

Peripheral Neuropathies
Clinical evaluation of peripheral neuropathies
Jul. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The authors review the clinical presentation, diagnosis, pathogenesis, and management of acute autonomic neuropathies. Research indicates that many patients with acute autonomic neuropathy test positive for elevated ganglionic acetylcholine receptor autoantibodies and present with a syndrome complex, including the sicca syndrome, orthostatic hypotension, lower gastrointestinal dysautonomia, and neurogenic bladder. Mouse models in which this antibody is induced also develop autonomic neuropathy. Other causes of acute autonomic neuropathy, including Guillain-Barré syndrome, which has been shown to be associated with Zika virus infection, and paraneoplastic syndrome, are described. COVID-19 may be associated with dysautonomia, but its association with Guillain- Barré syndrome is unclear, and a large epidemiological study does not support an association. Many patients with autoimmune autonomic neuropathy respond rapidly and well to immune-modulating therapy, including intravenous immunoglobulin. Givosiran is approved by the U.S. Food and Drug Administration for the treatment of acute hepatic porphyrias; however, there are considerable adverse effects associated with this medication that can be reduced by using a personalized medicine approach. The management and treatment of these disorders is discussed, including information on the management of infant botulism.
|
• All acute autonomic neuropathies share common clinical features that may include orthostatic hypotension, dry eyes and mouth, cold feet and hands with color or trophic changes in the skin, reduced or accentuated sweating, anorexia or early satiety referable to gastroparesis, erectile or ejaculatory failure, and defects in micturition stream or volume, but selective syndromes are known. | |
|
• Autoantibodies to nicotine ganglionic acetylcholine receptors likely have a pathogenic role in autoimmune autonomic neuropathy. | |
|
• The presence of dysautonomia is associated with a higher incidence of mortality from Guillain-Barré syndrome. | |
|
• Assessment of a patient with a suspected autonomic neuropathy includes a detailed history, blood pressure and heart rate measurement with the patient supine and after standing for 1 minute, and laboratory tests of the autonomic nervous system (eg, tests of cardiovagal function, tests of sympathetic function, and testing for gastroparesis). | |
|
• Some autonomic neuropathies are amenable to treatment of the underlying disease, and autoimmune autonomic neuropathies may be responsive to intravenous gammaglobulin or other immunosuppressive treatments. |
Acute autonomic neuropathy is an inclusive term used to describe diseases resulting from distinct etiologies but have in common pathology of the peripheral autonomic nervous system. Acute intermittent porphyria has been implicated in King George III of England's neuropsychiatric illness.
The specific clinical manifestations of acute autonomic neuropathies are dependent on the etiology of the neuropathy; however, all acute autonomic neuropathies share common clinical features. These include orthostatic hypotension, dry eyes and mouth, cold feet and hands with color or trophic changes in the skin, reduced or accentuated sweating, anorexia or early satiety referable to gastroparesis, erectile or ejaculatory failure, and defects in micturition stream or volume. The specific clinical manifestations are described below in relation to each cause of acute autonomic neuropathy.
Acute panautonomic neuropathy (pandysautonomia). This condition typically presents with an acute to subacute onset of autonomic dysfunction that is progressive over 1 to 2 months and is often preceded by a viral infection. Symptoms consist of severe orthostatic hypotension, anhidrosis, and parasympathetic failure with dry eyes, dry mouth, and dysfunction of bowel, bladder, and sexual performance. Patients can also experience abdominal pain, early satiety, bloating, nausea, vomiting, and alternating constipation with diarrhea as evidence of gastric and bowel paresis. Typically, the pupils are unresponsive to light, and the heart rate does not change with increased physical activity.
