





Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Waardenburg syndrome was first reported by van der Hoeve in 1916. In 1951, Waardenburg defined six main features: (1) dystopia canthorum, (2) prominent broad nasal root, (3) synophrys, (4) white forelock, (5) heterochromia iridis, and (6) congenital deaf-mutism. Four types of Waardenburg syndrome have now been delineated on the basis of clinical and genetic criteria. The molecular defective gene has been identified in all four forms. Patients with Waardenburg syndrome type I have a characteristic pleasant feline appearance. In Waardenburg syndrome type II, dystopia canthorum is absent. Deafness and all the facial and hypopigmentation features are due to a disturbance in the neural crest, the origin of melanocytes. The congenital anomalies plus neurosensorial deafness, neural tube defects, and other neurologic manifestations observed in Waardenburg syndrome justify its inclusion as a neurocutaneous syndrome. SOX10 mutations in Waardenburg syndrome type IV are associated with a more severe neurologic phenotype: peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, and Hirschsprung disease. It has been demonstrated that the same deletions at the SOX10 gene locus also may cause Waardenburg syndrome type II. Additional systemic findings have also subsequently been described.
|
• Waardenburg syndrome is a hereditary neurocristopathy that occurs in all ethnic groups, affecting mainly skin, hair, and audition. | |
|
• There are four clinical presentations of this syndrome. Waardenburg first described the syndrome. Type I is the most frequent and benign form. | |
|
• The four presentations manifest pigmentary and auditory deficits that can be identified at birth but require specific investigations and confirmation to enable early intervention and prevent further complications. | |
|
• Genetic defects have been identified in this hereditary condition in all four phenotypes. Consanguinity is frequent in some countries. | |
|
• In some instances early abdominal surgery is required, as in Waardenburg syndrome type IV with aganglionic megacolon. |
Waardenburg syndrome was first reported by the Dutch ophthalmologist Jan van der Hoeve in 1916 in two deaf twin girls with a special type of blepharophimosis (136). In December 1947, Dr. Petrus J Waardenburg, a Dutch ophthalmologist and geneticist, presented a deaf-mute man with "dystopia punctorum lacrimarum, blepharophimosis, and partial iris atrophy" to a meeting of the Dutch Ophthalmological Society (144). He acknowledged the report of the van der Hoeve twins with the same eye abnormalities who were “coincidentally” also deaf-mute. In August 1947, David Klein presented a 10-year-old girl with a severe auditory-pigmentary syndrome and dystopia canthorum to the Swiss Society of Genetics in Geneva (67). In addition, Klein’s patient had a severe musculoskeletal involvement. In 1948, Klein showed this patient to Waardenburg, who later did a search among 1050 patients of five Dutch institutions for the deaf. Other ophthalmologists who studied this ocular anomaly and confirmed its dominant inheritance were Halbertsma in 1929, cited by Waardenburg and Klein, and Gualdi in 1930 (68).
In 1951, Waardenburg published the first comprehensive review of this new syndrome (145) in which he fully acknowledged Van der Hoeve’s initial description and subsequent publications about this syndrome. Waardenburg defined six main features: (1) lateral displacement of the medial canthi combined with dystopia of the lacrimal punctum and blepharophimosis, (2) prominent broad nasal root, (3) hypertrichosis of the medial part of the eyebrows, (4) white forelock, (5) heterochromia iridis, and (6) deaf-mutism. Waardenburg characterized the syndrome as autosomal dominant with high penetrance of dystopia (159/161) but reduced penetrance of all other features.
Waardenburg observed several patients with heterochromia or isochromic hypoplastic irides but without canthus dystopia, but he did not investigate them further (109). In 1971, Arias drew attention to these patients as a separate division of the syndrome, which he named Waardenburg syndrome type II (05). Two of Waardenburg's original families had this variant, but both were small, and Waardenburg overlooked the familial "nonpenetrance" of dystopia. In 1981, Shah and colleagues described 12 infants with several characteristics of Waardenburg syndrome associated with Hirschsprung disease (113). For the congenital cutaneous lesions and the neurosensory deafness (plus other nervous system abnormalities), Waardenburg syndrome should now be included in the group of primary neurocutaneous syndromes (111). Waardenburg syndrome can be considered a disease because the etiology is known; however, it also represents a syndrome due to the constellation of clinical findings.
Four main types of Waardenburg syndrome have been delineated based on clinical and genetic criteria.
Waardenburg syndrome type I (WS1). This is the most frequent type; as a general rule, families characteristically are large.
The five cardinal features in Waardenburg syndrome type I, an autosomal dominant disease, are as follows:
|
• dystopia canthorum |
The first four are distinctive developmental abnormalities of this syndrome, whereas synophrys (confluent eyebrows), is a feature that may be found in normal people.
Waardenburg syndrome type I is the most benign of the four types. In the absence of deafness, these individuals are basically normal. In contrast to most genetic syndromes that cause cosmetic disadvantages, patients with Waardenburg syndrome type I usually have a pleasant physical appearance.
Dystopia canthorum (canthus dystopia, telecanthus). This was first described by van der Hoeve in 1916 in association with blepharophimosis (136). It refers to the lateral displacement of the medial canthus (internal canthus) and inferior lachrymal ducts with the punctate opposite the cornea, toward the limbus. This leads to increased distance between the inferior lacrimal points. Shortness of the nasal palpebral fissures with the appearance of blepharophimosis and fusion of the inner lids results in reduction of the medial sclerae.
The interpupillary distance usually is normal; however, in some patients the interpupillary and outer canthal distances are greater than normal, indicating a degree of hypertelorism. Dystopia canthorum is the most penetrant feature of Waardenburg syndrome type I, present in 99% of patients (06); it affects both eyes. Since van der Hoeve, different interocular distances have been used for determining the presence of canthal dystopia. A biometric index measuring the inner canthal, interpupillary, and outer canthal distances is used presently. Arias and Mota developed the W index for the diagnosis of lateral displacement of the inner canthi (06). A W index of more than 2.07 determines dystopia, whereas an index of less than 1.87 is normal. Other authors consider 1.95 or more to be indicative of a Waardenburg syndrome type I diagnosis (97; 109). Resetting the threshold of W index or novel index formulated with ethnicity-matched samples is necessary for clinical classification, which, in turn, is consistent with genetic classification for Waardenburg patients with distinct ethnicity (88).
