
Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
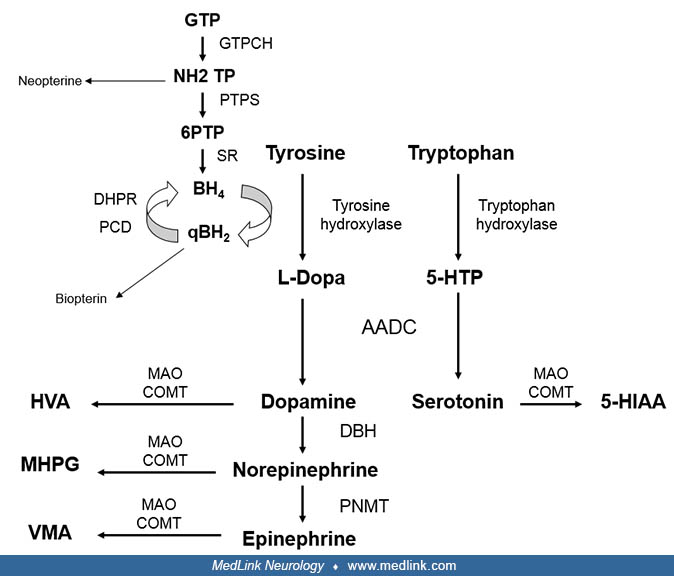
Tyrosine hydroxylase deficiency is an autosomal recessively inherited inborn error of metabolism that involves the biosynthesis of catecholamines (dopamine, epinephrine, and norepinephrine). The clinical presentation ranges from severe infantile parkinsonism to a phenotype reminiscent of dopa-responsive dystonia. Diagnosis is based on the pattern of monoamine metabolites in CSF and the molecular analysis of the TH gene.
• Tyrosine hydroxylase deficiency is an autosomal recessively inherited disorder that leads to a deficient production of catecholamines (dopamine, epinephrine, and norepinephrine). | |
• Patients usually exhibit developmental delay, infantile parkinsonism, dystonia, oculogyric crises, and features of autonomic dysfunction. Milder phenotypes may also occur. | |
• Tyrosine hydroxylase deficiency is diagnosed by detection of decreased CSF concentrations of the downstream metabolites of catecholamine degradation, homovanillic acid, and 3-methoxy-4-hydroxyphenylglycol. | |
• The treatment of choice is levodopa; alternatively, patients are treated with other dopaminergic drugs, mainly dopamine agonists and monoamine oxidase inhibitors. |
Tyrosine hydroxylase deficiency was first reported in 1971 in 2 brothers with early-onset progressive dopa-responsive dystonia (03). Subsequently, young infants with a more severe phenotype were recognized (12). In 2010, Willemsen and colleagues described the largest series reported to date (36 patients) and proposed 2 phenotypes: type A, which has an onset in infancy or childhood, is less severe, has a better prognosis, and is L-dopa responsive; and type B, which has an earlier onset (neonatal period, early infancy), is more severe, and is poorly L-dopa responsive (32). This classification was widely used by clinicians and researchers, although the authors acknowledge that the phenotype of tyrosine hydroxylase deficiency has a spectrum with overlap of clinical features between both groups (32). A standardized deep phenotyping study from the registry of the International Working Group on Neurotransmitter Related Disorders did not find clear differences in perinatal abnormalities, postnatal problems, and achievement of gross motor milestones; drug response varied regardless of the age of initial symptoms (16). Consequently, the authors proposed to abandon this classification. Currently, more than 70 cases have been reported worldwide (32; 07).
In 1996 the first mutations in the tyrosine hydroxylase gene were reported (21). The spectrum of mutations is heterogenous with no hot spots detected (15; 07). Two common mutations due to founder effects have been detected, one in the Greek population (c.707C> T) (25) and another in the Dutch population (c.698G> A) (31).
Nearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125