Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Vertical gaze palsies are symmetrical deficits in the upward or downward excursions of the two eyes. Lesions lie in the thalamus or midbrain, most often caused acutely or subacutely by ischemic or hemorrhagic stroke, neoplasm, aqueductal stenosis, or malfunction of ventricular shunts placed for hydrocephalus. Less common acute or subacute causes are demyelination, infection, malformation, and closed head trauma. Chronic causes include progressive supranuclear palsy, other parkinsonian states, Niemann-Pick type C, and Whipple disease. In focal thalamic or midbrain lesions, a constellation of manifestations may include skew deviation, third or fourth nerve palsies, convergence disorders, lid retraction, pupillary light-near dissociation, ataxia, and impairments in language and consciousness.
|
• Gaze palsies are impaired conjugate excursions of the two eyes. | |
|
• Both eyes should have the same degree of excursional impairment, which can occur in either the horizontal or vertical plane. | |
|
• This article covers vertical gaze palsies; horizontal gaze palsies are covered in a separate article. | |
|
• Vertical gaze palsies may be restricted to upgaze or downgaze or affect both directions. | |
|
• Associated clinical manifestations include skew deviation, third or fourth nerve palsies, convergence disorders, lid retraction, pupillary light-near dissociation, ataxia, and impairment in language or consciousness. | |
|
• Purely vertical gaze palsies are caused by intrinsic or extrinsic lesions of the thalamus or midbrain. | |
|
• Common acute or subacute processes are ischemic or hemorrhage stroke, neoplasm, aqueductal compromise, and malfunction of ventricular shunts placed for aqueductal compromise. | |
|
• Less common acute or subacute processes are demyelination, infection, malformation, and closed head trauma. | |
|
• The most common chronic process is progressive supranuclear palsy. | |
|
• Vertical gaze palsies can be mimicked by peripheral causes, including Graves disease and myasthenia gravis. |
The term “gaze palsy” refers to an impairment in ocular excursions that is of the same degree in the two eyes and limited to either the horizontal or the vertical plane. This article covers gaze palsies in the vertical plane. Horizontal gaze palsies are covered in a separate article.
Ocular excursions in the vertical plane may be partially reduced or completely absent. In severe unidirectional vertical gaze palsies, both eyes may be deviated away from the direction of the palsy. For example, a severe upgaze palsy may manifest as a downgaze deviation.
The only manifestation of a minor gaze palsy may be difficulty sustaining upward or downward gaze, such that the eyes repeatedly drift back toward straight ahead (primary, centric) gaze position. In an attempt to regain eccentric gaze, the patient will initiate repetitive saccades in the direction of the failing eccentric gaze. If the backward drift of the eyes is downward and the saccades are upward, the oscillations are called “upbeat nystagmus.” If the backward drift of the eyes is upward and the saccades are downward, the oscillations are called “downbeat nystagmus.” This vertical nystagmus may be the initial or resolving sign of a gaze palsy.
Vertical gaze palsies are caused by lesions of the thalamus or midbrain. The palsies are divided into those that arise from lesions that affect the thalamus or midbrain. Because thalamic lesions often damage the volitional subsystems (saccades and pursuit) and spare the non-volitional subsystem (the vestibulo-ocular reflex), they are called “supranuclear.” Because midbrain lesions often damage volitional and non-volitional ocular motor subsystems, they are called “nuclear.” Therefore, examination of a patient with a vertical gaze palsy or gaze deviation must include not only the testing of saccades and pursuit, but also the testing of the vestibulo-ocular reflex by means of the doll head (doll eye, oculocephalic) maneuver.
Lesions of the extra-axial ocular motor cranial nerves, neuromuscular junction, or extraocular muscles may appear to cause vertical gaze palsies if the eye movement deficits happen to be of the same degree in both eyes and limited to the vertical plane. Chronic progressive external ophthalmoplegia, a mitochondrial disorder, may at first selectively affect vertically acting extraocular muscles to produce limited excursions confined to the vertical plane. Myasthenia gravis may also fortuitously simulate a vertical gaze palsy, as may orbital trauma or inflammation of the extraocular muscles. A prominent example of extraocular inflammation is Graves disease, which preferentially affects the inferior rectus muscles, producing restricted upward eye movements. These mimickers, and the ways to distinguish them from CNS lesions, are reviewed later in the article.