Hypopigmentation. This abnormality is one of the cardinal signs observed in all four forms of Waardenburg syndrome. It affects the eyes, hair, and skin.
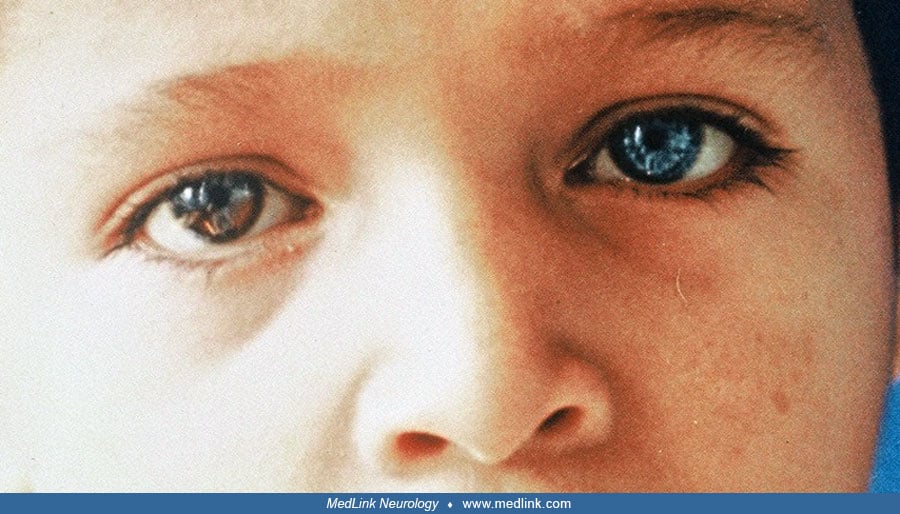
Eyes. Hypopigmentation of the iris results in a brilliant blue color that may be bilateral or unilateral (heterochromia, different color) and complete or partial. Partial heterochromia of the iris may be seen in the affected eye with a blue segment sharply demarcated from the rest of the iris. It is more common in Waardenburg syndrome type II than in type I (78). Bilateral congenital cataracts are an infrequent presentation (139). Unilateral cataract and glaucoma are unusual (63).
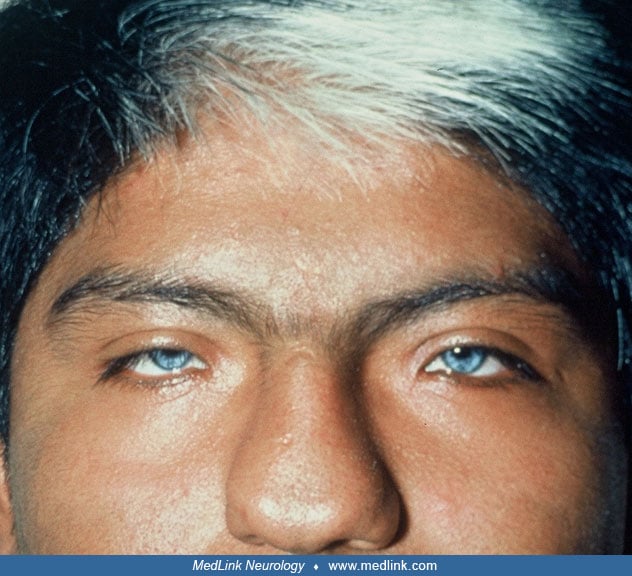
Hair. Poliosis, typically in the form of a white forelock, may be present in 20% to 40% of patients with Waardenburg syndrome. Less commonly, the forelock may be red or black (05). Hypopigmentation also may affect eyelashes and eyebrows. Poliosis may be present at birth then disappear and reappear later. Premature graying of the hair is frequent; it can be seen as early as 7 years of age (28). Complete depigmentation of the hair may occur in the teens (39). According to the Waardenburg Consortium, depigmentation occurs before 30 years, with the white hairs appearing in the midline (35).
Skin. Patchy hypopigmentation of the skin (leukoderma) is congenital and may be found on the face, trunk, or limbs. It is present in 8.3% to 50% of patients (29). Arias noted that hypopigmented areas frequently had hyperpigmented borders (05). Histological studies show absent or severely reduced melanocytes in the hypopigmented areas (29).
Deafness. Congenital deaf-mutism is frequent in patients with Waardenburg syndrome. The hearing loss may be unilateral or bilateral and varies in degree from slight to profound. Profound bilateral loss of hearing is the most common in both Waardenburg syndrome type I and type II. The auditory deficit is sensorineural and usually nonprogressive. Hageman and Delleman reported that 25% of subjects with type I and 50% of those with type II had a bilateral sensorineural hearing loss (46; 27). Newton found that the penetrance of sensorineural hearing loss varied from 69% in type I to 87% in type II (97). Variation between families was high, particularly in type I. Unusual audiogram shapes include low frequency and U-shaped losses, bilateral or unilateral, and sometimes a combination of a low frequency sensorineural hearing loss in one ear and a profound loss in the other (109). Unilateral sensorineural hearing loss is observed with less frequency (66).
Tubular nose. Patients with Waardenburg syndrome type I have a characteristic nose with a hypertrophic, broad, high nasal bridge usually with lack of frontonasal angle associated with hypoplasia of alae nasi, giving the nose a tubular appearance. Tubular nose is present in 80% of patients.
Synophrys. Joining of the eyebrows in the midline with hypertrichosis of the medial eyebrows is reported in 63.3% of patients with Waardenburg syndrome type I but only in 6% of those with type II (78). The later frequency is similar to that in the general population. In some patients, synophrys can be prominent (49).
