





















Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The author has a special interest in genetic malformations of the brain and has made significant contributions to this field. In this article, he updates our knowledge of Aicardi syndrome, including the newest findings by MR and genetics.
|
• Aicardi syndrome affects only girls, with rare exceptions in poly-x males. | |
|
• The original definition of Aicardi syndrome encompassed infantile spasms, agenesis of the corpus callosum, and chorioretinal lacunae (also known as pseudo-colobomata). | |
|
• Other additions to this core complex include other neuronal migration defects (eg, polymicrogyria, heterotopia); cerebral, cerebellar, costovertebral, and other ocular anomalies; distinctive facial traits; and a liability to develop intra- and extracranial neoplasms. | |
|
• The cause of the syndrome has yet to be clarified. | |
|
• Although the gene associated with this disorder is not known, it is believed to be located on the X chromosome. |
In 1965 Aicardi, Lefebvre, and Lerique-Koechlin reported eight cases of children with "spasms in flexion, callosal agenesis, and ocular abnormalities." In 1969 Aicardi, Chevrie, and Roussellie published their collective experience with 15 cases (including the previously presented eight) under the title "le syndrome spasmes en flexion, agenesies calleuse, anomalies chorioretiniennes" (03). In 1972 Dennis and Bower reported it as "the Aicardi syndrome."
Earlier cases with similar features were reported, but not recognized as specific entities. Sabin and Feldman reported "chorioretinopathy associated with other evidence of cerebral damage in childhood" (86). They considered it a "syndrome of unknown etiology separable from congenital toxoplasmosis."
Several internet websites now contain relevant information and links to parent support groups and other reference sources:
• Our Aicardi Life
• Genetics Home Reference: Aicardi syndrome
Criteria for the diagnosis first formulated by Aicardi and colleagues (03) are the classic triad of chorioretinal lacunae, callosal agenesis, and infantile spasms. In an epidemiologic study (38), the following entry criteria were added when only two of the classic criteria were fulfilled, at least any two of the following: cortical malformations (mostly polymicrogyria), periventricular and subcortical heterotopia, cysts around the third ventricle or choroid plexus, choroid plexus papillomas, and optic disc or optic nerve colobomas. Aicardi syndrome only exists in females, and, very rarely, in males with the XXY genotype.
Epilepsy. Seizures are often the presenting symptom and may start in the neonatal period. Of 186 patients whose data were collated in the first large-sized serial study, the seizure type was known in 156 (21). Ninety-seven percent had infantile spasms. Of these, 57% had only infantile spasms, and 43% had infantile spasms and other seizure types. Fifty-six percent had their first infantile spasm under 3 months of age, and 68% had their first seizure under 3 months of age. Thus, some had onset of seizures that were not infantile spasms. These were either unilateral hemitonic or focal. They either persistently lateralized or shifted in onset from side to side. In a study of 77 cases of girls with Aicardi syndrome, 71 had seizure activity (92%). Seizures occurred on a daily basis in 48 patients (92%). Only three patients were seizure-free with medication. Seizure frequency was not reported in 15 patients. Infantile spasms were the most common seizure type, reported in 17%. The remainder consisted of myoclonic (l4%), mixed seizure types (12.7%), generalized tonic-clonic (9.8%), focal and complex partial (7%), atonic (5.6%), tonic (1.4%), and atypical absence (1.4%). Seizures are generally intractable despite therapy, though seizure control is possible in some patients with severe epilepsy. The need for polytherapy and the use of the ketogenic diet underline the intractable nature of epilepsy in Aicardi patients (85). In a systematic analysis of 245 published cases, 98% of individuals had epilepsy, and 86% presented with infantile spasms at diagnosis (101). The median age at seizure onset was 2.2 months, and coexistence of multiple seizure types was common.
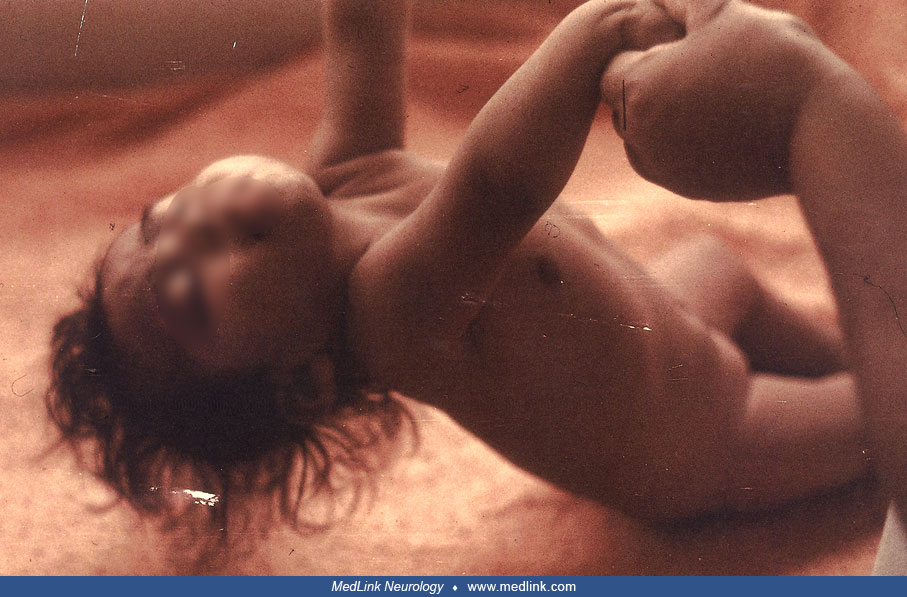
The spasms were often asymmetrical and even unilateral. The relatively lengthy duration of each spasm compared to idiopathic infantile spasms has been noted (23). All children had spasms in series, but some also had single spasms. The spasms could be flexor or extensor. Head turning was often noted during flexor spasms. Hemimegalencephaly can be observed in patients with Aicardi syndrome but may go unrecognized (34); the epilepsy starts in the neonatal period and usually is asymmetric and refractory.
The spasms persist in most patients, but the partial seizures disappear in infancy or middle childhood years. All types of seizures become less prominent over time. Other seizure types did not evolve from infantile spasms, but were present from onset (23). Agrawal and colleagues highlighted migrating focal seizures in early infancy as an initial presentation before the development of asymmetric flexor spasms (02). This finding expands the electroclinical spectrum of Aicardi syndrome and underscores the need to consider the diagnosis in infants with refractory migrating focal seizures.
Other authors suggest that evolution of the seizure types in Aicardi syndrome does occur (76). They suggest that a patient's early infantile epileptic encephalopathy evolved into West syndrome. Two other cases evolved into Lennox-Gastaut syndrome. It may be that what evolves is the EEG pattern, but the spasms persist and other seizure types either become less prominent or stop completely.
In those who have partial seizures as well as clusters of usually asymmetrical spasms, long-term polygraph recording has shown a consistent correlation. The focal EEG discharge usually precedes the partial seizures and the cluster of spasms (13).