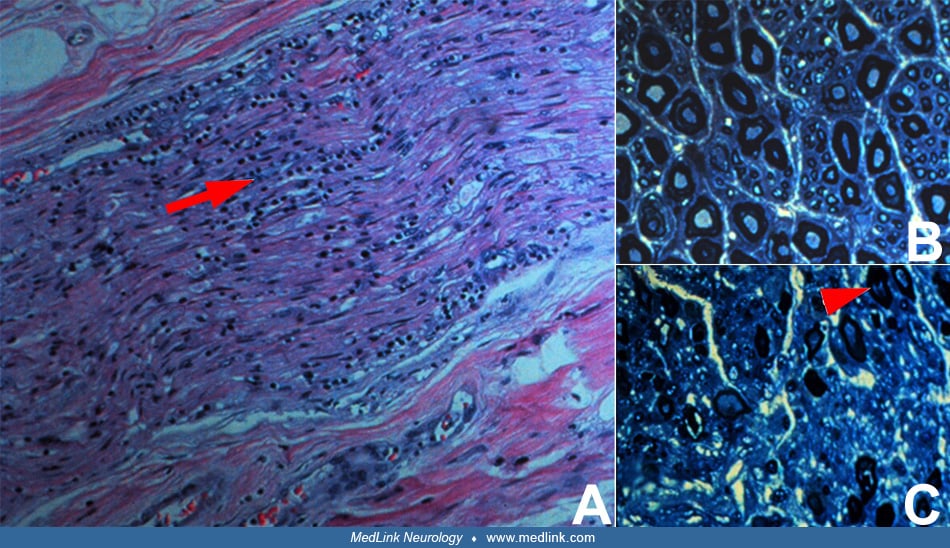
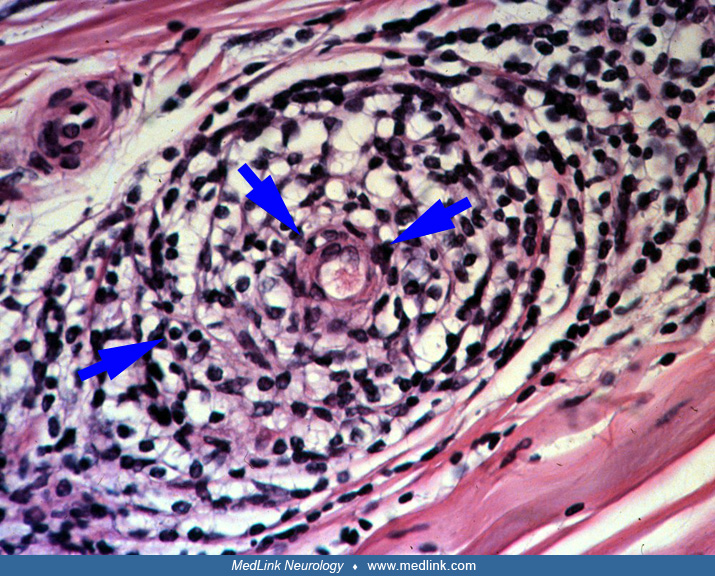
A viral infection, infectious mononucleosis, rubella, or Herpes simplex infection precedes the onset of acute panautonomic neuropathy in approximately 50% of subjects. Pandysautonomia can also be a paraneoplastic phenomenon. The frequent preceding infection, as well as the acute onset and presence of perivascular mononuclear cell infiltrate in the epineurium of sural nerve biopsies, suggests that the etiology is likely to be immune-mediated (40). In addition, this condition may be associated with an acellular, elevated CSF protein in up to 70% of patients.
One study found a positive correlation between high levels of autoantibodies to nicotine ganglionic acetylcholine receptors and the severity of autonomic dysfunction (46). These antibodies were found in about 40% of patients with idiopathic autonomic neuropathy and correlated with the severity of the neuropathy, suggesting that they are involved in pathogenesis.
Acute cholinergic neuropathy. This disorder is likely a subset of acute panautonomic neuropathy, affecting approximately 25% of patients (47). Patients with the restricted cholinergic form have a similar set of symptoms and findings (xerostomia, anhidrosis, constipation, urinary retention, and blurred vision), but do not have orthostatic hypotension. The abnormalities in this disorder are limited to the postganglionic cholinergic neurons. Gastrointestinal problems may be the primary manifestation of this illness.
Guillain-Barré syndrome. Guillain-Barré syndrome is an acute, acquired, monophasic autoimmune disorder of peripheral nerves. The term refers to a spectrum of acute idiopathic peripheral neuropathies. Guillain-Barré syndrome often develops in susceptible patients after an infection, most commonly Campylobacter jejuni, but it can also develop after infections with cytomegalovirus, Epstein-Barr virus, and Mycoplasma pneumoniae, or, in rare cases, after immunization. It has been shown that the Zika virus is associated with Guillain-Barré syndrome (31) and in an observational study it has been shown that Guillain-Barré syndrome is the most frequent neurologic manifestation in Zika virus postnatal infection (24). The association between COVID-19 and Guillain-Barré syndrome is unclear and is not supported by a large epidemiological study (21). However, disorders of autonomic function and specifically orthostasis may be observed post-COVID-19 (38). The European Academy of Neurology/Peripheral Nerve Society has provided new guidelines on diagnosis and treatment of Guillain-Barré syndrome that should be used in managing this complex disorder (43).
A review of the relationship between Guillain-Barré syndrome and exposure to influenza via infection or vaccination found that, except for the 1976 United States national immunization program against influenza A H1N1, influenza vaccine has resulted in extremely low rates of Guillain-Barré syndrome (25). In contrast, influenza infections are often a triggering event for Guillain-Barré syndrome. With regards to the human papillomavirus (HPV4) vaccine, postmarketing surveillance studies have provided inconsistent evidence about the incidence of Guillain-Barré syndrome following vaccination.