Other facial features such as patent metopic suture and square jaw have been described (37). Together, the peculiar development of the facial bones with the high nasal root, hyperplasia of the eyebrows, a small, narrow nose, the dystopia canthorum, and the color of the eyes give most patients with Waardenburg syndrome type I a pleasant feline appearance. All these facial features can be explained by a disturbance in the neural crest. In a family of 29 members evaluated for Waardenburg syndrome, 16 showed features of type I. Dental abnormalities were identified in three members, including conical teeth, taurodontism, and especially dental agenesis. Type I was transmitted in this family in an autosomal dominant pattern with variable expressivity and high penetrance. Dental manifestations resulted in considerable functional and aesthetic impact on affected individuals (118).
Neurologic complications are uncommon in Waardenburg syndrome type I and include neural tube defects such as spina bifida (104; 29; 80); a familial occurrence of this association is rare (94; 17). A report of a patient with type I was associated with meningomyelocele, Chiari malformation, and hydrocephalus at birth (48). These authors reviewed 32 cases of a variety of neural tube defects associated with Waardenburg syndrome, in particular meningomyelocele, but did not find any cases of meningocele. Epilepsy is infrequent. A report of a 5-month-old infant who presented multiple generalized seizures that responded to pharmacological treatment showed he had delayed myelination that later normalized (122).
Several associated abnormalities have been reported in Waardenburg syndrome type I (25), including high arched or cleft palate (42), blepharophimosis (type 3), glaucoma, hydrophthalmos, true esotropia (20% of the cases), unilateral or bilateral renal and urinary system anomalies (32) and rarely, anal atresia. Homozygous patients have more severe systemic complications (11; 152). The association of Waardenburg syndrome type I and laryngomalacia in a neonate had not been previously described (132). A 50-year-old female with congenital deafness and premature greying of hair presented defective vision in the left eye, broad nasal bridge, medial eyebrow flare with dystopia canthorum, hypochromic iris, irregular pupil, and cataract. Intraocular pressure was high (30 mmHg). Fundus was normal in the right eye, and the left eye showed albinotic glow and advanced glaucoma. A diagnosis of type 1 Waardenburg syndrome was established. She was started on topical antiglaucoma medications, and after 2 months, she underwent combined cataract and glaucoma surgery in the left. On follow up at 1 month, her vision had improved, and her intraocular pressure was normal (63).
Waardenburg syndrome type II (WS2). Waardenburg syndrome type II is less well defined than type I; it covers any auditory-pigmentary syndrome that does not clearly belong elsewhere. These patients have a normal face without midline anomalies. Dystopia canthorum is absent, and the nose is indistinct. However, the same pigmentation defects observed in type I are present in type II but with different frequency (78). In general, type II occurs sporadically. However, several familial cases have been reported (72; 92; 21). Heterochromia irides is present in 47%, which is more common than in type I; partial heterochromia is also common, in as many as 27.5% (109).
A unique pattern of non-uniform pigmentation in the ocular fundi has been reported (92). The hearing loss also is more frequent than in type I, observed in up to 77% of these patients (29). A white forelock and hypopigmented skin patches are more frequent in type I than in type II (78). As in type I, systemic and neurologic abnormalities are uncommon; however, Dr. Flores-Sarnat has seen two children with Waardenburg syndrome type II with intellectual disability and autistic features without other neurologic or systemic manifestations. In 2007, two other patients with type II and intellectual disability were described (15); one patient also presented severe congenital heart disease. Another child was reported with autistic-like behavior; he has more severe intellectual disability as well as hypotonia, unilateral cryptorchidism, and patchy depigmented areas at the thighs and in the abdomen (116). A 3‑year‑old girl with congenital bilateral sensory neural hearing loss and asymmetrical partial heterochromia of iris and fundus, showed normal development (70). Infrequently, sensorimotor polyneuropathy with distal demyelination has been reported (01). Unilateral congenital ptosis was reported in one member of a family with type II (18). Mandibular asymmetry and malocclusion were reported in a 14-year-old Romanian boy with a family history of Waardenburg syndrome in the mother and Usher syndrome in the father (120). An infant examined for speech delay was found to have a profound hearing deficit, Waardenburg syndrome type II, and overt cerebellar asymmetry due to right hemispheric hypoplasia (62). A report of a 6-year-old boy with bilateral anophthalmia, brain anomalies, and epilepsy also showed some overlapping features of type II, including neurosensorial deafness and multiple white lesions in the lower extremities (40).
Waardenburg syndrome type III (WS3, Klein-Waardenburg syndrome). This form is the least frequent of the four. It usually presents as a sporadic disorder; only occasional familial cases have been reported (131; 149). Patients with Waardenburg syndrome type III have the same features observed in type I (dystopia canthorum, depigmentation, and deafness) plus a musculoskeletal syndrome. Usually, the hypopigmentation anomalies are more severe than in type I (152). After Klein’s first report, a small number of other patients have been described who show musculoskeletal abnormalities similar to Klein's patient but in milder form (114). There are bilateral defects of the upper limbs, including hypoplasia, contractures, carpal fusion, syndactyly, and upper limb-pectoral girdle arthromyodysplasia. Other neurologic abnormalities such as microcephaly and cognitive impairment may be observed in these patients (29), but intelligence is generally normal. Meningomyelocele also has been reported in these patients (100). A rare presentation in a patient with cognitive deficit and multiple congenital malformations, including anophthalmia, low-set ears, and dental and leg anomalies, included self-mutilation, particularly in the oral region (84). Extensive scars on the tongue, lips, and hands were a consequence.