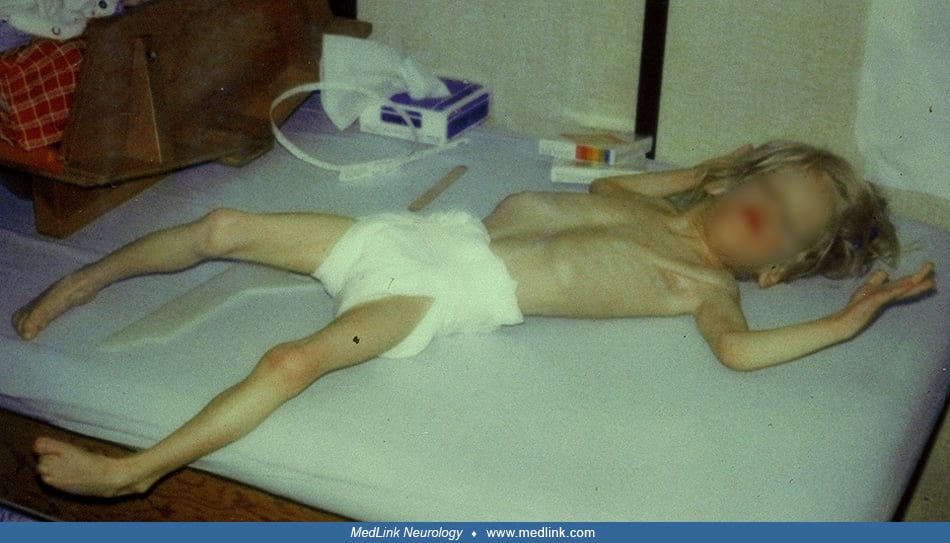
Developmental delay. Initially all children with Aicardi syndrome were described as severely delayed in both motor and cognitive abilities. Most were noted to show no evidence of communication or social contact. In a survey of Japanese institutions, all but one of 12 patients with typical Aicardi syndrome were bedridden without gaining milestones (110). However, milder forms of disability have been reported. In 1990, Abe and colleagues identified 11 such milder cases in the literature; the patients could walk independently and had some speech (01). In another report of 14 children diagnosed over 18 years, three were identified who could walk or crawl, and four had some language ability (68). A probable case of Aicardi syndrome with normal mental development, absence of infantile spasms, chorioretinal lacunae in one eye and callosal agenesis was described (81). Tuft and colleagues examined five girls with Aicardi syndrome by formal psychological testing and found cognitive disabilities varying between mild and profound, with poorest performance in patients with severe epilepsy (98). Quantitative natural-history modeling of Aicardi syndrome has provided a clearer understanding of developmental outcomes in contemporary cohorts (101). Intellectual functioning was available from 169 individuals and found that 96% exhibited intellectual disability, with severe impairment being the most common profile. For motor function, approximately one quarter of patients achieved independent ambulation, whereas more than half remained nonambulatory. Communication skills ranged from nonverbal status to limited single-word production in a minority (101).
Neurologic examination. Aicardi initially described "massive" hypotonia and pyramidal tract signs. Decorticate or decerebrate posturing have also been noted. Spasticity was reported in 20 of 74 patients collated (21). In 27 the only abnormality was the severe delay in acquisition of milestones and microcephaly. In another report, 42% of 43 patients had microcephaly (12). Infants and children with microcephaly usually had a normal head circumference at birth, but normal head growth did not occur subsequently. Some children with Aicardi syndrome are reported as examples of failure to thrive, probably of cerebral or hypothalamic origin, yet head circumference growth is spared. In a quantitative evaluation of 245 cases, corpus callosum abnormalities were present in 98%, cortical malformations in 21%, intracranial cysts in 35%, and nodular heterotopia in 18% of individuals (101).
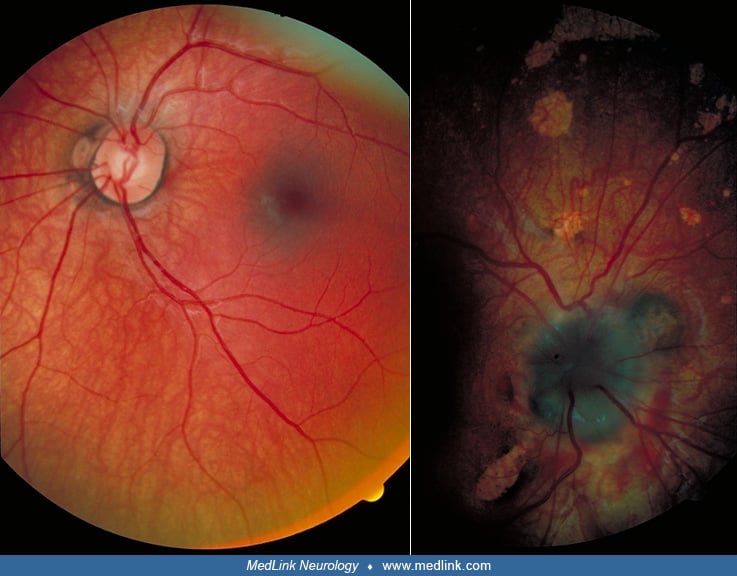
Ocular findings. Most of the children with Aicardi syndrome show no evidence of visual fixation or visual pursuit behavior. This is probably a cortical visual impairment in most. When there are major malformations of the eye such as microphthalmos or optic nerve anomalies, the blindness is more directly explained. Coloboma of the optic disc, usually unilateral, was noted in 80 of 184 cases, and optic nerve hypoplasia in five (21). Other abnormalities reported include retinal detachment, pseudogliosis of the optic disc, macular scars, pupillary membrane remnants, iris synechiae, and colobomata.
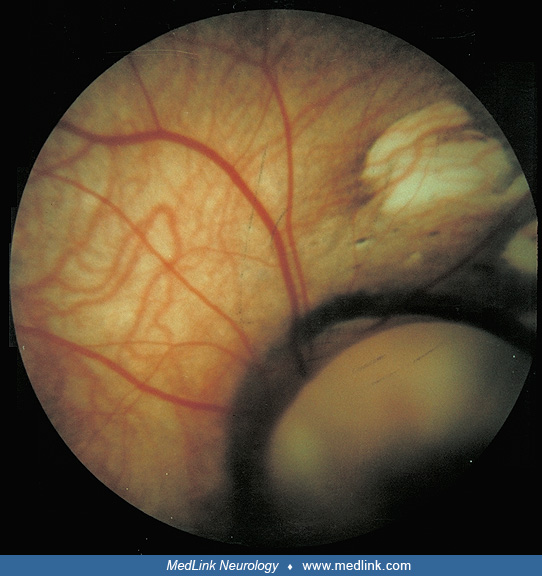
The virtually pathognomonic lesions are the chorioretinal lacunae. Absence of these lesions makes the diagnosis doubtful. These lesions are variable in size, borders, location, and color. Some features are more commonly noted. For example, these are defects of the retinal pigment epithelium and, hence, they are usually punched-out white lesions, though they could be yellowish or pinkish and shiny. They have rounded borders when they are close to the optic disc, but these become multilobulated, less regular, and ragged in more peripheral locations. Usually they cluster around the optic disc and they are at the same level as the rest of the retina, so retinal vessels cross them without bending at their borders. Pigmentary deposits usually hem the border of these lacunae, and it is rare to have pigment within. A small amount of pigment at the center of large peripapillary lacunae has been described. The lacunae do not change in appearance, size, or numbers over time. According to a systemic quantitative modeling of 245 cases with Aicardi syndrome, chorioretinal lacunae were identified in 99% of patients, whereas optic disc coloboma and microphthalmia were present in 31% and 24%, respectively, outlining the characteristic ophthalmologic profile of Aicardi syndrome (101).
Uniocular involvement with chorioretinal lacunae is rare, occurring in only 8% of documented cases. However, significant asymmetry, with one eye dramatically involved and the other showing only a few peripheral lesions, is common. Sometimes problems involving one eye's anterior segment do not allow assessment of the fundus oculi for lacunae. A single observation by fetal sonography described a unilateral cyst in the orbita causing proptosis at 25 weeks gestational age in a fetus with Aicardi syndrome (88). In a series of 40 cases seen by one ophthalmologist during the meeting of a family support group, chorioretinal lacunae were the most frequent abnormality (88%). Less common findings were persistent papillary membrane (5%) and anterior synechiae in one case (37). A thickened posterior lens capsule with intact nuclei has been described in postmortem studies of one case who died at 20 years old, a relatively long life duration for this syndrome (65). Ophthalmologic evaluation using fluorescein angiography under anesthesia of a 12-day-old baby girl with microphtalmia revealed a morning glory optic nerve and contralateral persistent fetal vasculature (PFV) besides chorioretinal lacunae typical of Aicardi syndrome (05).