There have also been case reports describing the development of Guillain-Barré syndrome as a complication of checkpoint immunotherapy (17; 48). This is an uncommon complication of this class of drugs, which are increasingly being used in the treatment of multiple types of cancer, but the resulting neuropathy is often severe and may present with profound autonomic dysfunction (48). In contrast to idiopathic Guillain-Barré syndrome, Guillain-Barré syndrome triggered by the use of immunotherapy agents is frequently characterized by a lymphocytic CSF profile, and it is often responsive to steroids (17). However, larger case series are needed to properly characterize this neuropathy and its treatment.
In contrast to acute panautonomic neuropathy, the clinical presentation of Guillain-Barré syndrome is primarily involved with the somatic nervous system. Patients present with acute to subacute ascending numbness and predominantly motor polyradiculoneuropathy as a monophasic illness. However, autonomic dysfunction is variably present in Guillain-Barré syndrome, and its presence is associated with a higher incidence of mortality (27; 04). Autonomic dysfunction in Guillain-Barré syndrome typically manifests as tachycardia and alternating orthostatic hypotension and hypertension (49). The cardiovascular findings occur in about one third to two thirds of patients and are usually mild (27). However, some patients with Guillain-Barré syndrome have severe autonomic dysfunction that can be life-threatening (10; 49). For example, serious bradyarrhythmias or even asystole can develop and may require the placement of a cardiac pacemaker. The severity of the dysautonomia seen in Guillain-Barré syndrome tends to parallel the degree of somatic impairment but can also develop in patients who are ambulatory (09). Other symptoms of autonomic dysfunction seen in Guillain-Barré syndrome include anhidrosis, diarrhea, gastroparesis, adynamic ileus, bladder dysfunction, hyponatremia, and syndrome of inappropriate antidiuretic hormone secretion. One simple bedside test for dysautonomia is bilateral ocular pressure for 25 seconds, resulting in significant bradycardia.
Abnormal laboratory studies seen in Guillain-Barré syndrome include CSF albuminocytological dissociation and an elevated protein in 65% within the first week and 85% in the second week. The CSF cell count is usually normal, and oligoclonal bands may be present in 10% to 30% of cases. Demyelination is observed on nerve conduction studies 1 week after onset, and over 80% the F-waves and H-reflexes are abnormal. Typically, sensory nerve action potentials are absent or reduced in the arms with preservation of the sural responses (sural sparing). Only 40% of patients recover to full premorbid status; substantial symptoms persist in approximately 10%. Evidence of residual neuropathy is found in approximately 50% of patients (08). The recovery of autonomic dysfunction parallels the improvement of motor function.
There is an axonal subtype of Guillain-Barré syndrome; acute motor axonal neuropathy (AMAN). This is the major subtype seen in Asia and Central and South America, where it can be seen in 30% to 65% of patients (23). Autonomic dysfunction is rare in acute motor axonal neuropathy, but this subtype tends to progress more rapidly and peak earlier than demyelinating Guillain-Barré syndrome. Patients with acute motor axonal neuropathy also have normal sensory examinations, and approximately 10% of patients have normal deep tendon reflexes. One of the main underlying mechanisms of acute motor axonal neuropathy is molecular mimicry of human gangliosides by Campylobacter jejuni lipooligosaccharides.
Autoimmune autonomic neuropathy associated with ganglionic acetylcholine receptor antibodies. Autoimmune autonomic ganglionopathy is a rare form of dysautonomia that is frequently associated with elevated titers of ganglionic acetylcholine receptor (G-AChR) antibody. More than 50% of patients with elevated ganglionic acetylcholine receptor autoantibodies have an autonomic neuropathy symptom complex with prominent cholinergic failure consisting of xerophthalmia, xerostomia, abnormal pupillary light response, lower gastrointestinal dysautonomia, and neurogenic bladder (36). The most common symptom is orthostatic hypotension. Studies have found a positive correlation between high levels of autoantibodies to ganglionic acetylcholine receptors and the severity of autonomic dysfunction (26), and this likely has a pathogenic role in many subjects with autoimmune autonomic neuropathy.
Autoimmune autonomic ganglionopathy typically affects previously healthy young or middle-aged individuals and has a 2:1 female predominance. It presents with a severe panautonomic failure that reaches peak severity in days to weeks and a slow recovery, with the majority of patients experiencing an incomplete recovery. On examination, patients have normal strength, reflexes, and sensation. Nerve conduction studies are normal, but autonomic testing reveals diffuse autonomic failure. Many cases have a preceding viral syndrome, but no specific infection has been implicated. Furthermore, similar to Guillain-Barré syndrome, autoimmune autonomic ganglionopathy may be associated with recent immunization or surgical procedure. Treatment typically involves immunosuppressive therapy, but approximately one third of patients recover spontaneously. Patients with symptoms of autoimmune autonomic ganglionopathy but who are seronegative for ganglionic acetylcholine receptor antibodies have also been described (14). The patients present with prominent sympathetic failure and sensory symptoms. They respond best to steroids rather than antibody-targeted immunotherapy.