Waardenburg syndrome type IV (WS4, Waardenburg-Shah syndrome, Waardenburg-Hirschsprung disease). Waardenburg syndrome type IV also is characterized by the association of type I features with congenital aganglionic megacolon or Hirschsprung disease. Type IV is rare, the less frequent type with an incidence of approximately 1 in 40,000 to 50,000 live births; it occurs in all races (23). The prevalence is less than 1 in 1,000,000 (16). This is also the most severe form of Waardenburg syndrome, with risk of mortality in early infancy. Mortality due to the abdominal complications and sepsis in the neonatal period and early infancy is high in type IV (143; 60; 02; 82; 45). It was described by Shah and colleagues in 1981 in 12 newborns from five families in Bombay who had Hirschsprung disease, white forelocks, and white eyelashes (113). Canthus dystopia was absent, hearing was not tested because all babies died in the neonatal period, and the description of the pigmentary disorder of the irides was nonspecific. The first report of the association between Waardenburg syndrome and Hirschsprung disease, suggesting that a neural crest defect was the common pathogenesis, appeared earlier (86). This is the only form of Waardenburg syndrome that may be transmitted as either an autosomal dominant or autosomal recessive trait, depending on the gene mutated. In a family with type IV distinct phenotypes among three different generations, hypopigmentation in the maternal grandmother, hearing loss in the mother, and Waardenburg syndrome type IV in the proband were observed (36). Neurologic involvement such as severe global developmental delay has been described since the early reports of this association (26). Dandy-Walker syndrome, congenital hypomyelinating neuropathy (56), and neonatal hypotonia and arthrogryposis (133) have been reported. A 4-year-old girl with the PCWH phenotype (peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease) showed also intellectual deficit, deafness, distal arthrogryposis, white hairlock, and growth retardation (138). The association of polydactyly, lumbosacral myelomeningocele, and agenesis of the corpus callosum reported in a female infant with type IV is an unusual presentation (112). A severe form in a child with deafness, bilateral complete agenesis of the semicircular canals, and mental retardation without Hirschsprung disease has been reported (123). Hirschsprung disease has a high prevalence in Kuwait, with an estimated consanguinity rate of 54%, and a strong male predominance; 2% of cases are associated with Waardenburg syndrome (151). The defective segment can be short, long, ultra-short, or total colonic aganglionosis (151). The clinical presentation is intestinal obstruction within 48 hours after birth; at times emergent surgery is required (60). Waardenburg syndrome type IV with total intestinal aganglionosis is rare. A neonate from Pakistan presented with bilious vomiting, refusal to feed, and failure to pass meconium, requiring several surgeries (65). Children with Shah-Waardenburg syndrome may develop short bowel syndrome and malabsorption that occurs after extensive intestinal resection (74). They present abdominal pain, diarrhea, and food intolerance because the decreased intestinal absorptive area leads to weight loss and malnutrition. Food allergy due to specific foods and eosinophilic infiltration of parts of the gastrointestinal system also can occur (74). Chronic constipation from the neonatal period was reported in a girl with heterochromia iridis, profound bilateral sensorineural hearing loss, inner ear malformations, and hypopigmentation of the hair without dystopia canthorum. She had a SOX10 mutation and was considered a clinical phenotype intermediate between type II and type IV syndromes (07). A female neonate with type IV presented a rare anomaly consisting of bilateral agenesis of the external ears (31). In a nonconsanguineous family from India, a 6-year-old boy was diagnosed with Waardenburg syndrome type IV after investigation for bilateral hypoacusis and the finding of brilliant blue eyes (16). He had a history of severe constipation at 6 months of age that required surgery and confirmed aganglionic megacolon. His 8-year-old sister and father were later diagnosed with type I. A 30-year-old male from India presented with elevated intraocular pressure, unusual iris morphology, and advanced glaucomatous optic atrophy in both eyes; he also had premature graying of hair. A diagnosis of Waardenburg syndrome type IV was confirmed with a mutation of EDN3 gene (103).
|
Major criteria: | ||
|
• Congenital sensorineural hearing loss | ||
|
- Two eyes of different color | ||
|
• Hair hypopigmentation: | ||
|
- White forelock | ||
|
• Dystopia canthorum: | ||
|
- W > 2.07 (Read and Newton suggest 1.95) | ||
|
• First degree relative with diagnosis of Waardenburg syndrome | ||
|
Minor criteria: | ||
|
• Congenital leucoderma | ||
|
| ||
|
At least two of the following criteria must be present: | ||
|
• Congenital sensorineural hearing loss | ||
|
- Complete or partial heterochromia of the iris | ||
|
• Pigmentary disturbances of the hair | ||
|
- White forelock from birth or in teens | ||
|
• A first- or second-degree relative with two or more of criteria 1–3 | ||
|
* Criterion 2c of Farrer and colleagues, corresponds to hypopigmented iris (35); however, it has been changed to hypoplastic eyes by Liu and colleagues (78) and Read and Newton (109). Patients with Waardenburg syndrome type I and type II do not have hypoplasia of the eyes; therefore, the correct description is hypopigmented iris as cited in the original reference. | ||
When deafness is absent, Waardenburg syndrome type I is a benign, nonprogressive condition that requires no treatment. Affected individuals have normal intelligence and they may have a normal life with normal life expectancy. Deafness is the most common associated complication; the impact of the disability depends on the severity of the auditory deficit. Waardenburg syndrome type II has a similar prognosis; however, deafness is more common in type II than in type I. The prognosis in Waardenburg syndrome type III and type IV is less favorable due to the associated anomalies, which also show different grades of severity.
A 15-year-old Mexican boy with Waardenburg syndrome type I had a negative family history and Mexican parents with brown eyes. He was the product of normal pregnancy and was delivered at term. He had a history of normal development except for lack of speech that lead to medical consultation in infancy. Audiometry revealed a profound bilateral deafness. Subsequent examination showed an alert, cooperative, teenager. He could not speak but understood and followed commands by signs or reading.
His skin was uniformly brown without areas of hypopigmentation. He had a median white forelock, present since birth; bilateral blue eyes with dystopia canthorum; synophrys; a tubular nose with a high, broad nasal bridge and hypoplasia of alae nasi; and a square jaw. Neurologic examination was normal with the exception of bilateral impairment of V3 auditory nerve without vestibular involvement. Repeat audiometry and auditory evoked potentials confirmed a profound bilateral neurosensorial deafness. His intelligence was normal. He attended special education for the deaf, had good support from his family, and was well adapted to his disability.