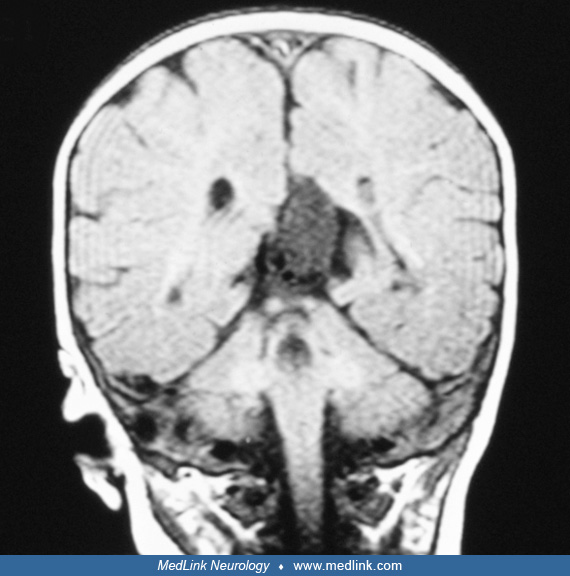
Brain imaging by MRI. In a large study including 23 girls with Aicardi syndrome, a typical profile has been found with polymicrogyria, predominantly frontal and perisylvian, which is often associated with “underopercularization” (48). All patients had periventricular heterotopia and the majority had intracranial cysts, either single or multiple. Cerebellar abnormalities were present in the majority. As a novel finding, tectal enlargement was found in 10 patients. Fetal MRI was applied in the case of a fetus with dilated ventricles picked up by fetal ultrasound at 18 weeks (45). Serial MRI confirmed callosal agenesis and migrational abnormalities consistent with Aicardi syndrome. The intrauterine MRI features of nine Aicardi syndrome confirmed cases have been described and compared with both those of postnatal MRI and intrauterine MRI cases with both corpus callosum agenesis-dysgenesis and cortical malformation (Aicardi syndrome mimickers) (91). In all cases with Aicardi syndrome, iuMRI was able to detect corpus callosum agenesis-dysgenesis and cortical development anomalies. A significant difference between Aicardi syndrome and Aicardi syndrome mimicker were found regarding sex, nodular heterotopia, posterior fossa abnormalities, coloboma, and cortical gyration abnormalities. These key fetal MRI features have been reported to suggest prenatal diagnosis of Aicardi syndrome.
In a study including 67 patients with Aicardi syndrome, 100% of the patients displayed corpus callosum malformations, 98% cortical dysplasia and nodular heterotopia, 96.36% intracranial cysts, and 63.63% posterior fossa abnormalities on brain imaging (64). This study reports the presence of basal ganglia dysmorphisms as a new feature.
Diffusion tensor imaging (DTI) and tractography, which identifies the presence and course of myelinated fiber tracts, was applied to two individuals with Aicardi syndrome with severe mental deficiency and two controls with callosal agenesis and relatively mild cognitive dysfunctioning (105). The main difference between patients and controls was the near-absence of normally present longitudinal intracortical connecting tracts in the former group, such as the superior longitudinal fasciculi, and partially retained presence of other intracortical connecting fiber tracts. The Probst- or cingulum bundles were present in both groups as the usual accompaniments of callosal agenesis. The findings may well help to explain the difference in cognitive function between both groups, despite the callosal agenesis in both groups.
Additional information on the extent of involved areas can be obtained by PET scanning, which is especially useful when surgical therapy is considered. In a study by Govil-Dalela and colleagues, PET scan showed a much more extensive area of involvement than suggested by the MRI scan, and it also showed more involvement in the hemisphere not considered for surgery than expected on the basis of the MRI alone (39).
Artificial intelligence enhanced diagnosis of Aicardi syndrome, by analyzing imaging data and integrating the findings with clinical data, is thought to assist in confirming the diagnosis more quickly and accurately (107).
Other clinical manifestations. The most common other manifestation involves the thoracic spine. Scoliosis may be present with or without vertebral anomalies identified on x-ray. In an orthopedic center, five patients with Aicardi syndrome were referred for scoliosis between 1.5 and 10 years, but only one had a vertebral malformation (40). Vertebral malformations found in Aicardi syndrome include abnormalities of segmentation with block vertebrae, fused vertebrae, hemivertebrae, or other variations in vertebral bodies or arches. These may be associated with rib changes, which are not seen without vertebral abnormalities.
Dysmorphic or asymmetric facies have been reported. No characteristic facial or other external appearance consistently suggests the syndrome, and the majority have a normal appearance. However, some common facial abnormalities have been quantified noninvasively in a facial morphometry study using 3D facial photographs through stereophotogrammetry (63). In 3D photogrammetric assessment of 11 Italian females with Aicardi syndrome (mean age 17.5), all patients showed shorter philtrum and right-side orbital height, shorter superior, middle, and inferior facial depths, and a smaller length of mandibular ramus; conversely, they sowed larger nasal and lower facial widths, and lower facial convexity.
Cleft lip and palate have been reported in Aicardi syndrome. It has been difficult to determine if this is more common than in other disorders affecting midline brain structures. It is noted in about 3% of the reported cases (100).
Aicardi syndrome patients have a propensity to develop tumors, both inside and outside the skull. Reported tumors include embryonic soft tissue carcinoma, hepatoblastoma, angiosarcoma, choroid plexus papillomas, posterior fossa medulloblastoma, oral extragonadal yolk sac (germ cell) tumor, pineal cyst, and late-onset retinoblastoma (95; 96; 97; 77; 72; 53; 17; 04). Anterior neural tube defects taking the form of intraorbital cystic encephalocele (46) and nasal cephalocele (67) have been described. The association of Aicardi syndrome and atypical dermal melanosis has been reported (24).
In a study compiled from 408 cases from multiple international sources, age at death ranges from 1 month to 33 years, with a peak at age 16 (56). A 2024 natural history model estimated survival at 94% at 1 year and 83% at 5 years, identifying age at onset (< 2.1 months) as a significant prognostic marker (101).
The prognosis for any infant diagnosed with Aicardi syndrome is poor for both quality of life and longevity. The complications are either a direct result of the features of the syndrome or the attempts at treating the epilepsy. There is no clinical evidence that a regressive neurologic disorder is a feature of the syndrome. In some the onset of the epilepsy is associated with some worsening, especially when the seizures are frequent on a daily basis and refractory to treatment. There is agreement that the seizures may be complex and bizarre (12), and different types coexist from the outset. In addition to asymmetric flexion spasms that are relatively prolonged (23), generalized clonic, hemiclonic, and hemitonic seizures coexist but are usually relatively brief (03; 21). The seizures may decrease with time (23; 68).
Although suggestions have been made that the children with more complete agenesis of the corpus callosum would have a more severe form of cerebral dysgenesis, this is not borne out by the documented cases (68). Two of four children who had some mobility and moderate or mild mental retardation had complete agenesis. Two of 10 with severe motor and mental retardation had partial agenesis. These authors could not identify any clinical, EEG, or neuroradiologic features as good early predictors of later outcome. Other authors, with less well-documented reports, have suggested that "better" response of the epilepsy to antiepileptic medication identifies a milder long-term prognosis (32; 01; 09).
Sudden death occurs in the syndrome. It is possible this is related to the epilepsy in a significantly handicapped infant. It could also be a result of the adrenocorticotropic hormone therapy used to treat the infantile spasms, or less likely a benzodiazepine. Certainly, longevity will be reduced in such immobile, severely handicapped children because of reflux with aspiration and pneumonitis.