Low titers of G-AChR antibodies have been found in patients with mild or chronic forms of autonomic dysfunction as well as in patients with other neurologic disorders, normal patients, or those with an underlying cancer. Antibody levels of less than 0.20 nmol/L have been found to have little clinical importance in the absence of symptoms of autonomic failure (06).
Other paraneoplastic autonomic neuropathies. Paraneoplastic autonomic neuropathy usually presents as a subacute panautonomic neuropathy and may be difficult to distinguish from pandysautonomic autonomic neuropathy. The disease may also be limited, for example, to the gastrointestinal system—paraneoplastic enteric neuropathy. The symptoms usually precede the diagnosis of cancer, and the paraneoplastic antibody level is not related to the severity of the clinical disease. Patients may have involvement of the dorsal root ganglia, as well as a variety of autonomic symptoms, including orthostatic hypotension, tonic pupils, anhidrosis, bladder paralysis, impotence, dysphagia, regurgitation, nausea, bloating, vomiting, and constipation that may be severe. Gastrointestinal symptoms typically affect the entire gut, but may be localized with achalasia, gastroparesis, reduced small intestinal transit, chronic pseudo-obstruction, and constipation that may be coupled with megacolon (16).
The condition may be associated with several types of cancer, including cancer of the lung (especially small cell) as well as lymphoma and cancer of the bladder and bowel. In up to 30% of subjects, there may be an increase in IgG levels or G-AChR antibodies. Multiple antibodies are associated with autonomic neuropathy, including Purkinje cell cytoplasmic antibody type 2 (PCA-2), antineuronal nuclear antibody (ANNA, also known as anti-Hu), collapsin response-mediator protein-5 antibodies (CRMP-5 or anti-CV2), and voltage-gated potassium channel antibodies (41). It is difficult to predict the type of antibody based on the clinical presentation, so diagnostic testing with a broad panel is often recommended.
Patients with this disorder may have absent sympathetic skin response, abnormalities of the R6 interval and Valsalva ratio. There is also evidence of axonal inflammation coupled with loss of peripheral axons or ganglion cells.
Another variant of paraneoplastic autonomic neuropathy is enteric neuronopathy, in which axonal degeneration, inflammation, or antibodies directed against the myenteric plexus are found. Affected patients may develop a pseudo-obstruction with severe nausea, abdominal pain, and bowel distention. Small-cell lung cancer and carcinoid tumors are the most common malignancies associated with paraneoplastic chronic intestinal pseudo-obstruction, and anti-Hu autoantibodies are often found to be present.
Lambert-Eaton myasthenic syndrome is an acquired presynaptic disorder of neuromuscular transmission frequently associated with autonomic neuropathy. A neoplasm is found in approximately 50% of cases. The most frequent association is with small-cell lung cancer, but it may also be seen with carcinoma of the breast, colon, stomach, breast, kidney, and prostate. Xerostomia is a frequent clinical manifestation (75% of patients). Other autonomic features include xerophthalmia, tonic pupils (60%), orthostatic hypotension, constipation, and bladder dysfunction. On autonomic testing, widespread dysfunction of both cholinergic and adrenergic function is described, suggesting a widespread transmitter defect. Abnormalities of the P/Q voltage-sensitive calcium channels are found in 100% of subjects with Lambert-Eaton myasthenic syndrome and cancer, and in over 90% of those with Lambert-Eaton myasthenic syndrome alone.
Treatment-induced neuropathy of diabetes (insulin neuritis). A small fiber and autonomic neuropathy may occur in individuals with chronic hyperglycemia due to a sudden improvement in glycemic control. This disorder has been defined as an acute onset of neuropathic pain or autonomic dysfunction within 8 weeks of a large improvement in glycemic control (decrease in HbA1c of > 2% points over 3 months). It was found that there was a strong correlation between the magnitude of the decrease of HbA1c and worsening of autonomic function, and individuals with type 1 diabetes were found to have greater autonomic dysfunction than those with type 2 diabetes (12). Compared to the distal sensory-motor polyneuropathy commonly seen in diabetes, treatment-induced neuropathy of diabetes presents with more pain and autonomic dysfunction and has an acute onset.
Connective tissue disease. Several connective tissue diseases may be associated with acute or chronic disturbances of autonomic function. In fact, neuropathy may be the presenting symptom of an underlying connective tissue disease.
It has been estimated that about 10% of patients with Sjögren syndrome have neuropathy, but more than half of patients with Sjögren syndrome report symptoms of autonomic dysfunction (15). Most patients are women, and a frequent complaint is dry mouth and dry eyes. Autonomic features are characterized by loss of sweating in the extremities and constipation. Upon testing, anti-Ro/SS-A and anti-La/SS-B antibodies are found in approximately 60% of patients with Sjögren syndrome. Other findings may include a positive antinuclear antibody, rheumatoid factor antibodies, and muscarinic (M3-type) acetylcholine receptors (45). Similar changes are described in other connective tissue disorders, and autoimmune mechanisms for the development of neuropathy are also suspected or known in rheumatoid arthritis and systemic lupus erythematosus.