The mutations of primarily six different genes responsible for Waardenburg syndrome have been identified (107):
Waardenburg syndrome type I. Waardenburg syndrome type I has been mapped to the distal part of chromosome 2q35, and the gene identified is PAX3 (paired homeobox) (38; 134). A different phenotype with mutations of PAX3 has been reported depending on whether homozygous or heterozygous inheritance occurs (149). Known mutations in PAX3 are etiologically associated with Waardenburg syndrome and syndromic neural tube defects. Mutations in the murine homologue, Pax3, are responsible for the phenotype of splotch mice, in which nullizygotes are 100% penetrant for neural tube defects (80). A novel missense c.788T>G mutation in PAX3 in a Turkish family with Waardenburg syndrome with intrafamilial phenotypic heterogeneity was identified (49). Previously unreported digenic mutations in PAX3/GJB2 Waardenburg syndrome type I were observed in two Chinese siblings (20). A novel truncating mutation in exon 3 of SOX10 that is associated with neurologic symptoms was demonstrated in a 5-month-old infant boy with type I who had multiple generalized seizures (122). An autosomal recessive mutation in EDNRB was described as the cause of Waardenburg syndrome type I in a 5-year-old patient (89). In another study, single-nucleotide variants and copy number variation detection was applied to investigate the genotype spectrum of Waardenburg syndrome in a large Chinese population (76). Five EDNRB variants were associated with type I in the heterozygous state, with a detection rate of 22.2%.
Waardenburg syndrome type II. Waardenburg syndrome type II has been mapped to chromosome 3p12.3-p14.1, and the causal gene is MITF (microphthalmia transcription factor, human microphthalmia) (53; 125; 128). Only 10% to 15% of patients with type II show the mutation in MITF gene (92; 21). The finding of SOX10 deletions in patients with type II confirmed a new gene of Waardenburg syndrome type II (15). Theses authors also reported neurologic phenotypes that have been observed in type IV (intellectual impairment, abnormal myelination) in some type II-affected patients with SOX10 deletions. In a boy with a neurologic variant of Waardenburg syndrome type II, a 725 kb deletion at the 22q13.1 chromosomal region, including SOX10 gene, was found (116). A novel Waardenburg syndrome-associated mutation was reported in a 19-year-old Chinese girl with type II at the stop codon of MITF (p.X420Y). This mutation resulted in an extension of extra 33 amino-acid residues in MITF. The mutant MITF appeared in both the nucleus and the cytoplasm, whereas the wild-type MITF was localized in the nucleus exclusively (121). In a screening of a Waardenburg syndrome type II cohort, six heterozygous EDNRB variations were disclosed; these variations are now estimated to be responsible for 5% to 6% of type II cases (57). A novel mutation (Lys344Ter) in exon 9 of the MITF gene has been reported in a 3-year-old girl that was pathogenic for Waardenburg syndrome type 2A (70). The diagnosis of a subtype of type II relies on identifying the genetic cause. For some subtypes, the general location (locus) of the gene responsible is known, but the specific gene responsible has not yet been identified. In 13 patients with Waardenburg syndrome type II from six unrelated Chinese families, the mutation profile of genes related to congenital deafness in those families (targeted sequencing with candidate variants validated by Sanger sequencing), mutations in SOX10 and MITF were two major causes of deafness associated with Waardenburg syndrome. De novo mutations were frequent in probands with SOX10 mutations but not in those with MITF mutations (110). A 28-year-old Chinese woman presented to hospital with a 6-month history of polyarticular pain (77). On exam she also had hearing loss, absence of puberty, blue irides, and premature graying hair. She was diagnosed with Waardenburg syndrome type 2. Whole exome sequencing revealed a de novo variant of SOX10 gene.
Waardenburg syndrome type III. Waardenburg syndrome type III (Klein-Waardenburg) is also associated to PAX3 gene (52). An infant born to consanguineous parents with Waardenburg syndrome type I presented the typical picture of type III. The three of them had mutations of PAX3 gene with a novel missense mutation. However, only the child presented a homozygous PAX3 mutation (149). A spontaneous mutant mouse line, Rwa, has been identified, which displays white spots on bellies and tail and white digits; spina bifida was also observed with lower penetrance (101). The defective allele of Pax3 was named Pax3 Rwa.
Waardenburg syndrome type IV. Waardenburg syndrome type IV (Waardenburg-Shah syndrome) is caused by mutations of three genes: the endothelin receptor B gene (EDNRB) on chromosome 20q13 (135), the endothelin-3 gene (EDN3) on chromosome 20q13 (09; 30; 51), and the SOX10 gene, also on chromosome 20q13 (124). When EDN3 or EDNRB genes are responsible for Waardenburg syndrome type IV, it is inherited as an autosomal recessive trait. If SOX10 mutations are involved, type IV is inherited as an autosomal dominant condition (133). The expression of SOX10 in the human embryo is not restricted to neural crest-derived cells but also involves fetal brain cells, most likely of glial lineage (133). Heterozygotes are usually unaffected or have isolated Hirschsprung disease (04; 10; 71). SOX10 mutations are specifically associated with a more severe phenotype called PCWH: peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease (138). SOX10 mutations, inherited in autosomal dominant way, are found in 45% to 55% of patients with type IV (36). These mutations are usually associated with a more severe phenotype, as in the case of a patient with congenital deafness, iris heterochromia, Hirschsprung disease, foot deformity, peripheral demyelinating sensorimotor neuropathy, and central dysmyelinating leukodystrophy with a novel heterozygous missense variant in the SOX10 gene (14).