Complications include developmental delay, intractable epilepsy, gastroesophageal reflux, recurrent pneumonia, and constipation. Respiratory infections are the leading cause of death in these patients. All patients have significant developmental delay, with milestones ranging from 2 to 36 months at the time of inquiry (85).
In a web-based survey of 795 people with more than 30 different rare epilepsy diagnosis groups, conducted using a questionnaire to collect data on various aspects, including comorbidities, the highest number of comorbidity classes reported per person were in Aicardi syndrome (n=61) (47). Comorbid classes and their prevalence in this syndrome were reported as vision/eye disorders (97%), brain abnormalities (92%), hyper/hypotonia (81%), learning/developmental disability (78%), mental health/behavioral issues (67%), bone/joint disorders (61%), sleep disorders (59%), gastrointestinal disorders (49%), oral/dental issues (47%), cardiology disorders (25%), endocrine disorders (23%), respiratory disorders (21%), hearing disorders (8%), blood/immune/lymphatic disorders (3%), and mitochondrial disorders (2%).
A study evaluating immunological abnormalities in patients with rare syndromes reported B-cell defect and low class-switched B lymphocyte count in an individual with Aicardi syndrome (41).
Although the prevalence of sleep disorders was determined to be 59% in this comorbidity and quality of life survey, the only study of sleep disorders in children with Aicardi syndrome is a case report of a 5-year-old girl who was found to have obstructive sleep apnea and treated with positive airway pressure (57). In a review of sleep disorders affecting children with congenital malformations of the CNS, it has been emphasized that sleep disorders are underrecognized and studied in this syndrome, and management of sleep disorders would help the outcome and quality of life (111).
Death from metastatic carcinoma and angiosarcoma has been reported from other centers as well, though these reports remain rare. In spite of the more frequent occurrence of choroid plexus papillomas in Aicardi syndrome, this has rarely led to the expected complication of high-pressure hydrocephalus.
Aicardi syndrome is almost exclusively limited to females. Rarely Aicardi syndrome occurs in males with XXY karyotype (49; 38; 113; 90). This has led to suggestions of an X-linked dominant disorder expressing its heterozygous form in females, but lethal in hemizygous males. Another explanation is that Aicardi syndrome is transmitted by a de novo mutation on the paternal X chromosome, which is only transmitted to the female offspring (38). Most females have random X-chromosome inactivation (XCI), defined as an equal likelihood for inactivation of the maternally or paternally derived X chromosome in each cell. Eble and colleagues found a significant increase of nonrandom X-inactivation in 23 girls with Aicardi syndrome, confirming the role of the X chromosome in this disorder (31). Rare cases are on record as males with a normal 46,XY karyotype (20; 06). A single case has been reported of a female with combined features of Aicardi syndrome and Turner syndrome carrying the mosaic 45,X/46,XX mosaicism (92). Array-based comparative genomic hybridization (array CGH) was used in a study of 18 patients to detect microdeletions and microduplications at high resolution (82 kb). No disease-associated aberrations were identified (112). Wang and colleagues screened the DNA of 38 girls with Aicardi syndrome by high-resolution, genome-wide array comparative genomic hybridization for copy number gains and losses (106). No significant copy number variants were found. Except for one report of occurrence in sisters that may have developed from germinal mosaicism (70), no familial recurrences have been reported to date. A de novo mutation in the postzygotic stage is the probable cause of monozygotic monochorionic female twins, one sister healthy and the other affected by Aicardi syndrome (80). Schrauwen and colleagues performed exome/genome sequencing of 10 children diagnosed with Aicardi syndrome and their parents and performed RNA sequencing on blood samples from nine cases, their parents, and unrelated controls (89). In two families, there were de novo mutations: one nonsense mutation in TEAD1 and one possibly damaging variant in OCEL1. A possible role of these genes in Aicardi syndrome was independently tested in 38 patients with Aicardi syndrome (108). No mutations or variants were found in either gene. Introducing a new line of research with epigenetic DNA modification as a possible causal mechanism, Piras and colleagues compared DNA methylation sites in trios (parents vs. their affected child) (79). Different methylation patterns between parents and probands with Aicardi syndrome were found for a small number of genes with a neurodevelopmental role. However, no single site of aberrant methylation was generally shared.
Considering the overlapping phenotypic and inheritance features, Moog and Dobyns proposed oculocerebrocutaneous syndrome (Delleman-Oorthuys) and Aicardi syndrome; both may be pathogenetically related or caused by mosaic mutations of the same gene or genes in the same functional pathway (71).
Although no significant genetic etiology has been identified yet, Wong and colleagues noted that new technologies detecting low-level mosaicism and balanced rearrangements, as well as platforms examining changes at the DNA and chromatin level affecting regulatory regions, are all potential avenues for future studies, which may solve the mystery of the etiology of Aicardi syndrome (109).
Ha and colleagues reported rare and predicted damaging variants in four autosomal genes (WNT8B, SLF1, KMT1B, and SZT2), in four unrelated individuals with classical and suspected Aicardi syndrome (42). Based on in vivo findings and murine expression studies, the de novo WNT8B and SLF1 variants were reported to be good candidates to explain Aicardi syndrome in these individuals. Both individuals with the WNT8B and SLF1 variants displayed the classical triad of Aicardi syndrome. Like other previous reports, this study suggests that the causative alleles in individuals diagnosed with Aicardi syndrome will not be all X-linked (89; 61). Aicardi syndrome has a wide spectrum and includes classical, likely, and suspected cases, each with variable clinical and imaging phenotypes. Ha and colleagues report that the genetic heterogeneity identified by this study is reflective of Aicardi syndrome not being a single malformation syndrome (42).
Neuropathologic findings.
Cerebral hemispheres. Several reports identify gross asymmetry that may include both cerebral and cerebellar hemisphere size, with features suggesting hemimegalencephaly (25; 76), with variable degrees of agyria. Although a normal cerebral hemisphere surface is sometimes reported (43), polymicrogyria is the more common finding. An underlying unlayered cortex is considered characteristic for the syndrome (33; 11). But there have been reports of the usual 4-layered cortex more commonly identified with nonspecific polymicrogyria (76). In some reports, the cortex was definitely layered (25), but the number of layers was not ascertained, and a normal 6-layered cytoarchitecture with mature neurons has also been described (43). Some authors present an argument that the unlayered polymicrogyria with corpus callosum agenesis is a specific entity and that their cases with these findings but without the clinical criteria to fit Aicardi syndrome may be "variants" (11).
Variable size and distribution of nodules of heterotopic neurons in the cerebral hemispheres are described in the majority of reports. These are most often in a periventricular location or in the white matter in the centrum semiovale. Nodular heterotopia is common in association with corpus callosum agenesis (59; 78). Its presence simply identifies a disorder of neuronal migration occurring before 18 weeks’ gestation.
Van den Veyver and colleagues studied eosinophilic inclusions in cortical astrocytes, previously reported (16) in autopsy material from two patients with Aicardi syndrome (103). The inclusions were negative for glial fibrillary acidic protein but strongly positive for vimentin and filamin, the latter being a microfilament associated protein, involved in neuronal migration and deficient in X-linked periventricular heterotopia (36). Hyaline protoplasmic astrocytopathy, as the phenomenon came to be called, is also found in other disorders. The common denominators appear to be chronic epilepsy and cortical dysplasia (82). Filamin may be part of the stored proteins in protoplasmic astrocytopathy in cases of neocortical dysplasia not linked to Aicardi syndrome (44).
Cerebellum. Abnormalities of the cerebellum are noted in only 8% of the imaging studies (21). Of the few available autopsy reports, half describe cerebellar abnormalities (25; 76; 43; 93). These are more likely to be at a microscopic level only when the gross macroscopic aspect is reportedly normal. Dorsal fusion of the cerebellar hemispheres and underdevelopment of the tonsils and inferior vermis have been reported (25; 19). The findings included cerebellar heterotopia and variably decreased ganglion cell number in the Purkinje cell layer with an increase in Bergman glia. In the molecular layer broadened dendrites of Purkinje cells and patches of a persistent external granular layer were reported (76).