Drug-induced acute autonomic neuropathy. Various drugs can induce both acute and chronic autonomic neuropathies. These include cisplatin, vincristine, amiodarone, metronidazole, perhexiline, and paclitaxel. For clinical and pathological findings, see the MedLink Neurology article, Chronic autonomic neuropathies.
Botulism. Ingestion of improperly canned foods containing Clostridium botulinum toxin can result in an acute cholinergic neuropathy coupled with ptosis, cranial nerve paralysis, bulbar weakness, and neuromuscular paralysis. The cholinergic abnormalities include anhidrosis, xerophthalmia, xerostomia, orthostatic hypotension, gastrointestinal paralysis, and urinary retention. Symptoms usually begin within 48 hours of ingestion of contaminated food. Another increasingly recognized source of infection is wound botulism in drug users (35). The toxin blocks neurotransmitter release at peripheral cholinergic nerve terminals. Type A, B, and E may cause symptoms. Symptoms may be worse with type A>B>E. Electrodiagnostic studies show low-amplitude motor responses that decrease in 50% of patients with 2 Hz stimulation and increase with 20 to 50 Hz stimulation. Even in the absence of abnormalities on electrodiagnostic testing, there may be abnormalities of autonomic cardiovagal function.
Infant botulism is the most common clinical form of botulism in the United States and typically presents with hypotonia, bulbar weakness, and flaccid paralysis. Although injection of contaminated honey has been implicated in a number of cases, most cases of infant botulism do not have a definitive source. In these cases, infection is presumed to be due to swallowing spores adhered to dust particles in the air. Diagnosis is made by the presence of at least one of the following symptoms: blurred vision, double vision, difficulty speaking, slurred speech or hoarseness, dysphagia, pooling of secretions, or drooling (34). At least one of the following signs is present: ptosis, extraocular palsy or fatigability, facial paresis, fixed pupils, or descending paralysis starting with cranial nerve paralysis. Treatment should be based on clinical features and should not be delayed to complete testing. The CDC should be contacted prior to submitting testing.
The gold standard method and only FDA-approved method for identifying botulinum neurotoxin, used in specialized public health laboratories, is the mouse bioassay. Specimens are injected intraperitoneally into the mice with and without antitoxin. The mice are then observed for up to 96 hours by expert technicians for signs of botulism. Low levels of toxin might not produce signs in mice. Other methods include a real-time polymerase chain reaction (PCR) test, used by some reference laboratories, which detects bont genes A to G. The PCR detects DNA and not the actual toxin; thus, another method, such as a mouse bioassay, is required to determine toxin toxicity. The mass spectrometry method for detecting botulinum neurotoxin (Endopep-MS) is highly sensitive and specific and can differentiate among botulinum neurotoxin (34).
The most important treatment for botulism is supportive care, intubation, and mechanical ventilation when necessary. Intensive care has reduced the mortality to less than 5%. Health care providers who suspect botulism should immediately call their local or state health department to arrange for an emergency clinical consultation and shipment of antitoxin, if indicated. The only specific therapy for botulism is botulinum antitoxin. When administered within 24 to 48 hours of symptom onset, antitoxin may stop respiratory failure and progression of paralysis but does not affect existing paralysis. The antitoxin is toxin type–specific and binds and neutralizes botulinum toxin that is not irreversibly bound to synaptic receptors. Botulinum antitoxin is a mixture of antibodies to botulinum toxin types A, B, C, D, E, F, and G and can be obtained from the CDC at https://www.cdc.gov/botulism/hcp/clinical-overview/?CDC_AAref_Val=https://www.cdc.gov/botulism/health-professional.html. In most cases, the antitoxin should exceed the circulating amount of any specific toxin.
Porphyria. There are various forms of porphyria, but the two types of porphyria that are usually associated with autonomic neuropathy are acute intermittent porphyria and porphyria variegata.
In acute intermittent porphyria, there is an abnormality of porphobilinogen deaminase and overproduction of porphyrin precursors such as aminolevulinic acid and porphobilinogen. Abdominal pain is usually the first sign of an attack and often precedes neuropathy. Attacks are paroxysmal and key symptoms include intense abdominal pain, acute peripheral neuropathy, and encephalopathy. Autonomic dysfunction is the major presentation of acute attacks; its pathophysiological mechanisms are unknown. Autonomic abnormalities include tachycardia, hypotension, urinary retention, nausea and vomiting, or gastrointestinal symptoms including either diarrhea or constipation (33). Other clinical features include facial weakness, dysphagia, psychosis, depression, dementia, and seizures. The condition may progress over days to weeks and usually results in complete recovery, although recurrence may occur. Nerve conduction studies show small compound motor and sensory nerve action potentials, and EMG may show myopathic potentials. Pathology in the nerve biopsy shows axonal degeneration, and demyelination may occur in the CNS. There are often substantial delays in diagnosis, and patients may be misdiagnosed as Guillain-Barré syndrome. Small fiber neuropathy and allodynia with sparing of large fiber sensory modalities may result in early recognition of acute intermittent porphyria in patients presenting with acute polyneuropathy (01).