Conjugate and disconjugate gaze. The term “conjugate gaze” refers to the movements of the two eyes in the same direction. Gaze palsies are disorders of conjugate gaze. They may be congenital or acquired, unidirectional, or bidirectional.
The term “disconjugate gaze” refers to movements of the two eyes in opposite directions. It is not pertinent to gaze palsies, in which the impairment is always conjugate. The term “convergence” is used to describe a specific type of disconjugate eye movement in which the eyes move inwardly (toward each other) as the patient alters fixation from a distant to a near target. When vertical conjugate eye movements are impaired, the brain may automatically activate convergence, although that movement will not assist in bringing the eyes into eccentric gaze.
The term “disconjugate gaze” is often misapplied to eyes that are out of alignment. The proper term here is “ocular misalignment.” (The term “strabismus” refers to ocular misalignment discovered at birth or in early childhood.) Ocular misalignment is a common feature of vertical gaze palsies. If the eyes are vertically misaligned, possible features in addition to the gaze palsy are skew deviation, third nerve palsy, and fourth nerve palsy. If the eyes are horizontally misaligned, possible additional features are third nerve palsy, convergence weakness, and convergence excess.
The term “ocular ductions” refers to the excursions of each eye examined one at a time (the unexamined eye is covered). Ocular ductions should be equally impaired in all gaze palsies. However, in vertical gaze palsy, third or fourth nerve palsies may be superimposed on the gaze palsy, creating ductional asymmetries in the two eyes, ocular misalignment in some fields of gaze, and diplopia.
Gaze palsies manifesting as impaired saccades or pursuit. Gaze palsies may include impaired saccades and pursuit. Impaired saccades can be hypometric or slow. Impaired pursuit will appear as cogwheel rather than smooth ocular movements as the more robust saccadic system substitutes for the damaged pursuit system to allow the eyes to catch up to a moving target.
Gaze palsies manifesting as jerk nystagmus. Mild gaze palsies may consist of the inability to maintain eccentric gaze. The eyes will repeatedly drift back toward straight-ahead position. The drift will be met by a repeated saccadic effort to take the eyes into eccentric gaze, setting up an oscillatory cycle called “gaze-evoked jerk nystagmus.”
Gaze palsies manifesting as gaze deviation or gaze preference. Unidirectional vertical gaze palsies are often accompanied by eccentric positioning of the eyes in the opposite direction. The term used to describe this phenomenon is “gaze deviation.” If the gaze deviation can be overcome by imploring the patient to look in the opposite direction, the term “gaze preference” is used.
Supranuclear gaze palsy versus gaze apraxia. Gaze apraxia has been applied to patients with Balint-Holmes syndrome owing to biparietal dysfunction, in which poor initiation of saccades and defective pursuit are present with sparing of the vestibulo-ocular reflex. That condition also features disorders of visual spatial function. To avoid confusion, it is best to reserve the term “gaze apraxia” for delayed initiation of volitional conjugate eye movements and sparing of spontaneous saccades, a phenomenon that occurs in a wide variety of neurologic diseases. It differs from a gaze palsy, in which even spontaneous saccades are defective.
Vertical gaze palsies can be caused by bilateral lesions of the cerebral hemispheres, but in those cases, horizontal gaze is frequently impaired as well. Vertical gaze palsies that occur without horizontal gaze palsies are always caused by unilateral or bilateral lesions of the thalamus or midbrain. Familiarity with the ocular motor pathways helps to understand why lesions in these locations cause such gaze palsies.
Volitional vertical gaze is initiated jointly by signals originating in the frontal and parietal cortex of both cerebral hemispheres. It is mediated by two ocular motor subsystems: saccades and pursuit. The saccadic pathway governs refixational eye movements that occur spontaneously, to command, and if the patient desires to view an eccentric target. Pursuit is initiated by the desire to follow a moving visual target.
Saccadic signals pass from cerebral cortex to the diencephalon and eventually to the midbrain rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF), which moves the eyes from straight-ahead gaze into upward or downward gaze. Maintenance of upgaze and downgaze is mediated by the midbrain interstitial nucleus of Cajal (INC). The riMLF and INC send their signals to the vertically acting extraocular muscles for upgaze (superior rectus, inferior oblique) or downgaze (inferior rectus, superior oblique) through separate pathways (10; 08).