Basal ganglia. Basal ganglia dysmorphisms, ranging from mild to more severe forms, associated with hypertrophic thalamic adhesion or agenesis of the anterior limbs of the internal capsulae have been reported (in 76.36%) in a large cohort of 67 patients with Aicardi syndrome (63). It was not considered pathognomonic but as a new feature over the well-known ones.
Brainstem. Unilateral brainstem hypoplasia, not clearly reflecting predominant involvement of the contralateral cerebral hemisphere, was reported by Steffensen and colleagues (93).
Cysts. A true interhemispheric glioependymal cyst was described (14) in a case that, in retrospect, was an example of Aicardi syndrome decades before the disease acquired its eponymous name. Also, later literature including MRI findings underscores the association of glioependymal cysts with Aicardi syndrome. Such cysts, however, are not specific for Aicardi syndrome and may be seen in other cases of callosal agenesis (08; 07).
Cysts of the choroid plexus are mentioned (21) and a neuroradiologic illustration given, but no specific description is found in autopsy reports.
In a study describing the neurologic phenotype of 36 children with agenesis of corpus callosum and interhemispheric cysts, severe intellectual disability and epilepsy were associated with type 2b cysts (multiple cysts different from CSF with malformations of cortical development), always due to Aicardi patients (n=6) (99). The most important factor correlating with an adverse neurologic outcome in children with corpus callosum agenesis was the diagnosis of Aicardi syndrome.
Choroid plexus papillomas. Choroid plexus papillomas have been commonly found in autopsy material in reports of Aicardi syndrome (25; 95; 83; 35). Although choroid plexus papillomas tend to be solitary, documented cases with multifocal chord plexus tumors have usually occurred in children with Aicardi syndrome (69). They have been considered a specific tumor of Aicardi syndrome. They are usually in multiple locations and of variable size and may or may not be associated with hydrocephalus (97; 69).
Other neuropathological findings. One patient has been reported with Aicardi syndrome and Dandy-Walker malformation (52).
Ocular pathology. The chorioretinal lacunae have received special attention in various reports because they are a necessary feature of the syndrome. However, another less frequently present abnormality, coloboma of the optic disc, is also a defining feature of the disease.
Chorioretinal lacunae. The earliest pathologic description of the chorioretinal lacunae is attributed to R. Barry in a presentation to the European Ophthalmic Pathology Society in 1973 (25). These were described as "gaps" in the retinal pigment epithelium with depigmented zones of the intact retinal pigment epithelium. In some areas, this lined a scleral ectasia extending as a tube to the scleral exterior. Hamano and colleagues also described zones of the retinal pigment epithelium that were hypopigmented or depigmented (43). They noted that pigment granules also dispersed into the rod and cone layer. There were, on electron microscopy, broadly extending cytoplasmic processes containing numerous melanin granules mainly in the rod and cone layers, whereas the choroidal layer was thin.
In some descriptions, the retina overlying the chorioretinal lacunae may be histologically abnormal, with retinal photoreceptor degeneration (55) and atrophy of outer retinal segments (66). One group described the formation of photoreceptor folds at the edges of the lacunae (26). The electron microscope appearance at the edge of the chorioretinal lacunae has been described (35). There was an abrupt ridge of proliferated retinal pigment epithelium surrounding a flat depressed area corresponding to atrophic pigment epithelium cells with ill-defined cobblestone appearance. There were numerous papillary structures lined by nonpigmented proliferated epithelium.
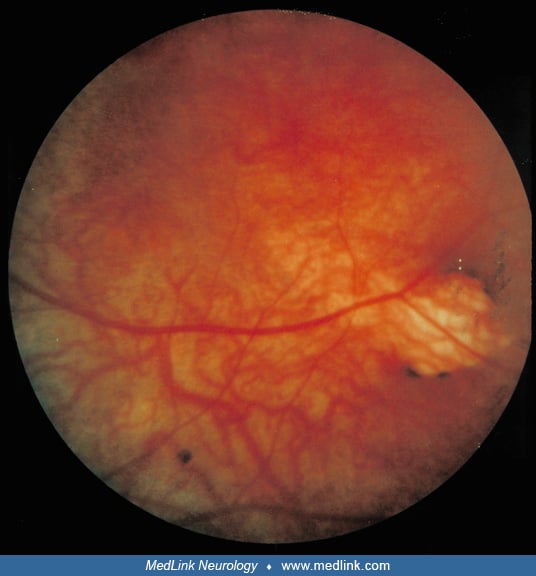
Optic disc coloboma. The coloboma of the optic disc is different from the chorioretinal lacunae and arises in a different manner. It is a relatively frequent abnormality on clinical examination of the fundus oculi in over 35% to 40% of the reported cases collated (27; 21).
In the histologic examination reported, the retinal pigment epithelium had proliferated at the edge of the optic disc. Extensive tubular and papillary projections extended within gliotic areas and along the meninges of the malformed optic nerve (35).
Though the optic disc coloboma is not as specific to Aicardi syndrome as chorioretinal lacunae, the failure of closure of the choroidal fissure can be more precisely timed to 7 weeks’ gestation. It may involve the iris, ciliary body, and retina as well as choroid.
Pathogenesis. The unique complex of findings that constitutes Aicardi syndrome has been juxtaposed to other complex malformations with X-linked inheritance.
The phenotypic and genetic relationship of Aicardi syndrome to another disorder, microphthalmia with linear skin defects, has received attention. Like Aicardi syndrome, microphthalmia with linear skin defects is X-linked dominant, observed only in females, and apparently lethal in otherwise normal XY males. The genetic basis of microphthalmia with linear skin defects may be related to that of Aicardi syndrome, as it involves deletions at Xp22 in the human. Research efforts have centered around the search for candidate genes in this region (58), although one study has identified a novel gene at the breakpoint of the microphthalmia with linear skin defects critical region that is not mutated in Aicardi syndrome (102).
Genetic control of regional specification. The pathogenetic relationship of maldevelopment of the corpus callosum, neuroblast migration, optic papilla, retina, thoracic vertebral bodies, and ribs is unclear. The thoracic sclerotomes divide transversally and form the primitive vertebral bodies in the fourth week of gestation. Other sclerotomic cells migrate ventrolaterally to establish primordia of the ribs during the fifth week. Segmentation of the vertebral bodies is not complete until the eighth to ninth week. This coincides with the timing for choroidal fissure closure at 7 weeks. The corpus callosum formation spans 3 to 20 weeks’ gestation. The unlayered polymicrogyria implies an insult at 6 to 9 weeks, but insults to cause layered microgyria, heterotopia, and chorioretinal lacunae may span a longer period of gestation.
The role of impaired neuroblast migration in the pathogenesis of Aicardi syndrome invites comparison to similar disorders. Cytoskeletal proteins are involved in neuroblast migration, eg, filamin in X-linked subependymal heterotopia (36). One study underscores the possible involvement of cytoskeletal organization in the pathogenesis of Aicardi syndrome (103).
Gene studies making use of whole exome sequencing (WES) or whole genome sequencing (WGS) so far have been negative, including trio studies of parents and their affected offspring to exclude de novo mutations (61; 79).
In a comprehensive study, knockdown of tead1, wnt8b, and slf1 produced similar zebrafish morphant phenotypes, with the highest frequency of eye-related defects observed in wnt8b morphants (42). The defects in tead1, wnt8b, and slf1 morphants demonstrate the involvement of these genes in the development of tissues (eye and brain) in which Aicardi syndrome pathologies are observed. Wnt8b and Slf1 had similar expression patterns in embryonic mouse brain. Ha and colleagues reported the individuals with the WNT8B and SLF1 variants displayed the classical triad of Aicardi syndrome with accompanying features including ventriculomegaly, atrophy of the cerebellar hemispheres, scoliosis, and skeletal abnormalities (42).