Treatment during an acute attack of acute intermittent porphyria includes (1) treatment with heme preparations; (2) symptomatic treatment of autonomic dysfunction, polyneuropathy, and encephalopathy; (3) exclusion of precipitating factors; and (4) adequate nutrition and fluids. Givosiran is an FDA-approved siRNA molecule directed against hepatic delta-aminolevulinic acid synthase 1 and induces a sustained decrease in this enzyme (03). A dose of 2.5 mg/kg monthly by subcutaneous injection administered to acute hepatic porphyria (including acute intermittent porphyria) patients with chronically elevated porphobilinogen and delta-aminolevulinic acid levels resulted in a 74% reduction in the mean annual attack rate and a 77% reduction in the annualized number of days of intravenous hemin use. The daily worst pain score was also significantly reduced in the givosiran group compared with placebo and was well tolerated (37). Givosiran is now an approved treatment for those over 12 years of age. Patients on givosiran can develop chronic kidney disease, cerebral venous thrombosis, pulmonary embolism, and pancreatitis (32). These serious adverse events could be explained, at least in part, by excessive elevations of homocysteine (up to 400 µmol/L). In most patients with active porphyria, there is already a mild disturbance of homocysteine metabolism. To prevent adverse effects, 80 mg of vitamin B6 per day is recommended, and the givosiran dosing should be personalized to reduce adverse effects.
A defect of the enzyme protoporphyrinogen oxidase exists in variegate porphyria. The clinical features are similar to acute intermittent porphyria, but in variegate porphyria, skin photosensitivity is frequent. The most common clinical manifestation of variegate porphyria is adult-onset cutaneous blistering lesions of sun-exposed skin and acute attacks, which may include autonomic neuropathy and can occur any time after puberty. The disorder is common in South Africa due to a founder effect dating back to early Dutch settlement of the subcontinent. An increase in fecal coproporphyrin and protoporphyrin occurs between attacks.
Panautonomic neuropathy has a range of possible outcomes. A review of 27 patients from the Mayo experience suggests that one third of patients have a good outcome, one third improve but are left with substantial deficits, and one third does not improve (40).
Subarachnoid hemorrhage is an uncommon but serious complication of Guillain-Barré syndrome, referable to autonomic instability and hypertension (10). Poor prognostic factors include the need for mechanical ventilation, rapid disease progression, bulbar dysfunction, dysautonomia, increasing age (especially greater than 40 to 60 years), severe weakness, early complete areflexia in the acute stage, and axonal loss on EMG.
A 14-year-old girl developed a flu-like illness associated with high fever and aching of her joints. Two weeks later, she developed burning in her hands and feet, coupled with allodynia. One week later, she started to develop severe orthostatic intolerance, episodic diarrhea, instability of her cardiac rhythm, a decrease in peripheral sweating, and gastric stasis. The condition continued to worsen, and she became confined to bed with severe hypotension on standing and required intravenous nutrition. There was no history of rash, xerophthalmia, or xerostomia. The past medical history, social, and family histories were unremarkable. At presentation, the patient was taking no medications.
General examination was normal except for evidence of severe weight loss. There was severe hypotension on standing, with no corresponding increase in the heart rate. The cranial nerve examination, muscle tone, limb strength, and reflexes were normal. On sensory examination, there was a decrease in cold perception and pinprick in the distal lower extremities, with normal vibration and joint position sense. The remaining neurologic examination was normal.
Laboratory studies were notable for Epstein-Barr virus titers that became positive 1 week after presentation. Nerve conduction studies were normal except for mild prolongation of the sural sensory responses bilaterally. Autonomic studies showed a resting tachycardia of 140 beats per minute; a heart rate range with deep breathing of 0; absent sympathetic responses in the upper and lower limbs; and the R6, Valsalva, and 30:15 ratios were severely abnormal. A sural nerve biopsy showed mild axonal degeneration, but no inflammatory infiltrates.
The patient was treated with high-dose prednisone, midodrine, and fludrocortisone. The autonomic abnormalities improved significantly within 2 months and were essentially normal after 6 months. A presumed diagnosis of viral-induced pandysautonomia was made.
The remainder of the autonomic neuropathies discussed here are either idiopathic, toxin -induced, or secondary to another underlying disease state.
The pathogenesis and pathophysiology of each of the autonomic neuropathies are described under the specific disease.
The incidence of Guillain-Barré syndrome is approximately 1:100,000 to 2:100,000 per year.
