



































































Neurobehavioral & Cognitive Disorders
Mental status examination
Jun. 17, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The topic of alcohol abuse related to acute and chronic exposure covers a wide spectrum of neurologic syndromes involving the central and peripheral nervous systems. This article discusses historical perspectives, clinical manifestations, clinical vignettes, etiology, pathogenesis, pathophysiology, epidemiology, prognosis, complications, and management. In addition, the authors discuss select mechanisms of cellular injury by alcohol.
|
• Alcohol intoxication has an acute and chronic symptomatology. | |
|
• The lethal dose of alcohol varies widely and depends on many external and internal factors. Tolerance develops with repeated exposures. | |
|
• Wernicke-Korsakoff syndrome is particularly found in the alcoholic population. | |
|
• Alcohol affects almost all areas of the nervous system. | |
|
• Alcohol affects the functioning of many systems in the body (heart, liver, muscle, nerve). |
Hominids adapted to metabolize ethanol long before human-directed fermentation, which began at least 11,000 years ago in the early Mesolithic (Middle Stone Age) period (78; 51; 207; 193; 11; 81).
The term “alcoholism” was first used by a Swedish physician in 1849 to describe the adverse systemic effects of alcohol. Also, early psychiatry texts described a syndrome of alcohol-related deterioration characterized by intellectual and behavioral abnormalities (13).
The Diagnostic and Statistical Manual of Mental Disorders, 4th edition, text revision (DSM-IV-Tr) defined alcoholism as “maladaptive pattern of drinking, leading to clinically significant impairment or distress” (14).
The definition of alcoholism by the National Council on Alcoholism and Drug Dependence and the American Society of Addiction Medicine is “a primary, chronic disease characterized by impaired control over drinking, preoccupation with the drug alcohol, use of alcohol despite adverse consequences, and distortions in thinking” (217).
The role of alcoholism in the development of cognitive and functional decline was known in Ancient Greece (148) and has received serious study in Western medicine for more than 250 years.
In Greek mythology, Silenus was the frequently drunken companion and tutor of the wine god Dionysus. Artistic depictions of the drunken Silenus show him supported by satyrs or carried by a donkey.
Some Renaissance artistic depictions of the drunken Silenus show features of alcoholic cirrhosis, perhaps most fully in the painting Drunken Silenus (1626) by Spanish painter Jusepe de Ribera (1591-1652): evident signs of liver disease in this painting include parotid gland swelling, spider angioma (left parasternal area), gynecomastia, and ascites.
A clinic-pathologic description of hepatic encephalopathy from alcoholic cirrhosis was described by Italian anatomist and pathologist Giovanni Battista Morgagni (1682-1771), Professor of Anatomy at the University of Padua, in his De sedibus et causis morborum per anatomen indagatis (Seats and Causes of Diseases Investigated by Anatomy) (216).
Alcoholic neuropathy was documented at least as early as 1787 by English physician and philanthropist John Coakley Lettsom (1744-1815) (189), but other neurologic complications of alcoholism were probably not recognized until the end of the 19th century or later.
Alcohol-related deficits in memory and intellectual ability were reported in 1878 by British psychiatrist Robert Lawson (1846-1896) of the Wonford House Lunatic Asylum in Exeter (185; 184).
Subsequently, in a series of three articles from 1887 to 1889, Russian neuropsychiatrist Sergei Sergeievich Korsakoff (sometimes spelled Korsakov; 1853/1854-1900) gave a comprehensive description of the persistent amnestic confabulatory state now known as Korsakoff psychosis, occurring in conjunction with peripheral polyneuropathy, a combination he initially labeled either as “psychosis associated with polyneuritis” or “polyneuritic psychosis” (170; 169; 168; 339; 334; 335; 333; 177; 180; 181). Korsakoff based his conclusions on at least 46 patients, about two-thirds of whom were alcoholics, whereas the remainder suffered from a diverse group of disorders associated with protracted vomiting.
Korsakoff was apparently unaware of the syndrome incorporating a confusional state, ophthalmoparesis and other oculomotor findings, ataxia, and neuropathic features, which had been described by German neuropsychiatrist Carl Wernicke (1848-1905) in 1881 and labeled as “Die acute, hämorrhagische Poliencephalitis superior” (acute hemorrhagic superior polioencephalitis) and is now generally called Wernicke encephalopathy (344; 163; 164). The clinico-pathologic overlap between Wernicke encephalopathy and Korsakoff psychosis was ultimately recognized in the late 1920s and early 1930s (107; 151; 46), and the two terms became linked as Wernicke-Korsakoff syndrome.