The syndrome has been reported from such diverse areas as India, Australia, and Japan, as well as Europe and North America. It has also been seen in children of North African origin.
Several epidemiologic studies yield birth incidence rates approximating 1:100,000 females or less. Kroner and colleagues found 1:105,000 live births in the Netherlands and 1:93,000 in the United States (56). A prevalence of 0.63 per 100,000 females under 29 years of age was found in Norway (60).
In another study, among 180 infants presenting with infantile spasms, 2% had Aicardi syndrome; 5% of those identified with a prenatal cause had the syndrome (74).
Aicardi syndrome cannot be prevented. No particular risk factors have been identified, although the association with advanced parental age has been recognized. There is no risk for recurrence in affected families.
Due to the highly specific findings, differential diagnosis is limited.
Uncertainty may exist when the ocular fundus is inaccessible because of anterior ocular defects. In the presence of severe ocular malformations precluding visualization of the fundus in a young female infant with neuroimaging findings including nodular heterotopia, corpus callosum agenesis, and a midline cyst, the problem is how to counsel the family. A diagnosis of Aicardi syndrome is assurance of unlikely future recurrence. If the clinical seizure features, EEG, brain, and skeletal imaging studies are suggestive of Aicardi syndrome, some reassurance can be provided. No molecular genetic test currently exists to definitively diagnose the syndrome when the necessary clinical parameters, including chorioretinal lacunae, cannot be documented.
When the chorioretinal lacunae are not accompanied by corpus callosum agenesis, or when the chorioretinal findings are "atypical," specific chromosome studies looking for translocation may be considered (30). Should the infant be a male with chorioretinal lacunae and corpus callosum agenesis, chromosome studies looking for the XXY pattern should be considered.
The presence of pigmentary changes within a large peripapillary lesion may raise the issue of an in-utero toxoplasmosis infection. Pigmentation within the lesion does occur in peripapillary chorioretinal lacunae in Aicardi syndrome. Such a solitary lesion is unlikely in Aicardi syndrome, and the presence of other typical rounded, clear-centered, punched-out lesions coexistent in the same or the other eye settles the diagnosis. These clinical findings preclude the cost of any diagnostic investigations besides neuroimaging studies.
Several groups have encountered children who were female, had early onset of infantile spasms, and on exam had chorioretinal lacunae, but the neuroimaging did not show agenesis of the corpus callosum (22). In a collated report, Chevrie and Aicardi identified six such cases (21). Four had abnormalities on cranial CT, and two were normal. According to Darwish and colleagues, in one such case the cranial CT was normal and the child was clinically better, developing the ability to walk (23). She had a fundus suggestive of Handmann anomaly, the "morning glory" syndrome, in addition to the lacunae, which were bilateral but dramatically asymmetrical. Such children may have a different condition or represent an incomplete Aicardi syndrome.
Differential diagnosis might be complicated during the evaluation of intrauterine brain abnormalities. Venkatesan and colleagues reported a female with fetal and postnatal MRI findings of agenesis of corpus callosum and type 2b interhemispheric cysts, characteristically found in Aicardi syndrome, but was found to have oral-facial-digital syndrome type 1 (OFD1) (104). They also presented three other companion cases with pre- and postnatal imaging of patients with Aicardi syndrome. These cases highlight the importance of widening the differential diagnosis to also include OFD1 for female patients with callosal anomalies.
Chevrie and Aicardi state that the chorioretinal lacunae have not been encountered in other conditions (21). Carney and colleagues remind us that chorioretinal lacunae have rarely been observed in two other conditions: the amniotic band syndrome and oral-facial-digital syndrome type VIII (19). In the female infant with infantile spasms but none of the other features of these syndromes, chorioretinal lacunae indeed are pathognomonic for Aicardi syndrome.
The diagnosis is mainly clinical, based on early-onset epilepsy, callosal agenesis, and chorioretinal lacunae. In this context, the neuroimaging finding of agenesis of the corpus callosum is necessary to make the diagnosis of "complete" Aicardi syndrome. Chevrie and Aicardi accept partial forms of agenesis within the complete syndrome (21). Most of the reports to date of Aicardi syndrome are based on cranial MRI. There is too much variability in electrodiagnostic studies such as EEG, auditory brainstem response, and visual evoked potential, for them to be used as markers of the syndrome. It is argued that some of the features of the EEG in the proper clinical context assume significant weight when there is uncertainty with the other parameters. In a large systematic analysis, only 60% fulfilled the classic triad, whereas 40% were diagnosed based on revised criteria incorporating cortical malformations or other major features (101).
Brain imaging.
Cranial ultrasound. A combination of findings in the same neonatal cranial ultrasound may be specific to Aicardi syndrome. This triad includes multiple cysts of the choroid plexus, corpus callosum agenesis, and widened atria and occipital horns of the lateral ventricles (84; 73).
Magnetic resonance imaging. MRI is superior in defining the extent of callosum agenesis and identifying gyral anomalies and abnormalities in myelination. Routine MRI of Aicardi syndrome patients may show callosal agenesis, asymmetric brain development, intrahemispheric neuroepithelial cysts, neocortical dysplasia, especially polymicrogyria in the frontal and perisylvian regions, periventricular nodular heterotopia, and cerebellar hypoplasia.
MRI in 20 patients with Aicardi syndrome showed a high frequency of migratory disorders to the cerebral cortex (94%). Wide variability is demonstrated in the callosal defect and migratory abnormalities, among other findings (94).
A study on the laterality of brain and ocular lesions in 26 patients showed asymmetry of ocular lesions in 18% and asymmetry of brain lesions in 58%, with a preponderance of right-sided brain lesions (18). A significant correlation was found between the most severe involved sides of the brain and ocular lesions (18).
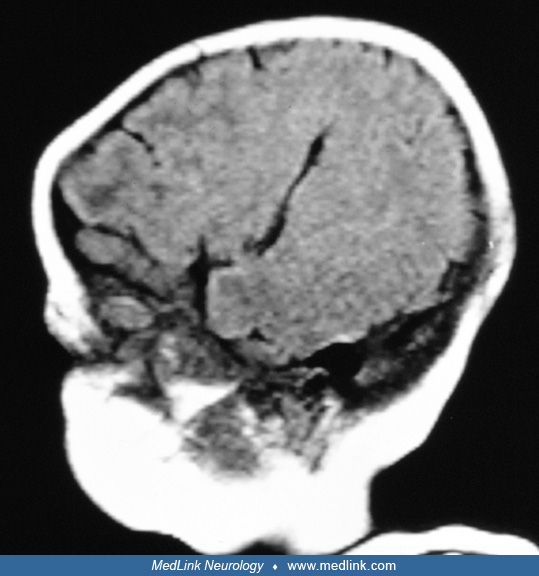
Cranial computed tomography. The bulk of the literature on neuroimaging was in the collated report (21). Ninety percent of 152 cases for which detailed data were available had total agenesis of the corpus callosum by cranial CT-documented criteria. Other cranial CT findings are less well documented. Forty-four percent of 184 cases had specific mention of irregular lateral ventricular contours.
A supratentorial cyst other than porencephaly was mentioned only 4% of the time. Our experience is different (Table 1), attesting to the inaccuracies of the collated derived report. Posterior fossa abnormality including cerebellar hypoplasia, or a Dandy-Walker variant, was mentioned in 6% of the cases, but this may again be an underestimate. Abnormalities of the choroid plexus were mentioned in 6%.
