease: angiomyolipoma rupture, pneumothorax, and chylous effusions. Renal failure, preeclampsia, and fetal growth restriction can occur (106; 67). Evaluate renal status with ultrasound or CT (risk‑benefit assessed).
Reproductive genetics. Prenatal testing and preimplantation genetic testing are available when a familial pathogenic variant is known.
Medications. Antiseizure medications require teratogenic risk counseling and therapeutic drug monitoring. Everolimus and sirolimus may be teratogenic; use effective contraception during treatment and for approximately 8 weeks after discontinuation. Estrogen‑containing contraceptives are generally avoided in women with lymphangioleiomyomatosis. Smoking cessation is imperative. Avoid nephrectomy if possible.
Fetal assessment and perinatal care. Detailed prenatal ultrasound is indicated for cardiac rhabdomyomas and CNS anomalies (most evident in third trimester). Fetal brain MRI can be considered when indicated. Maternal sirolimus to target trough 10 to 12 ng/mL has been used prenatally to manage rhabdomyomas in select cases (42). After delivery, consider cardiac ultrasound, ECG, brain MRI, and prompt referral to pediatric neurology (127).
• Estrogen use in teenage and adult females | |
• Smoking at all ages | |
• Nephrectomy |
General anesthesia requires heightened vigilance for intracranial hypertension (eg, from SEGA), status epilepticus, renal dysfunction, respiratory failure (eg, lymphangioleiomyomatosis), large‑vessel aneurysms, and cardiac arrhythmias. Pre‑procedure planning should address airway, hemodynamics, intracranial pressure, renal dosing, and seizure prophylaxis.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Hema R Murali MD MBBS
Dr. Murali of Geisinger Health System has no relevant financial relationships to disclose.
See ProfileNarayana S Murali MD
Dr. Murali of the Marshfield Clinic and Geisinger Health System has no relevant financial relationships to disclose.
See Profile
Bernard L Maria MD
Dr. Maria of Thomas Jefferson University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 06, 2026
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Developmental Malformations
May. 08, 2026