The term “alcoholic hallucinosis” (also known as “alcohol hallucinosis” and “alcohol-related psychotic disorder”) refers to a disorder of acute onset, with a predominance of auditory hallucinations (although delusions and hallucinations in other sensory modalities may also be present), no disturbance of consciousness, and a history of heavy alcohol consumption (111).
This syndrome has often been attributed to Swiss psychiatrist Paul Eugen Bleuler (1857-1939), who labeled it alcoholic hallucinosis (Alkoholhalluzinose) and considered it as an alcoholic madness (Alkoholsahnsinn) (35); Bleuler was also responsible for other psychiatric terms, including schizophrenia, schizoid, and autism. However, in his textbook, Bleuler acknowledged the earlier description by Wernicke, who labeled the disorder as “chronic hallucinosis in alcoholism” (chronische Halluzinose beim Alkoholismus) (345). Even earlier, in 1847, the French author Claude-Nicolas- Séraphin Marcel described a similar symptom complex under the label of folie d'ivrogne (ie, drunken madness) (201).
Although alcoholic intoxication was long recognized in art, portrayals of delirium tremens in caricatures (305) and health propaganda posters became particularly prominent in the early 20th century, in the years before and during Prohibition (1920-1933) in the United States.
Marchiafava-Bignami disease is a progressive alcoholism-related neurologic disease characterized by corpus callosum demyelination and necrosis and subsequent atrophy. The disease was first described in 1903 by the Italian pathologists Ettore Marchiafava (1847-1935) and Amico Bignami (1862-1929) in an Italian Chianti drinker (202). In the autopsy of this patient, the middle two-thirds of the corpus callosum were necrotic.
In the last century, through careful observation and description, American neurologist and neuropathologist Raymond Delacy (“Ray”) Adams (1911-2008) made the most prodigious contributions in understanding the neurologic complications of alcohol abuse, in conjunction with various protégés, notably Boston-born neurologist Joseph Michael (“Joe”) Foley (1916-2012) and, subsequently, Canadian-American neurologist Maurice Victor (1920-2001) (02; 03; 04; 05; 101; 06; 336; 335; 337; 331; 338). Foley began to work with Adams on the neurologic manifestations of liver disease in the late 1940s after Foley returned from his military service during World War II (183; 178). In a series of reports from 1949 to 1953, Adams and Foley described the clinical (neurologic), electroencephalographic, and neuropathologic features of alcoholic liver disease, including the clinical and electrophysiologic features of asterixis (02; 03; 04; 05; 101).
Social commentary on the neurologic degeneration and social decay from alcoholism. The neurologic disorders and social decay resulting from alcoholism were targeted by social reformers in many countries since at least the 18th century.
Drunkard with death. From Images of death (1547), by Hans Holbein the younger. The German-Swiss painter and printmaker Hans Holbein the younger (c1497-1543) created a famous sequence of images in his Imagines mortis... [Images of death], now typically referred to as The Dance of Death. In one of these images, death is pouring alcohol down the throat of an alcoholic while another man next to him vomits from drinking too much.
Other 16th century images of the adverse effects of alcohol. Among the profusion of prints in the 16th century were many that showed the adverse effects of alcohol abuse: vomiting, ataxia, stupor, and coma.
“Venus, Podagra, and Bacchus.” In the 17th century, there were printed images that were meant to convey the dangers of venereal or alcoholic excess. Among these is the frontispiece of Scriptum apologetico-politicum de podagra oder das so benahmte ... Zipperlein (1687) by Johann Andreas Schlegel (fl 1657-1681). Sitting under grapevines heavy with fruit is Podagra, who, though looking like the wise philosopher, personifies the excess represented by Venus and Bacchus, who are sitting beside him.
William Hogarth and Gin Lane. English engraver, pictorial satirist, and social critic William Hogarth (1697-1764) depicted the effects of alcoholism in his famous engraving, Gin Lane (1751).
Hogarth's engraving, published according to Act of Parliament on February 1, 1751 in support of what would become the "Gin Act," shows a poor London street (the area depicted is St. Giles, London) strewn with hopeless drunka...