The zebrafish, lacking functional SOX10, is considered a good animal model for human Waardenburg syndrome type IV (34). Peripheral neuropathy and lack of Hirschsprung disease has been reported in type IV with a SOX10 gene sequencing identified "de novo" splice site mutation (c.698-2A > C). (123). The first characterization of SOX10 deletions in patients with Waardenburg syndrome type IV was reported in 2007 (15). The SOX10 gene, encoding a transcription factor, is essential for neural crest development and myelin formation both in the central and peripheral nervous systems (147). Mutations in this gene are associated with two distinct “neurocristopathies.” A milder, more restricted spectrum trait, Waardenburg-Hirschsprung disease (WS4, OMIM: 277580), combines Waardenburg syndrome and Hirschsprung disease (106). A more severe and complex neurologic trait, “peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease” (PCWH, OMIM: 609136) reveals additional de- and dysmyelinating phenotypes in the peripheral and central nervous systems (108; 133; 56; 54). The report of a type IV family, carrying an insertion of 19 nucleotides in exon 5 of SOX10, showed distinct phenotypes among three different generations (36). The vast majority of disease-associated SOX10 mutations result in premature termination codons (PTCs), causing either Waardenburg syndrome type IV or PCWH depending on the position of the mutations. Failure to properly terminate SOX10 translation causes the generation of a deleterious functional domain that occurs because of translation of the normal 30-UTR; the mutant fusion protein causes a severe neurologic disease (55). Bilateral agenesis or hypoplasia of the semicircular canals or both, associated with an enlarged vestibule and cochlear deformity, strongly suggest a diagnosis of Waardenburg syndrome linked to a SOX10 mutation (33). A novel variant site in the SOX10 gene in Waardenburg syndrome type 4 was found for the first time in a Chinese family in an 8-year-old boy with bilateral pale blue irides (141). He had a history of congenital deafness and severe constipation. At 6 months of age, he required surgery that confirmed aganglionic megacolon (Hirschsprung disease). His 8-year-old sister and father were later diagnosed with Waardenburg syndrome type I. A study to investigate Waardenburg syndrome type 4 in an underrepresented Mongolian population with whole-exome sequencing revealed a 5-year-old girl with profound sensorineural hearing impairment (SNHL), dystopia canthorum, and a white forelock (43). A 9-year-old boy also had profound sensorineural hearing impairment, dystopia canthorum, and in addition, Hirschsprung disease. Both patients had bony abnormalities in the inner ear and mutation in SOX10.
Pathogenesis. Waardenburg syndrome results from a defect in the migration and differentiation of neural crest cells. Three derivatives of the neural crest are related to Waardenburg syndrome: melanocytes, ganglion cells and membranous bones of the face (particularly the forehead, the orbits, and the nasal bones). Waardenburg syndrome has been recognized as a neurocristopathy since 1959 (37).
Physical absence of melanocytes from the skin, hair, eyes (choroid and iris) and the stria vascularis of the cochlea cause the auditory-pigmentary manifestations of Waardenburg syndrome. The absence of the plexuses of Meissner and Auerbach that derive from the neural crest results in aganglionic megacolon or Hirschsprung disease. The cartilages of the larynx (thyroid, cricoid, and arytenoids) and the intrinsic muscles also derive from neural crest, which might explain the rare association of Waardenburg syndrome and laryngomalacia (132). Microtia and severe micrognathia were observed in a neonate with Waardenburg syndrome and multiple malformations, also due to defective neural crest (150). Several mechanisms may contribute. Motor trigeminal nerve sheaths derive from neural crest; if they are defective, innervation of the mandibular muscles is impaired and the muscular stimulation of bone growth is deficient during the period of most rapid growth in the late 2nd and 3rd trimesters. Neural crest forms the membranous bone of the mandible itself. Microtia can occur if neural crest cells that differentiate in the mesenchyme of branchial arches are defective. The rare finding of bilateral agenesis of external ears is also the result of defective neural crest (31).
Melanocytes originating in the neural crest produce all the melanin that pigments the skin, hair, and eyes, with the exception of the retinal melanocytes that are derived from neural ectoderm. In addition, melanocytes are required for the development of auditory structures such as the stria vascularis of the cochlea (119). In the absence of melanocytes, the stria is abnormally thin, no endocochlear potential is generated, and later in development Reissner membrane collapses, leading to destruction of the organ of Corti (119). Absence of melanocytes could be caused by a failure in differentiation of the neural crest, a migration failure of melanoblasts, or a failure in survival at their final destination (109). In Waardenburg syndrome type II, both skin and retinal melanocytes are affected; therefore, this is a melanocyte-specific disorder, whereas type I, type III, and type IV are neurocristopathies involving neural crest derivatives such as the frontal bone, limb muscles, and enteric ganglia (109). The PAX3 gene is involved in neural crest development; it is expressed in the developing somites, dorsal spinal cord, mesencephalon, and neural crest derivatives (44; 129; 130; 41). This gene controls some aspects of the development of the face and inner ear and is expressed in the mesenchyme of the limb buds, explaining the phenotype of type III (12). Malformations include a lack of muscle in the limb, a failure of neural tube closure, and dysgenesis of numerous neural crest derivatives (83). At present, over 50 PAX3 mutations have been identified in patients with type I or type III (35; 130; 127; 109).
The MITF gene is the human homologue of the mouse microphthalmia (mi) gene (08; 50) and is an important factor for differentiation, function, and survival of melanocytes (142). It is expressed in adult skin and in embryonic retina, otic vesicle, and hair follicles. It has been suggested that in dominant families with Waardenburg syndrome type II, haploinsufficiency is the most likely cause (98). Mutations in the MITF gene have also been found in Tietz syndrome, which is related to Waardenburg syndrome (22; 117).
The SOX10 gene is expressed during human embryonic and fetal development and has an important role in early development of neural crest-derived tissues related to Waardenburg syndrome, such as melanocytes and the enteric nervous system. Sox10 is also particularly important for development of the inner ear, and its mutation in Waardenburg syndrome type II and IV explains the reason for neurosensory hearing loss (148). The study of 13 patients with Waardenburg syndrome type II from six unrelated Chinese families showed that mutations in SOX10 and MITF are two major causes of deafness associated with Waardenburg syndrome, and de novo mutations were frequently found in probands with SOX10 mutations but not in those with MITF mutations (110). The common etiology of SOX10 in Kallmann syndrome and Waardenburg syndrome is significant, and it has been suggested that they share a common pathway in the development of neural crest cells (140).