When there is corpus callosum agenesis, asymmetric ventriculomegaly affecting particularly atria and occipital horns, with irregular margins and a supratentorial midline cyst, the combination is suggestive of Aicardi syndrome (23; 21).
Skeletal imaging. The presence of a thoracic vertebral or rib anomaly is common and was documented in 75% of 90 of the 184 collated patients reviewed by Chevrie and Aicardi in 1986 (21). Ohtsuka and colleagues found "vertebral malformations" in all their six patients, but this included scoliosis or kyphosis as well as vertebral body fusion, butterfly vertebra, and spina bifida (75). Scoliosis, absent or malformed ribs, and hemivertebrae were reported in 39% of 17 patients by Donnenfeld and colleagues (29). We found these anomalies in three of our six cases. In the multi-institutional survey of Aicardi syndrome in Japan, four of 12 "typical" cases had block vertebra or hemivertebrae and two had scoliosis, but only one of eight "atypical" cases had vertebral body abnormalities (110).
Visceral malformations. A case of Aicardi syndrome, together with bronchopulmonary sequestration and hepatoblastoma, was reported by Kamien and Gabbett (53).
Electrophysiologic studies.
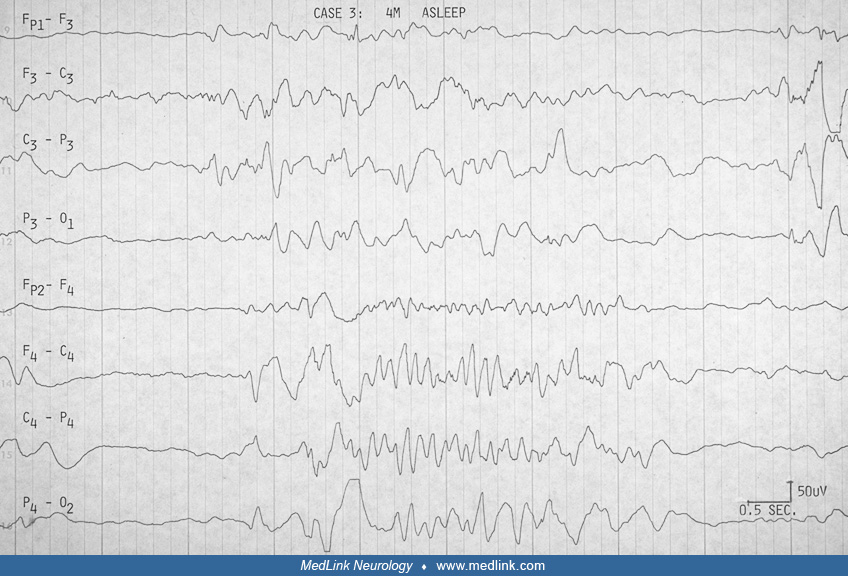
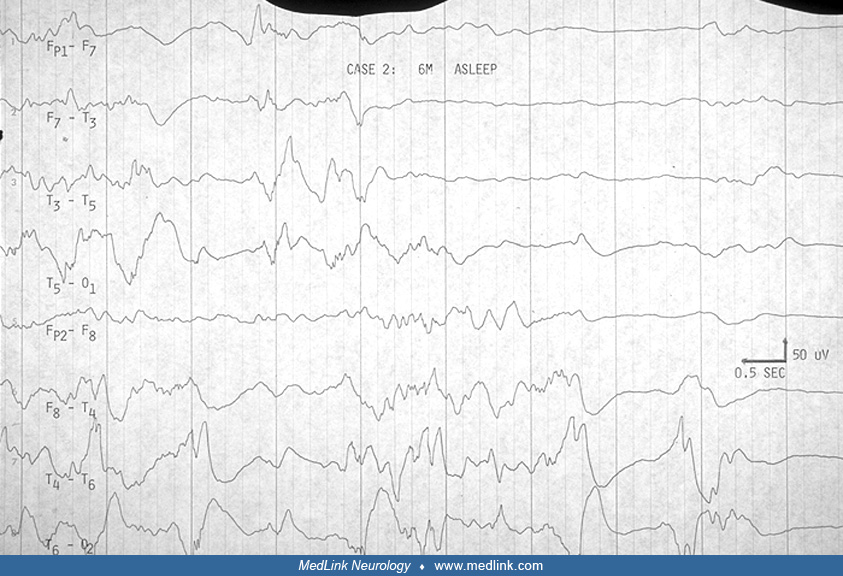
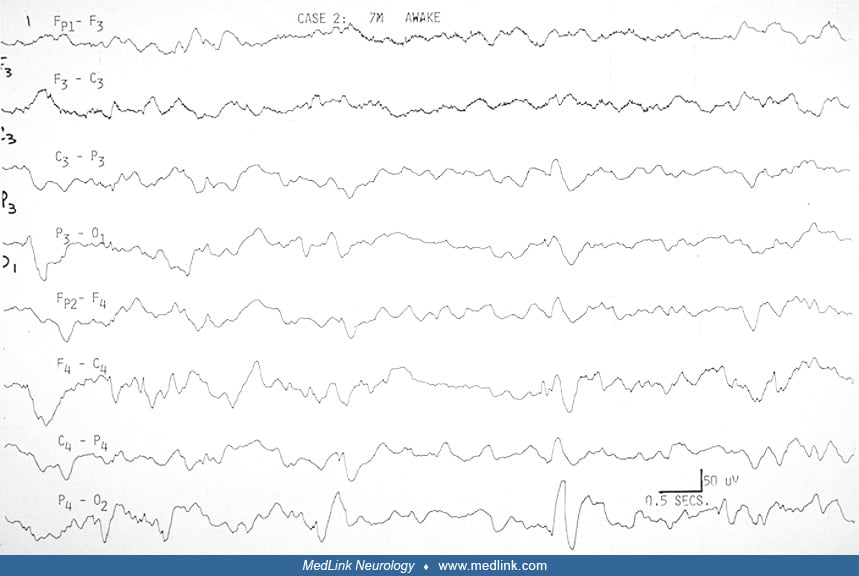
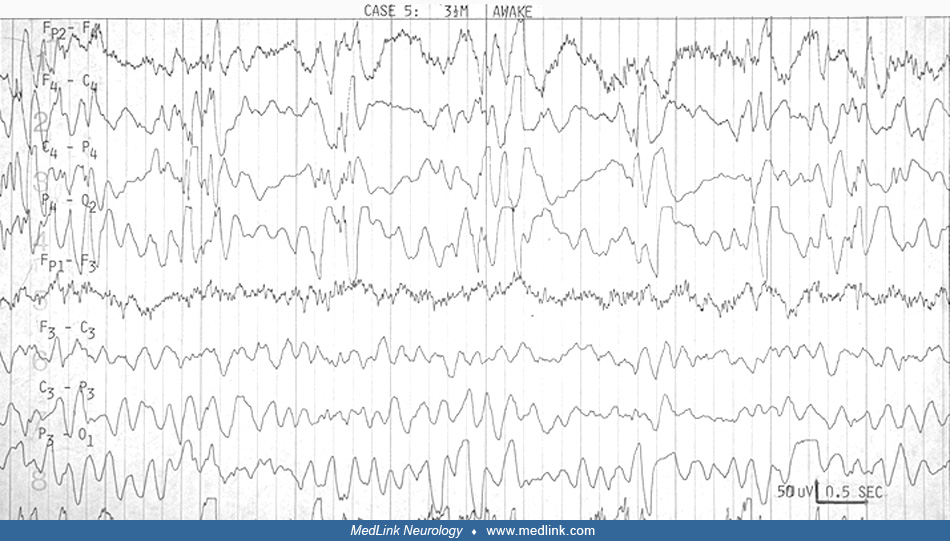
EEG. The EEG in Aicardi syndrome is always abnormal. It has been scrutinized for its value as a marker for the disorder. It is accepted that each EEG finding is not specific. The findings change with the age and state of the child, but a constellation of findings in the specific clinical context of the young female infant with spasms can be suggestive of Aicardi syndrome. The findings probably reflect the more profound neuronal migration disorder, complete corpus callosum agenesis, and asymmetry in the hemispheres that characterize the syndrome.