Hogarth's engraving, published according to Act of Parliament on February 1, 1751, in support of what would become the "Gin Act," shows a poor London street (the area depicted is St. Giles, London) strewn with hopeless drunkards and lined with gin shops and a flourishing pawnbroker. The inhabitants of Gin Lane are being destroyed by their addiction to the foreign spirit of gin, with the engraving illustrating shocking scenes of child neglect, starvation, madness, drunken brawls, and death. Hogarth's illustration is filled with satirical humor: the pawnbroker's shop depicted is "S. Gripe pawnbroker;" the distillery is "Kilman distillery;" a gin shop sign reads "drunk for a penny, dead drunk for two pence, clean straw for nothing;" and a drunkard's paper is headed "the downfall of Mdam. gin."
A poem below the engraving reads as follows:
|
Gin cursed Fiend, with Fury fraught, Virtue and Truth, driv'n to Despair, Damn'd Cup! that on the Vitals preys, |
At that time, there was no quality control whatsoever, and gin was frequently adulterated (eg, with turpentine). When it became apparent that copious gin consumption was causing social problems, social reformers and the government made efforts to control the production of the spirit. The Spirit Duties Act (commonly known as the Gin Act of 1736) imposed high taxes on sales of gin, forbade the sale of gin in quantities of less than two gallons, and required an annual payment of £50 for a retail license. These measures had little effect beyond increasing smuggling and driving the distilling trade underground. The act was repealed by the Gin Act of 1743, which set much lower taxes and fees.
Alcohol addiction. Beginning in the 18th century, several images illustrated the addictive potential of alcohol. For example, English caricaturist and satirical poet John Collier (1708-1786), writing under the pseudonym Tim Bobbin, published two separate illustrations titled "They cry, who want it; having it they laugh," (1773) illustrating the craving for alcohol among addicts. Other later images emphasized the extent alcoholics will go to so that they can have alcohol, like one in which a bum drinks the alcohol from the cigar lighter. Some 19th-century images took this issue more seriously and emphasized the struggle that alcohol addicts have with their "enemy."
Caricaturing the debate over the health effects of alcohol (1799-1902). British caricaturist and printmaker James Gillray (1756-1815), who was famous for his etched political and social satires, published an engraving entitled "Punch cures the Gout, the Colic, and the Tisick" (1799). Two men and a woman are sitting around a small table on which is a large punch bowl; each is holding a glass. On the right is seated a fat man with both feet and his left hand bandaged; in the center, a young woman is holding her stomach; and on the left, a man appears to be near death. The fat man with gout claims that "PUNCH cures the GOUT," to which the woman adds "the COLIC," and the sickly man adds "and the TISICK." Note that "tisick" in British English is archaic for a splutter (a short explosive spitting or choking noise) or a cough.
In another caricature with a similar theme, artist William Heath (1795-1840) published an engraving entitled "Palatable Physic" (1810), in which "physic" meant medicine, and alcohol was being promoted as a palatable medicine. Two men and a woman are sitting around a table, drinking to their health. The man on the left, holding a glass of alcohol, claims, "This good Appetite gives [sic]." The woman in the middle similarly holds a glass of alcohol and adds, "Cures the vapors also." The snarling man on the right, also holding a glass of alcohol, asks, "Will it Cure the Cursed Pain I have got in my toe?"
Another illustration from this period, "The Rival Majicians [sic] or Raising the Spirit" (1808) by Samuel De Wilde (1748-1832), raises concern about the powerful effects of rum. Black men, representing the West Indies, distill sugar and molasses, while spirits representing the effects of alcohol emanate from a large vat, and representatives of agriculture, holding corn and barley, fall back in fear.
In 1842, British caricaturist and book illustrator George Cruikshank (1792-1878) presented an illustration in which people are reaching for alcoholic drink falling from a pile of barrels of liquor, likened to the upas tree; skeletons litter the ground. Alcohol is implicitly compared to the upas tree, a poisonous tree allegedly fatal to approach with sap that is used as arrow poison. A Dutch version of this image appeared a quarter century later, then with a warning against binge drinking.
In the print "King Alcohol, and his Prime Minister" (1872), designed and engraved by John Warner Barber (1798-1885), a man identified as "King Alcohol" and a skeleton representing Death stand on barrels labeled "Strong Beer, Wines, Rum, Gin, Brandies, Whiskies, compounded liquors," with "social class" and "moderate drinkers" on the left, as well as the "suicide and murderous fighter," and on the right, the "dead and drunken," their broken families, ruined homes, and their graves. "Hope" at lower left mourns the "60,000 annually in the U.[S.] States." "King Alcohol" holds a tankard in his raised right hand with a snake coiled around his arm. Printed above is reference to Proverbs XXIII 32: "In the end, it will bite like a snake and poison like a viper."