The pathway that subserves upgaze passes through the posterior commissure in the pretectum and dorsal midbrain to reach those nuclei. The pathway that subserves downgaze reaches the riMLF and INC more ventrally. Hence, lesions extending caudally from the thalamus and affecting the pretectum or dorsal midbrain may damage upgaze selectively, whereas lesions that affect the midbrain tegmentum may damage downgaze selectively.
The pursuit pathway originates in the visual cortex, which sends signals to the parietal cortex and its links to the frontal cortex. Signals leave both cortex regions to course to the cerebellum and eventually to the ocular motor nuclei for vertical gaze in the midbrain. Within the midbrain, the pursuit pathway generally follows the same course as the saccadic pathway.
Non-volitional (reflex) vertical gaze originates in the semicircular canals and otoliths and connects to medullary vestibular nuclei. Signals pass via central tegmental brainstem pathways directly to the midbrain ocular motor nuclei that subserve upgaze and downgaze. This vertical vestibulo-ocular reflex keeps the eyes stable in space when the head moves up or down.
Thalamic lesions lie rostral to the ocular motor centers that mediate vertical gaze so that they interrupt vertical saccades and pursuit and spare the vestibulo-ocular reflex pathway, which approaches midbrain nuclei ventrally through the tegmentum. The dissociation between impaired volitional gaze and spared reflex gaze yields the term “supranuclear.” Lesions that lie in the midbrain itself interrupt rostral input for saccades and pursuit and caudal input from the vestibulo-ocular reflex. Therefore, there is no dissociation between volitional and reflex gaze, and the term “nuclear” is applied.
Vertical gaze palsies consist of upgaze deficits or downgaze deficits, or a combination of both (09). The excursional deficits may be complete or incomplete, but to qualify as gaze palsies, the deficit in the amplitude of excursions should be of equal degree in the two eyes. Although it may appear that saccades or pursuit are selectively affected, detailed testing reveals that both subsystems are usually defective (42). Sparing of the vestibulo-ocular reflex in diencephalic lesions depends on the nature and extent of the lesion. This feature is not reliable in separating thalamic from midbrain lesions.
A mild form of vertical gaze palsy may manifest with normal amplitude of eye excursions, but the excursions are marked by slow saccades, saccadic pursuit, or by a jerk nystagmus with its fast phase in the direction of gaze.
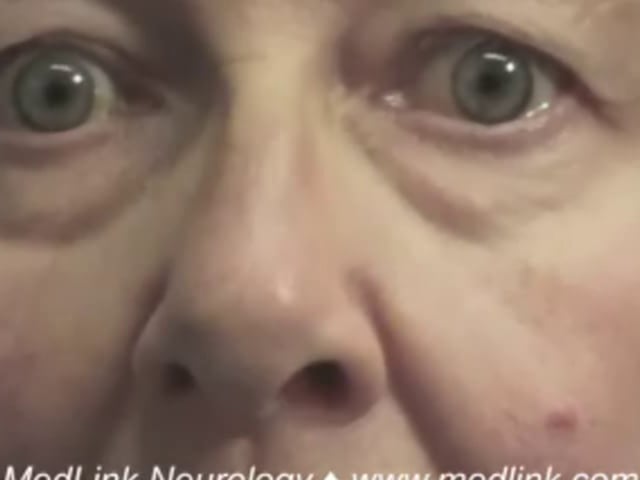
Selective upgaze palsy. Lesions of the caudal thalamus, pretectum, and dorsal midbrain tend to impair upward gaze and spare downward gaze. If the damage is severe, the eyes will deviate into downward gaze (see Downgaze deviation). If the lesion is large enough to extend into the tegmentum, vertical gaze palsies will occur in both directions.