Food-borne botulism is most common in California, Colorado, New Mexico, and Illinois. The geographical areas most usually involved are type A, Rocky Mountains and the West; type B, East, especially the Allegheny range; type E, Baltic, Alaska, and Great Lakes states.
The incidence of acute intermittent porphyria is 1:10,000 to 1:100,000.
Many of the autonomic neuropathies have no known prevention. Avoidance of toxins and medications known to produce autonomic neuropathy can help prevent some forms.
The diagnosis of acute autonomic neuropathies is usually straightforward. The differential diagnosis includes the following:
|
• Autonomic seizures |
The proper approach to assessment of the patient with a suspected autonomic neuropathy begins with a detailed history directed towards a review of symptoms referable to autonomic dysfunction. This includes orthostatic symptoms, abnormal sweating, erectile dysfunction or ejaculatory failure, nausea or early satiety, dry eyes and dry mouth, cold hands and feet, and urinary or fecal incontinence. The bedside evaluation of the patient focuses on examination of the vascular system, skin color and texture, moisture of the mucous membranes, abdominal exam looking for gastric distension, and pupillary reactivity.
Blood pressure and heart rate should be recorded while the patient is supine and after standing for 1 minute. Adrenergic failure is characterized by orthostasis without an appropriate reflex elevation of the pulse. Dehydration or diuretic use may cause hypovolemia, suggested by orthostasis with an appropriate rise in pulse.
Laboratory tests of the autonomic nervous system are designed to produce objective and reproducible measurements of any autonomic dysfunction. They can also be used to monitor disease progression and can serve as markers for when to adjust therapy. The best tests of cardiovagal function are tests of heart rate variability, for example, the R6 interval and heart rate range to deep breathing. The Valsalva ratio is another well-established test. Measuring the Valsalva ratio, as opposed to beat-to-beat blood pressure variation during the Valsalva maneuver, may be less reliable than the R6 ratio. Tests of sympathetic function include beat-to-beat change in blood pressure with tilting, tests of sudomotor function, such as quantitative sudomotor axon reflex testing, and sympathetic skin potentials.
Sinus arrhythmia is the term used to describe the normal variability of the heart rate in response to respiration. Normally, the heart rate increases with inspiration and decreases with expiration. Several different measures of sinus arrhythmia are available. One method is to measure the response to respiration ratio of the maximal to the minimal heart rate during respiration. The patient breathes in and out six times per minute and is instructed using one of the following methods: (1) the technician asks them to breathe in and out deeply so that one cycle of inspiration and expiration takes 10 sec; and (2) the patient follows a sinusoidal computer-generated wave or an oscillating bar. The five largest consecutive responses are averaged, and a ratio of the response to respiration interval during inspiration compared to expiration is calculated. It takes about 30 seconds to reach stable values, so initial responses should not be used.
The Valsalva maneuver causes the heart rate to vary as a function of the changes in intrathoracic pressure and blood pressure. It consists of four phases, which can be helpful in assessing autonomic function. Phase 1 consists of a brief increase in blood pressure without a change in heart rate. Phase 2 consists of falling blood pressure and an increase in heart rate as the strain continues. Phase 3 begins with the abrupt release of strain and results in a transient further dropping of blood pressure. Phase 4 consists of an increase in blood pressure and decrease of the heart rate below the baseline rate. Phases 2 and 4 are the most important for measuring reflex autonomic responses. The patient is instructed to blow into a mouthpiece (with an air leak to prevent closure of the glottis) connected to a pressure-monitoring device, and the patient maintains a pressure of 40 mm Hg for 10 to 15 seconds. The heart rate is monitored for 30 seconds, and the Valsalva ratio is calculated by comparing the highest heart rate to the lowest heart rate recorded during the 15 to 20 seconds after the release of strain. The ratio is then expressed as a fraction comparing the lowest heart rate divided by the highest heart rate.
Quantitative sudomotor axon reflex testing is based on the axon reflex sweat response mediated by the postganglionic sympathetic sudomotor axons activated by acetylcholine. The stimulus is a constant electrical current applied for 5 minutes, and then the sweat response is recorded for a subsequent 5 minutes. There are several recognized abnormal patterns of this test, and the results are sensitive and reproducible. The response may be normal, reduced, absent, excessive, or persistent. A reduced or absent sweat response indicates postganglionic sympathetic sudomotor failure. The persistent response consists of continued sweating even when the stimulus ceases, also known as the “hung up” response.
Sympathetic skin potentials are the voltage differences measured between two skin surfaces that occur after direct or indirect stimulation. Sympathetic skin responses can be performed using EMG equipment and are easily recorded from the skin surfaces of the hands and feet. Reflex stimuli such as auditory, visual, tactile, pain, thermal, startle, and respiratory stimuli can all produce sympathetic skin responses (07). Commonly, a random loud sound is used, such as a dropped metal trash can or a cymbal crash. The amplitude of the sympathetic skin response is measured for comparison against normal.