In a late-19th century sketch, a man holds up a glass of alcohol and says "It's killing me but I like it."
Cartoons about the adverse effects of alcohol appeared in national magazines, including Harper's Weekly in the United States and other magazines in Europe.
Death, appearing as a doctor with a top hat and cane, is standing before a stout man sitting in a chair. The man is holding a glass, and on the table next to him are the remnants of overindulgence. Cartoon by French artist Jose...
“The Drunkard's Progress.” The 4-part sequence of "The Drunkard's Progress" (1826) by American engraver and historian John Warner Barber (1798-1885) depicts the neurologic and social decay from alcoholism, complete with biblical admonitions. In the first image, "The Morning Dram," the father is drinking at 8 AM, ignoring his wife and children.
Hand-colored engraving by John Warner Barber (1798-1885), printed in New Haven, Connecticut. Biblical quotation above the image: "Wo unto them that rise up early in the morning that they may follow Strong Drink... Isa 5 C. 11v....
In the second image, "The Grog Shop," two men are drinking at a saloon, while two are brawling, one is vomiting, and one is unconscious on a bench.
Hand-colored engraving by John Warner Barber (1798-1885), printed in New Haven, Connecticut. Biblical quotation above the image: "Wo unto them that are mighty to drink wine, and men of strength to mingle Strong Drink... Isaiah ...
In the third image, "The Confirmed Drunkard," the father is intoxicated on the floor, while his wife and children are afraid, and the home is falling apart.
Hand-colored engraving by John Warner Barber (1798-1885), printed in New Haven, Connecticut. Biblical quotation above the image: "Who hath wo? Who hath sorrow? Who hath contentions? Who hath wounds without cause? ... They that ...
In the final image of the sequence, "Concluding Scene," there is an auction sign on the house, ordered by the sheriff. The family is evicted and departing with the wife in tears.
Hand-colored engraving by John Warner Barber (1798-1885), printed in New Haven, Connecticut. Biblical quotations above the image: "The drunkard shall come to poverty. Proverbs 23 Chap. 21 v." [Proverbs 23:21] and "The wages of ...
Many other versions of "The Drunkard's Progress" were published in the 19th century, some of which showed as many as nine steps in the drunkard's gradual decline in society, including such adverse outcomes as poverty and disease, criminal activity, becoming a bum, and his eventual death by suicide, sometimes also highlighting the devastation wrought on the drunkard's family. There were also staged photographs of some of these adverse effects once photography was more readily available in the late 19th century.
Conceptually similar illustrations were "The Bottle" (1847) by British caricaturist and book illustrator George Cruikshank (1792-1878) of a man ruined by alcohol and placed in an institution and "A family losing their possessions after the husband turns to drink" (c1830-1860).
The hangover. In an interesting caricature from the 1830s, two cartoon panels illustrate the effects of overindulging in alcohol. "Night" depicts a laughing man with friends seated at a table set with decanters labeled "Port" and "Sherry," holding a goblet of wine in his raised right hand and fruit in his left hand. "Morning" depicts the same man lying in bed tormented by two imp-like figures personifying wine: "Sherry" hammers the man's head while "Port" prods his stomach with a red-hot poker. This indicates some of the symptoms of a hangover: headache and nausea.
The responsibility of purveyors of alcohol for the negative effects. Social critics and reformers blamed both the alcoholic and those that manufactured or sold alcohol for the adverse outcomes on individuals and society. The responsibility of purveyors of alcohol for the negative effects was portrayed in various ways, but often with religious overtones. For example, the engraving titled "Illustration of the Rumseller's just doom and final exit" (1835).
A Latin expression serves as an epigram: "Facilis descensus Averni: sed revocare gradum, Hoc opus, hic labor est." ("The descent to Perdition is easy—but to retrace one's steps—this is labor—this a task severe.") The engraving depicts a man who is praying the rosary, about to be impaled by the devil, and struck by lightning bolts held in the hand of God. He is descending into Hell, portrayed by fire, smoke, a snake, and a fire-breathing serpent or dragon. A poem below the engraving reads as follows:
|
Behold! the soulless wretch—destroyer of his race— Companion of the worm—the victim of despair— |
Prohibition in the United States. Leading up to Prohibition in the United States, numerous forms of propaganda were used to convince people of the harms of alcohol (or of its relative safety by opponents of prohibition). One political cartoon of that era, "A sample room and its samples" (1902), for example, depicted a saloon keeper in front of a morbid saloon window display.