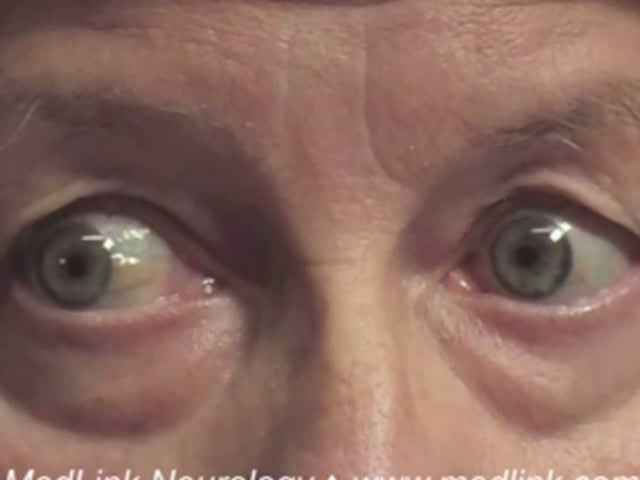
Accompanying signs. Lesions of the caudal thalamus and dorsal midbrain produce a constellation of signs known as the dorsal midbrain, pretectal, Parinaud, or Koerber-Salus-Elschnig syndromes (49). In many cases, the eyes converge and retract repetitively as the patient attempts upward saccades. This “convergence retraction” is a clonic substitution movement attributed to aberrant signaling within the midbrain. It simultaneously activates all extraocular muscles. As the muscles contract, they pull the eyes back into the orbit. Because the medial rectus muscles are most powerful, the retracting eyes also converge. Convergence retraction is often misdiagnosed as a nystagmus, but it consists of cycles of intrusive saccades.
Other manifestations associated with upgaze palsy include lid retraction (Collier sign), pupillary light-near dissociation, skew deviation, fourth nerve palsy, third nerve palsy, convergence excess (causing esotropia), convergence weakness (causing exotropia), ataxia, aphasia, amnestic dementia, and somnolence (12; 16; 09; 36; 07; 43; 49). When these signs are caused by thalamic or midbrain stroke, they are called the “top of the basilar” syndrome, referring to ischemia in the distribution of proximal arterial branches of the posterior cerebral artery (11).
Selective downgaze palsy. Selective impairment of downward gaze, which is much less common than selective upgaze palsy, results from tegmental midbrain lesions that do not affect the posterior commissure. The eyes may deviate into upgaze. Selective downgaze palsy may be accompanied by ataxia, nystagmus, skew deviation, third nerve palsy, fourth nerve palsy, convergence weakness, convergence excess, or somnolence (14; 33). Patients often report that they cannot see clearly through their multifocal spectacles because they are unable to shift their eyes toward the reading correction. The downgaze deficit may also impair their ability to see where they are walking.
Paroxysmal upgaze deviation. Paroxysmal upgaze deviation has been reported in developmentally normal children from the age of 1 week to 4 years. The episodes are relieved by sleep and usually cease spontaneously after 1 year (17; 28; 39). Lasting from seconds to minutes, the episodes are marked by sustained upgaze and head down position, during which the children maintain normal consciousness but cannot execute downward saccades and often display downbeat nystagmus and limb incoordination.
The tonic upgaze episodes have uncommonly followed vaccination or brief upper respiratory infectious manifestations, but in most cases, there is no precipitating event. Neurologic examination is usually normal, as are electroencephalography, serum metabolic studies, and brain imaging.
However, paroxysmal upgaze deviation may be accompanied by cognitive impairment of varying degrees (28). Antiepileptic treatment has not had a clear impact. In rare cases, epileptic abnormalities, periventricular leukomalacia, dorsal midbrain neoplasms, demyelination, or Vein of Galen malformations have been encountered in these cases. Hence, although paroxysmal upgaze deviation is often benign and temporary, that presumption can only be made after a thorough evaluation has excluded developmental delay, epilepsy, and structural lesions. The discoverable lesions are centered in the tegmental midbrain, presumably impairing downgaze and eliciting upgaze deviation and downbeat nystagmus. Even so, most cases will end up being nonepileptic, self-limited, dyskinetic manifestations of delayed brain development.
In older patients, favored causes of paroxysmal upgaze deviation are systemic hypotension, tic disorder, factitious causes, and oculogyric crisis (44). Considered a dystonic reaction, oculogyric crisis is usually provoked by initiation or amplification of dopamine-blocking antipsychotic agents, but other medications, genetic disorders, and neurodegenerative, tardive, and encephalitic states that lower brain dopamine levels have been implicated (44).
Downgaze deviation. Downgaze deviation may occur when the forces that drive the eyes downward overwhelm the forces that drive them upward. That phenomenon, known as “sunset eyes,” is particularly prominent in acute shunt malfunction and in thalamic infarction or hemorrhage. Paroxysmal downgaze of variable duration has also been rarely described in infants (19; 47). During the episodes, the infants may have “stiffening limbs” but normal consciousness. EEG and brain imaging have been normal, and the children have usually developed normally. These episodes may reflect delayed maturation of the dorsal midbrain or its incoming connections. But because they have also been reported in neurologically impaired children, a full investigation is warranted (48).