The ligand-receptor interaction between EDN3 and EDNRB is important for the development of epidermal melanocytes and enteric neurons.
Pathologic findings. Inner ear histological studies in patients with Waardenburg syndrome have revealed cochleosaccular degeneration (37). Absent organs of Corti, atrophy of the spiral ganglion, and reduction of the nerve fibers have been reported. A computed tomographic study in patients with Waardenburg syndrome type I and type II presenting profound, bilateral deafness showed several abnormalities in the temporal bone (81). They included narrowing of the internal auditory canal porus and hypoplasia of the modiolus. A third case of Waardenburg syndrome type IV, which did not have myenteric ganglion cells in the sigmoid colon and rectum, was reported (146).
Autopsy reports of individuals with Waardenburg syndrome are rare. A male infant born at 29 weeks’ gestation with Waardenburg syndrome (type not confirmed) had gross tetraphocomelia and multiple organ malformations (lungs, cardiac, renal, liver) and died 10 minutes after birth. His brain showed hydrocephalus and malformation of the temporal lobes (150). Cochleosaccular degeneration and a complete absence of pigmentation was reported at necropsy in a 3-year-old child with Waardenburg syndrome type IV (93). Another case, a 2.5-year-old boy with congenital deafness, patchy cutaneous hypopigmentation, bilateral blue irides, patchy white eyelashes and eyebrows, a broad nasal root, and Hirschsprung disease also showed Dandy-Walker malformation, which was confirmed by autopsy (143). The colon was absent, and the small intestine showed an absence of ganglia. Bilateral complete semicircular canal agenesis was reported in a child with Waardenburg syndrome type IV (123). An unusual demyelinating peripheral neuropathy characterized by excessive focal folding of myelin sheaths was described in a patient with Hirschsprung disease and a diagnosis of Waardenburg syndrome type II (probably corresponded to type IV) (59). An infant boy with lethal congenital hypomyelinating neuropathy and Waardenburg syndrome type IV who had a heterozygous SOX10 mutation (Q250X) showed in the histopathological studies an absence of peripheral nerve myelin despite normal numbers of Schwann cells and profound dysmyelination in the central nervous system (56).
Waardenburg syndrome is observed at all ages, from newborns to elderly people. Both sexes are affected equally. It occurs in all ethnic groups without any predominance. The estimated prevalence in the Netherlands in the Waardenburg review (145) was 1/42,000 of the general population and 1.43% in the congenitally deaf. Fraser studied 2355 deaf children and estimated a prevalence of 1.44 to 2.05/100,000 in the general population (39). Consanguinity is a risk factor (47). The most common types are Waardenburg syndrome type I and type II, and according to the diagnostic criteria, they are about equally common. Waardenburg estimated a mutation rate of 0.4/100,000 gametes (145). A high incidence in Africans from Kenya has been reported (29). A computerized literature review found sporadic cases of the syndrome in many ethnic groups, including Japanese, Taiwanese, and Middle Eastern families (95). The first reports of Waardenburg syndrome type I and type II in China with novel mutations in PAX3, MITF and SOX10 genes have appeared (18). The authors identified, for the first time, SOX10 mutations in cases of Waardenburg syndrome type II with an estimated frequency similar to that of MITF mutations. A screening program for Waardenburg syndrome in Colombia disclosed 95 affected individuals belonging to 95 families, giving a frequency of 5.38% of Waardenburg syndrome among the institutionalized deaf population and 45 noninstitutionalized affected relatives. They classified 62.1% of the propositi as type II and 37.9% as type I (126).
Folic acid has been recommended in high-risk pregnancies for Waardenburg syndrome (61; 31; 45). Rare patients with Waardenburg syndrome who present hyperpigmented regions in their fundi should be monitored closely for the development of choroidal malignant melanoma (58).
Tietz syndrome (75)
A rare association of Waardenburg syndrome type II and Kallmann syndrome was reported in a man who carried a novel insertion of a 20 bp fragment into the coding sequence of the exon4 of the SOX10 gene. The patient exhibited anosmia and hypogonadotropic hypogonadism (characteristics in Kallmann syndrome) and also bilateral sensorineural deafness, blue irises, and white forelock (19).
In another case associated with Kallmann syndrome, an aplastic olfactory bulb was demonstrated (140). Tietz syndrome is also related to Waardenburg syndrome (22; 117).