The window display sign says: "SHOW WINDOW EXHIBIT. WHAT OUR LIQUORS CAN DO. GENUINE SPECIMENS MADE ON THE PREMISES." The implications of this sign are displayed in the window in the form of five people damaged by alcohol, four of whom are labeled with their alcohol-induced problems: madman, tramp, convict, and idiot child (fetal alcohol syndrome). The fifth person, a woman, appears depressed or confused.
American poet Will Carleton (1845-1912) published "The serpent of the still" first in 1887 in Harper's Weekly and then in a collection of his poems directed at "young Americans" in 1906 (49; 50).
|
The Serpent of the Still The tempter, as God's legends tell— He comes not now in subtle mood— | |
|
(49; 50) |
Both versions were accompanied by an arresting illustration, "He twines about her trembling life," by popular pen and ink illustrator Jessie Curtis Shepherd (1842-1907): A mother is holding off a boa constrictor wrapped around her and her three small children, with flames surrounding them, a liquor store is in the background, and a still is behind them with the inscription "The worm of the still."
A mother is holding off a boa constrictor wrapped around her and her three small children. Flames surround them. A liquor store is in the background on the right. A still is behind them on the right, with the inscription "The w...
Even before Prohibition, some laws restricted very high alcohol content spirits. For example, the anise-flavored spirit absinthe was banned in the U.S. in 1912 and in several European countries around that time due to its alleged dangerous properties, being 45% to 74% alcohol by volume or 90 to 148 proof in the U.S.
Motion picture poster for "Absinthe" shows a man seated, with hand on head, at table with three glasses of absinthe. Metropolitan Division, U.S. Lithograph Company. (Source: Courtesy of the U.S. Library of Congress, Washington,...
The 18th Amendment to the U.S. Constitution banned the manufacture, transportation, and sale of intoxicating liquors. The Volstead Act was ratified by the states on January 16, 1919, and went into effect on January 17, 1920, marking the beginning of the period in American history known as Prohibition. Despite an army of agents of the Bureau of Prohibition, Prohibition proved difficult to enforce; eventually, the illegal production and sale of liquor (“bootlegging”), the proliferation of illegal drinking spots ("speakeasies"), and the accompanying rise in gang violence and other crimes led to waning support for Prohibition by the end of the 1920s.
The 21st Amendment was ratified on December 5, 1933, repealing the 18th Amendment and ending Prohibition.
20th century public health messages concerning alcohol. A wide variation in public health messages were employed in the United States, but also in other countries with significant problems with alcoholism, like Russia. In the United States, anti-alcohol messaging was promulgated by the U.S. National Institute on Alcohol Abuse and Alcoholism in a variety of posters: "Getting drunk doesn't make you -- tall -- rich -- strong ... just drunk" (1973), "The typical alcoholic American" (1975), and "If you drink a lot of beer, you drink a lot" (1975).
|
• Based on the temporal relationship between alcohol abuse and onset of neurologic symptoms, the diverse neurologic manifestations of alcohol abuse may be subdivided into three main categories: (1) acute intoxication; (2) a withdrawal syndrome from sudden abstinence; and (3) a varied group of acute, subacute, or chronic disorders secondary to chronic alcohol abuse. | |
|
• In contrast to the acute and (usually) reversible pharmacological effects of ethanol intoxication, prolonged alcohol abuse leads to persistent and potentially irreversible neurologic deficits, potentially affecting any level of the nervous system. | |
|
• Alcohol withdrawal seizures usually occur after years of alcohol abuse with a single generalized tonic-clonic seizure or a brief cluster of such seizures occurring 6 to 48 hours after the last drink. | |
|
• Alcohol withdrawal seizures typically occur as blood alcohol approaches zero, or shortly thereafter, and are rarely seen after more than 2 days of abstinence. | |
|
• The initial phase in Wernicke encephalopathy is classically characterized by the triad of mental confusion, oculomotor disturbance, and gait ataxia. | |
|
• Oculomotor disturbances in Wernicke encephalopathy can include nystagmus, abducens or conjugate gaze palsies, and ptosis. | |
|
• Wernicke encephalopathy may progress to hypotension, stupor, coma, hypothermia, and death if the underlying thiamine deficiency is unrecognized or untreated. | |
|
• Approximately 80% of untreated patients with Wernicke encephalopathy subsequently evolve to a Korsakovian state. | |
|