In toddlers, common causes of upgaze palsies are aqueductal stenosis and cerebrospinal fluid shunt malfunction. Aqueductal stenosis leads to dilation of the third ventricle, which compresses the dorsal midbrain. An identical phenomenon occurs with malfunction of a ventriculoperitoneal, ventriculoatrial, or third ventriculostomy used to treat aqueductal stenosis (03). Both conditions may present with worsening headache owing to increased intracranial pressure. In aqueductal stenosis, there may also be papilledema. But if shunt malfunction is of recent onset, papilledema is often absent because intracranial pressure has not been elevated long enough (26). In these conditions, the upgaze palsy becomes an important clinical indicator. It may be overlooked because the patient avoids looking up or adopts a chin-up head position.
In teenage children and adolescents, an important cause of upgaze palsy is an enlarging pineal tumor, which exerts pressure on the subjacent dorsal midbrain. Alternative causes include head trauma, primary demyelinating disorders, infections, vascular malformations, and neoplasms.
In adults, the principal cause of an acute upgaze palsy is a thalamic or midbrain ischemic stroke. Impaired perfusion in the perforating branches of the first portion (P1) of the posterior cerebral artery is the underlying mechanism (“top of the basilar syndrome”). As in younger patients, head trauma, demyelination, infections, vascular malformations, and neoplasms may be causes.
In elderly adults, an important chronic cause is progressive supranuclear palsy, which affects volitional gaze in all directions, but preferentially vertical gaze. Downgaze tends to be more affected than upgaze. In progressive supranuclear palsy, the vestibulo-ocular reflex is spared, earning the term “supranuclear.” Vertical supranuclear palsy is also reported in other parkinsonian disorders and in other tauopathies, such as corticobasal degeneration (25). Vertical gaze palsies also frequently occur in Niemann-Pick disease type C (40) and to a lesser extent in Whipple disease (35; 24). The many accompanying features of progressive supranuclear palsy, Niemann-Pick disease type C, and Whipple disease will allow distinction.
Vertical gaze palsy may also be caused by infections or para-infectious conditions, paraneoplastic encephalitis (05), prion disease (27; 22), HIV encephalitis (21), and Creutzfeldt-Jakob disease (34).
Rare causes include encephalitis, brain abscess, trauma, transtentorial herniation, and Wernicke encephalopathy (20). Iatrogenic vertical gaze palsy has been described after implantation of depth electrodes for stimulation, presumably due to damage to the riMLF (01).
Disorders that affect the ocular motor cranial nerves, neuromuscular junction, and extraocular muscles may fortuitously produce a pattern that looks like a true vertical gaze palsy. The five major considerations include the following.
(1) Inflammatory orbitopathy. Inflammatory disorders that affect the extraocular muscles may involve both eyes and happen to cause symmetrical eye movement deficits limited to the vertical plane. The ocular adnexal tissues (eyelids, conjunctiva, lacrimal glands) may show congestive features. Lid retraction or ptosis may be present. Examples are Graves disease and idiopathic orbital inflammation. The deficits in eye movements are due to cicatricial shortening of extraocular muscles that restricts excursions. Diagnosis is based on identifying the clinical features and noting inflammatory imaging signs. Passive ductions, in which the eye is anesthetized and moved by means of grasping the sclera with a forceps, will disclose the restriction.
(2) Chronic progressive external ophthalmoplegia (CPEO). This is a mitochondrial disorder affecting the extraocular muscles that may be evident in childhood or later. It affects ocular excursions in all directions, but in some cases, vertical excursions may be preferentially weakened. Accompanying features include ptosis, retinopathy, neck weakness, and cardiac conduction defects. Orbital MRI typically discloses small-caliber extraocular muscles. Included among the mitochondrial causes of CPEO are Kearns-Sayre syndrome and MELAS. Myotonic dystrophy and oculopharyngeal dystrophy are rare causes, but like other genetic or metabolic disorders, they do not cause selective damage of vertical gaze. Moebius syndrome, a congenital disorder featuring aplasia of brainstem motor nuclei, may rarely cause gaze palsies isolated to the vertical plane (38).