The initial descriptions of Aicardi in 1965 and 1969 have only been confirmed by the many subsequent reports. His initial observations about the effect of age on the EEG findings in Aicardi syndrome were extended (32). The ictal EEG features were reviewed (75).
Aicardi and colleagues pointed out the "split-brain EEG," the suppression burst feature, and the augmentation of its periodicity in sleep in the young infant before 3.5 months (03). Between 4 and 9 months, hypsarrhythmia evolves in the awake record, whereas the pseudoperiodic feature becomes prominent in sleep. Ohtsuka and colleagues have used "BIBs" to denote the bilateral independent bursts that Aicardi referred to as "split-brain suppression burst" and the pseudoperiodic aspects of the EEG in the syndrome (75). Menezes and colleagues used "quasi periodic" as a better term for what was referred to as "pseudo periodic" earlier in describing the EEG in Aicardi syndrome (68). Asymmetry is frequently seen (Table 2), and should not be confused with asynchrony. It was always mentioned in Aicardi and colleagues' cases (03).
|
Hypsarrhythmia Modified |
Asymmetry |
Asynchronous Burst Suppression or Bilateral Independent Bursts |
|
10 of 15 |
8 of 15 |
15 of 15 (03) |
|
| ||
Fariello and colleagues suggested that the record is less periodic and less abnormal in drowsiness before it becomes more periodic in deeper sleep (32). They did not find sleep spindles and other transient activities in sleep. Most other authors agree these are present, however, though brief or poorly organized or asymmetric. We have been impressed that the degree of asynchrony does not change in the awake and sleep recording (23). Like Bertoni and colleagues (12), Darwish and colleagues did not find absolute asynchrony (23). This is not specifically addressed by other authors (see Table 2 below).
|
Complete asynchrony |
Quasi periodicity |
Sleep spindles* |
|
15 of 15 |
15 of 15 |
15 of 15 (03) |
|
| ||
Several authors agree that the highly suggestive bilateral independent bursts, or split-brain pattern, evolve with time into a much less specific pattern of multifocal spike and spike-wave discharges on a slow and disorganized background. Bursts of sequential focal spikes were seen in two of our six cases, although multifocal spikes were seen in four of six, and four of six had bursts of beta activity that were asymmetric.
A retrospective study described the long-term EEG evolution in Aicardi syndrome cohort (n=12), followed for up to 23 years, and identified possible early predictors of the clinical and EEG outcomes (62). In addition to the “classical severe phenotype” already described in the literature, this study identified a new “mild phenotype.” The two phenotypes show completely different EEG features at onset of epilepsy and during its evolution, which correspond to different clinical outcomes; these findings might help to predict long-term clinical outcomes (62).
Evoked potential studies. Evoked potential studies are usually normal for the sensory end organ. For example, the ERG is normal, in spite of numerous lacunae, in most reports. This lends support to contentions that there is only involvement of the retinal pigment epithelium and atrophy of the underlying choroid. It is also accepted that a normal ERG is generated as long as 50% of the retina is normal. The visual evoked potential may be abnormal. In this case, the optic nerve, tracts, or radiations must be involved.
The auditory brainstem response wave I is usually normal. Later parts of the auditory brainstem response responses have rarely been reported abnormal.
Management of the infant with Aicardi syndrome is based on three principles; (1) the epilepsy is treated pharmacologically; (2) the development and physical well-being of the infant is maximized through proper intervention, physiotherapy, and, where milder forms exist, developing a communication system; (3) proper counseling, training, and support of families regarding the syndrome is necessary. It carries a long-term burden for the family unit, yet encouragement can be given in regard to future pregnancies, especially when the infant is the first for the couple. There is no cure for Aicardi syndrome. Standard treatment is symptomatic and generally involves management of seizures and interventional special educational programs for mental retardation.
The response of the epilepsy to antiepileptic drug management is poor. Although pyridoxal phosphate was effective in 28% of infants with idiopathic infantile spasms, it was effective in 13% of those with prenatal causes, including Aicardi syndrome (74). Response to clonazepam, valproic acid, adrenocorticotropic hormone, and phenobarbital was poor in all cases in one series (23). In one report, nine of 14 patients were the same or worse and five showed some improvement, though in only two were the seizures recurring less frequently than daily (68). It is unclear whether the reports of improved outcome in association with good seizure control with vigabatrin (09) or adrenocorticotropic hormone (01) simply identify the initially less involved infant. Hamano and colleagues did document an improvement in auditory brainstem response after use of adrenocorticotropic hormone to control otherwise refractory seizures (43). An open-label study investigating the efficacy of highly purified cannabidiol on convulsive seizures (tonic-clonic, tonic, clonic, atonic, and focal seizures with prominent focal features) of Aicardi syndrome was published (28). In this study, the median percent decrease of convulsive seizures from baseline (n=14) was 58.3% (n=11, IQR: 40%-87%) by week 12, and there was a decrease of 59.2% by week 48 (n=7, IQR: 45%-86%). The percentage of 50% or more responders for the Aicardi subgroup was 71% for week 12 and week 48.
Studies on ketogenic diet for these patients are limited (85; 87). In a study of 15 patients with Aicardi syndrome, ages 4 months to 34 years, ketogenic diet therapy was reported helpful, although seizure freedom was rare (87). It was especially helpful for those who were more drug-resistant and did not have infantile spasms at ketogenic diet therapy onset (87).
Some girls may respond favorably to surgery, or a vagal nerve stimulator (85; 54).
Kasasbeh and colleagues resorted to callosotomy in two patients with Aicardi syndrome and intractable epilepsy with multiple seizure types (54). Both patients had partial callosal agenesis, made total by callosotomy. In one patient, a substantial and sustained reduction in seizure frequency was obtained. Results with vagal nerve stimulation in two patients were satisfactory in one and disappointing in the other. A case of a 12-year-old girl with Aicardi syndrome who underwent corpus callosotomy has revealed a seizure reduction of over 50%, 4 months after the procedure (10). Although published data on neurosurgical management of patients with refractory epilepsy due to Aicardi syndrome is scarce, corpus callosotomy has been underscored as an effective, well tolerated method of controlling seizures, particularly for patients with partial agenesis of the corpus callosum (10). A patient with Aicardi syndrome who had total agenesis of corpus callosum underwent right subtotal hemispherotomy at the age of 3 months (51). After the procedure, her epileptic spasms dramatically improved, which may suggest disconnecting the thalamocortical and subcortical pathways in the epileptic network plays a role in controlling epileptic spasms generation.
Artificial intelligence is thought also to potentially transform the neurosurgical management of Aicardi syndrome by improving diagnostic accuracy, personalizing surgical planning, and enhancing intraoperative and postoperative care. From the early identification of the disorder to the long-term monitoring of treatment outcomes, artificial intelligence may offer tools that can significantly enhance patient care and quality of life (107).
Prenatal sonographic findings are reported in two children with Aicardi syndrome (15).
Anesthetic management in patients with Aicardi syndrome may be complicated by difficult venous access in the limbs due to severe generalized hypotrophy of subcutaneous tissues and difficult caudal block due to severe vertebral malformation, among other complications (50).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Nesibe Gevher Eroglu-Ertugrul MD
Dr. Eroglu-Ertugrul of Hacettepe University has no relevant financial relationships to disclose.
See Profile
Haluk Topaloglu MD
Dr. Topaloglu of Hacettepe Children's Hospital in Ankara, Turkey, has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neurogenetic Disorders
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Developmental Malformations
Apr. 24, 2026