Identification of the clinical picture, including a history of consanguinity and categorization of the type of Waardenburg syndrome is the first step for confirming the diagnosis. Conventional audiometry and the use of otoacoustic emissions are recommended in order to provide optimum fitting of hearing aids, especially in children (102). Early detection of hearing deficits, as early as the newborn period, may be established with auditory evoked potentials. In children with unilateral sensorineural hearing loss, a rigorous screen for pigmentary abnormalities is recommended (66). Computed tomographic scanning of the temporal bone may disclose abnormalities of the inner ear, such as malformation or absence of the semicircular canals and cochlea (96). The rare report of a young infant with Waardenburg syndrome type I and seizures revealed bilateral hypoplasia of the semicircular canals and cochlea by CT (122). His brain MRI demonstrated delayed myelination, which showed recovery to normal myelination on his follow-up MRI. Molecular genetic tests are important to determine the specific genes involved and may be essential in some patients without a classical picture. The biometric index based on three measurements is particularly helpful in distinguishing type I and type II and may be used as a guide to look for a PAX3 mutation (109). A W index of 1.95 or more is diagnostic of dystopia canthorum and, therefore, of type I. It is important to pay attention to the characteristic dysmorphology of Waardenburg syndrome; otherwise, it could be misdiagnosed as nonsyndromic deafness (115). Molecular analysis is necessary in doubtful cases (59) and essential to define the most severe cases in Waardenburg syndrome type IV (133; 56; 138). The detection of both partial and whole gene deletions of PAX3/MITF increases the mutation detection yield by at least 6% and supports integrating MLPA into clinical molecular testing primarily for patients with Waardenburg syndrome type I and type III (87). Analysis of the 3-dimensional (3D) structure of PAX3 helps verify the pathogenicity of a missense mutation. Multiple ligation-dependent probe amplification (MLPA) analysis of PAX3 has increased the sensitivity of genetic diagnosis in patients with Waardenburg syndrome type I, particularly in those patients with isolated, mild, or nonspecific symptoms (85). In those cases that present with neuropathy, electrodiagnostic studies and nerve biopsy are required (01). In patients with the PCWH phenotype, MRI is essential to investigate central hypomyelination (138). Studies in large samples, including familial and isolated probands, have the advantage of finding novel causative mutations in different types of Waardenburg syndrome. In a large Brazilian sample of patients with Waardenburg syndrome type I and type II, 10 novel causative mutations were found (13). In special cases, prenatal diagnosis of spina bifida can lead to the diagnosis of Waardenburg syndrome type I in the mother and other members of her family (69). Neonates and young infants with difficult breathing and inspiratory stridor require direct laryngoscopic examination to rule out laryngomalacia (132). Bilateral temporal bone abnormalities associated with agenesis or hypoplasia of greater than one semicircular canal, an enlarged vestibule, cochlear hypoplasia, or agenesis of cochlear nerve have been demonstrated with MRI (33). In addition, agenesis of the olfactory bulbs, hypoplastic or absent lacrimal glands, hypoplastic parotid glands, and white matter signal anomalies were found by the same authors (33). A scoring system designed to predict the severity of Hirschsprung disease, isolated or associated with Waardenburg syndrome, was applied at the time of diagnosis (99). The authors’ goal was to evaluate a possible clinical-genetic correlation. In the severe group, most mutations were located in the RET proto-oncogene.
Management is directed to the treatment of associated complications. The hearing deficit is the most common impairment; therefore, early diagnosis and intervention to improve this problem are important steps for the psychological development of these children and help to reduce their sense of isolation (29). Special education is essential to enhance normal development in children with hearing deficits. Cochlear implants are indicated in these patients. In one study, the average age at implantation was 37 months (range, 18 to 64 months) and children accepted and regularly used their devices. In general, patients with Waardenburg syndrome have above-average performance after cochlear implantation (24) and their cognitive development gradually improves to a normal level (146).
In another study, cochlear implantation at the age of 2 years or younger had similar good results, but the presence of additional disabilities requires special counseling (137). Surgical correction of congenital megacolon should be performed early; in many cases it is successful. Understanding of the syndrome and tolerance of associated disabilities are important for the integration of these patients into society (29). In patients with Waardenburg-Shah syndrome, the Ziegler operation is recommended when there is inadequate ganglionic bowel length to gain time for the child to grow and to decrease total parenteral nutrition complications (60). Patients with Shah-Waardenburg syndrome who develop food allergy and eosinophilic gastrointestinal diseases with abdominal pain, nausea, vomiting, and chronic diarrhea after intestinal resection might benefit from a trial of strict dietary avoidance of suspect allergenic foods (74). In a female infant with Waardenburg syndrome type IV who underwent 14 surgical interventions from birth to 3 years of age, including a skin grafting of a nonhealing ulcer from malnutrition, parenteral nutrition was required for more than 20 months due to recurrent enterocolitis and poor oral intake (91). Self-evacuation improves with daily glycerin enemas; probiotics are also recommended (146). Ptosis is rare in Waardenburg syndrome; it has been reported in Waardenburg-Sha syndrome. Abnormalities in the eyelid ptosis can be corrected by plastic surgery, which is beneficial to children's physical and mental development (79). Cataract and glaucoma are uncommon; however, a 50-year-old female with Waardenburg syndrome type 1 had surgery with good outcome (63).
No complications have been reported during pregnancy on Waardenburg syndrome type I. Fertility is normal in type I and type II. A reduction of fetal movement was noted during the last month of pregnancy; the proband had Waardenburg syndrome type IV, arthrogryposis, and meconial ileus and died on day 11 of life (133). In the case that both parents are affected by Waardenburg syndrome, complications in the pregnancy including polyhydramnios, intrauterine growth restriction, and decreased fetal movements associated with multiple anomalies in the fetus also affected by Waardenburg syndrome were reported (73). Folic acid has been recommended in women with pregnancies at risk of Waardenburg syndrome (61; 31). Patients with Waardenburg syndrome requiring obstetrical anesthesia have not been reported in the literature, suggesting that there is no increased risk of obstetrical complications (03). Two cases of Waardenburg syndrome type III were diagnosed in the first trimester of pregnancy, due to a homozygous mutation in PAX3. The ultrasound revealed increased nuchal translucency and lack of active movements, with bilateral wrist contractures and club feet (90).
Patients with Waardenburg syndrome may require surgical treatment, particularly in types 3 and 4, but very little has been published about the anesthetic implications (02). Characteristic facial features, muscle contractures, and difficult airway may cause difficulties in both direct laryngoscopy and tracheal intubation, often require cochlear implant at an early age, and require anticipation and special equipment (105). The anesthetic management of a child with Waardenburg syndrome type IV undergoing multiple surgical procedures was complicated by perioperative problems, such as malnutrition, electrolyte imbalance, and communication difficulties due to congenital deafness and blindness (91). In a report of two neonates with type IV who presented with constipation since birth, the second neonate was lost to follow-up (02). Pudendal nerve block was applied in a child with Waardenburg syndrome who required surgery for cleft palate and spina bifida (64). Meticulous attention is required with preoperative evaluation, coexistence of other system abnormalities, airway management, and perioperative nutrition strategies (risk of malnutrition increasing with the extended aganglionic segment of the intestine).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Laura Flores-Sarnat MD
Dr. Flores-Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 20, 2026