(3) Fisher syndrome and Bickerstaff encephalitis. These two disorders are attributed to antiganglioside antibody myelin destruction. Fisher (also called “Miller Fisher”) syndrome is characterized by impaired ocular excursions, ataxia, and areflexia, but all three components may not be present. In some cases, the gaze deficits preferentially affect vertical gaze. The excursional deficits in the two eyes are often so symmetric that a gaze disorder of CNS origin is suspected (06). That inference becomes even more credible in a variant called Bickerstaff encephalitis, which manifests altered consciousness, nystagmus, ataxia, and other signs of brainstem dysfunction (23). Serum anti-GQ1b antibodies are often present (13; 46).
(4) Myasthenia gravis. This antibody-mediated post-synaptic disorder may rarely impair eye movements symmetrically in the two eyes and selectively affect vertical ocular excursions. Weakness of the orbicularis oculi and bulbar and proximal extremity muscles is likely to be present. Acetylcholine receptor, muscle-specific kinase, or lipoprotein receptor-related protein 4 antibodies are often present. In antibody-negative cases, a decremental response may be demonstrated on repetitive stimulation electromyography, and jitter may be demonstrated on single-fiber electromyography.
(5) Botulism. A usually food-borne toxic post-synaptic disorder, this condition usually limits ocular excursions. Horizontal eye movements are apt to be more limited than vertical eye movements. Diagnostic clues are autonomic effects, including copious lacrimation and salivation, as well as dilated pupils and accommodative paralysis. Diagnosis is confirmed by detection of botulinum toxin in food or stool.
A 69-year-old orthopedic surgeon suffered a stroke 6 months prior to evaluation. He presented with somnolence, imbalance, and diplopia. His initial examination notes documented a right third nerve palsy and ataxic gait. His symptoms had improved but persisted. The drowsiness responded to methylphenidate.
He had no anisocoria in bright light or darkness, and light reactions were intact. He had no ptosis but developed lid retraction on attempted upgaze (Collier sign). Neither eye elevated more than 10% above midline, whether by saccades, pursuit, or the oculocephalic reflex. Downgaze was also absent. The right eye had only 50% of normal adduction range, which was not improved by convergence. Horizontal saccades and pursuit were otherwise normal. He was intermittently drowsy during examination, and his gait showed a wide-based ataxia requiring support.
His ocular signs represented a combination of right third nuclear palsy and vertical gaze palsy. MRI showed bilateral lesions of the thalamomesencephalic junction, consistent with a “top of the basilar” stroke affecting both paramedian thalamic arteries.
MRI is preferred and will detect most cases with vascular or neoplastic origin. Gaze palsy in parkinsonian conditions like progressive supranuclear palsy correlate with midbrain atrophy, creating the “morning glory sign” (02).
In cases with gradual or subacute onset, disorders of muscle, neuromuscular junction, or peripheral nerves should be considered.
Progressive supranuclear palsy is a neuropathologically defined disease of tau inclusions in glia and neurons (18). The phenotypes are broad, with some sparing eye movements or having late-onset ocular motor abnormalities (25; 37). MRI features have been proposed, but these are based on small studies and have not been validated against pathological diagnoses (04). Reduced midbrain volume is significantly associated with greater ocular motor dysfunction (32).
Niemann-Pick type C can be diagnosed with plasma biomarkers and genetics (15). Widespread signs of motor neuron dysfunction are usually present in patients with the amyotrophic lateral sclerosis subtype.
Patients with progressive supranuclear palsy often have reduced blinking. The resulting dry eyes can be palliated with tear substitutes. Progressive lenses and bifocals often fail in progressive supranuclear palsy because patients cannot move their eyes downward to use the appropriate part of the lens. They may find single-vision glasses for different tasks to be more effective. Spectacle prisms may help eliminate diplopia in those with ocular misalignment. Base-in prisms may be useful in convergence weakness.
Miglustat therapy has been associated with a stabilization of neurologic manifestations in Niemann-Pick disease type C (30; 41; 29). Animal model studies have shown that hydroxypropyl-beta-cyclodextrin appears to reduce cholesterol and lipid accumulation in the CNS and prolongs survival times (31; 45).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Jonathan D Trobe MD
Dr. Trobe of the University of Michigan has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Neuro-Oncology
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026