Gastroparesis with autonomic disorders can be diagnosed using one of several techniques, such as gastric scintigraphy, radiopaque markers, transabdominal ultrasound, MRI with gadolinium, SPECT, stable isotope breath tests, swallowed capsule telemetry, antroduodenal manometry, and electrogastrography.
Some of the autonomic neuropathies are amenable to treatment of the underlying disease. Some patients with autoimmune autonomic neuropathy have been found to be responsive to intravenous gammaglobulin (18; 39). Sometimes multiple therapies may be required; there is evidence that patients may respond with combined therapy with prednisone and mycophenolate with plasmapheresis even if they had not responded with either treatment alone (13; 28).
Acute inflammatory demyelinating polyradiculoneuropathy is responsive to treatment with plasma exchange or intravenous immunoglobulin. More recently, efgartigimod has been reported to be effective in case reports in which intravenous immunoglobulin or plasma exchange were not effective.
Early detection and treatment of the underlying cancer is the primary treatment for paraneoplastic autonomic neuropathy. In addition, there are various anecdotal reports of the successful use of IVIG for the symptoms of autonomic neuropathy (42). Lambert-Eaton myasthenic syndrome usually responds to treatment of the underlying malignancy, but immunosuppressive agents such as prednisone and azathioprine, as well as plasma exchange and IVIG, have been successful in improving symptoms (22). High doses of 3,4-diaminopyridine and guanidine hydrochloride improve the neuromuscular block and may also improve autonomic function. 3,4-diaminopyridine in divided doses up to 60 mg/day is currently the therapy of choice. However, the drug is not FDA-approved in the United States, but it is available under compassionate use protocols. Acetylcholinesterase inhibitors are usually of little benefit when used alone but are helpful in augmenting the benefit of 3,4-diaminopyridine.
Midodrine, an alpha agonist, used at 2.5 to 10 mg orally three to four times per day, is safe and effective for treating impaired cardiovascular autonomic function. The value of fludrocortisone, a mineralocorticoid, in acute autonomic neuropathy is less clear (44). In theory, pyridostigmine, an acetylcholinesterase inhibitor, should improve ganglionic function; however, the effect is more limited in practice. The usual dosage is 30 to 60 mg orally three times a day or 180 mg ER in the morning. Droxidopa is a precursor of norepinephrine, and the usual dosage is 100 to 600 mg three times a day. Atomoxetine, a norepinephrine transporter inhibitor, and octreotide, a somatostatin analog, may be useful in patients unresponsive to other therapies. Among other adverse effects, many of these medications can significantly worsen hypertension, particularly supine hypertension. It is very important to treat supine hypertension, particularly at night, because it can induce nocturnal pressure natriuresis that aggravates daytime orthostasis (19). Potential treatments include clonidine, angiotensin 2 receptor antagonists, angiotensin-converting enzyme inhibitors, calcium channel blockers, sildenafil, eplerenone, and continuous positive airway pressure (30).
Botulinum toxin has been studied and found to be successful in treating some of the symptoms of autonomic dysfunction, including bladder symptoms related to detrusor overactivity and axillary hyperhidrosis (29).
In a randomized, double-blind, placebo-controlled trial of botulism immune globulin (BIG) for infants with suspected infant botulism, Arnon and colleagues demonstrated that administration of botulism immune globulin within 3 days of hospitalization decreased the length of stay from 5.7 to 2.6 weeks (02). There are also reports of successful treatment of wound botulism with botulinum antitoxin even days after presentation (05), although earlier treatment appears to be more effective than later treatment. Otherwise, treatment is supportive and includes respiratory support as well as debridement and penicillin in botulism associated with wound infection.
The optimal management of treatment-induced neuropathy of diabetes is unknown. It has been suggested that symptoms improve with discontinuation of insulin therapy and allowing for worsening of metabolic control, but this has not been studied. Autonomic dysfunction can be managed symptomatically and has been found to improve over time (11).
In porphyria, hematin can suppress the symptoms of the acute attack, phenothiazines may help with the abdominal pain and psychotic disorders, and meperidine may help with pain. Givosiran is an siRNA against delta-aminolevulinic acid synthase 1, the rate-limiting enzyme of heme biosynthesis (20), and can significantly reduce the use of hematin and improve symptoms (03; 37).
In some acute autonomic neuropathies, only symptomatic treatment is required.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

James W Russell MD MS
Dr. Russell of CAMC Center for Academic Medicine and West Virginia University has no relevant financial relationships to disclose.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University received a consultant honorarium from Roche and Ovid Therapeutics.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jul. 16, 2026
Peripheral Neuropathies
Jul. 14, 2026
Peripheral Neuropathies
Jun. 26, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
May. 12, 2026
Neuromuscular Disorders
Apr. 23, 2026