• The Korsakoff syndrome is a memory disorder that typically emerges as the acute symptoms and signs of Wernicke encephalopathy subside; the amnesia is characterized by an inability to recall events for a period of a few years before the onset of illness (retrograde amnesia) and an inability to learn new information (anterograde amnesia). | |
|
• The common manifestations of alcoholic pellagra are encapsulated in “the four Ds”: diarrhea, dermatitis, dementia, and death. | |
|
• Most commonly, hepatic encephalopathy is a chronic disorder that occurs in the setting of alcoholic cirrhosis with portosystemic shunts. | |
|
• Hepatic encephalopathy has a wide range of presentations, progressing successively through stages: (1) unimpaired cognitive function with intact consciousness; (2) lethargy and confusion or delirium; (3) somnolence and disorientation; and (4) coma. | |
|
• Alcoholic cerebellar degeneration is a disorder of progressive cerebellar degeneration that can be seen in patients with severe alcoholism; it preferentially affects the anterior and superior vermis, giving rise to a remarkably stereotypic syndrome of ataxic stance and gait. | |
|
• In acute alcoholic myopathy, massive muscle injury triggered by acute alcohol abuse may result in multiple organ failure and death, but alcoholic rhabdomyolysis generally has a good prognosis if renal failure is avoided or is aggressively treated. | |
|
• With chronic alcoholic myopathy, only half of the sober patients recovered to normal strength over 5 years, indicating that chronic alcoholic myopathy is only partially reversible. | |
|
• Secondary disabilities (eg, school dropouts, criminal behavior, and substance or alcohol abuse problems) are common in young adults with fetal alcohol spectrum disorders. |
The consumption of alcohol and alcohol use disorder are associated with injuries, violence, cancers, nonmalignant conditions of the gastrointestinal system, infections, adverse effects on the cardiovascular system, and neurodegenerative diseases (01).
The effects of alcohol on the central and peripheral nervous system are varied, and overuse of alcohol can have serious medical and neurologic consequences, even death. Sustained alcohol intake can predispose to generalized cortical and cerebellar atrophy, amnesic and dementia syndromes (eg, Korsakoff syndrome, alcoholic dementia), and specific white matter disorders (eg, osmotic demyelination syndrome and Marchiafava-Bignami syndrome) (347).
Based on the temporal relationship between alcohol abuse and the onset of neurologic symptoms, the diverse neurologic manifestations may be subdivided into three main categories: (1) acute intoxication; (2) a withdrawal syndrome from sudden abstinence; and (3) a varied group of acute, subacute, or chronic disorders secondary to chronic alcohol abuse.
Acute alcohol intoxication. Ethanol enters and distributes rapidly throughout the body after ingestion. Intoxication occurs because ethanol readily crosses the blood-brain barrier. It acts directly on neuronal membranes and interacts with numerous neurochemical receptors. Behavioral effects may include euphoria, dysphoria, social disinhibition, drowsiness, belligerence, and aggression. In nonalcoholic individuals, these may occur at serum concentrations as low as 50 to 150 mg per dL. Even a moderate dose of alcohol substantially impairs prospective memory function (83). Lethargy, stupor, coma, or death from respiratory depression and hypotension occur at progressively higher concentrations. The lethal dose varies widely because tolerance develops with repeated exposures. Some alcoholics may appear sober at a serum level as high as 500 mg per dL, whereas this same level can be fatal in nonalcoholic individuals.
As alcohol is metabolized, acute inebriation often leads to a hangover, a condition characterized by headache, nausea, fatigue, and drowsiness.
Chronic alcohol abuse. In contrast to the acute and (usually) reversible pharmacological effects of ethanol intoxication, prolonged alcohol abuse leads to persistent and potentially irreversible neurologic deficits, potentially affecting any level of the nervous system (58; 77). The sites of neuronal injury and, hence, the clinical presentations are governed by genetic, nutritional, and other environmental factors. Individuals with alcohol use disorder typically have impaired memory and smaller precentral frontal and hippocampal volumes (92). Neurologic disorders in individuals with chronic alcohol abuse may occur in isolation, or, more commonly, multiple syndromes may be present in a single patient.
Alcohol withdrawal. Sudden cessation of drinking in a chronic alcoholic typically produces a withdrawal syndrome of central nervous system hyperexcitability and may, if very severe, produce delirium tremens. The earliest symptom is a generalized tremulousness. Insomnia, agitation, delirium, hallucinations (auditory, visual, or tactile), or other perceptual disturbances may follow. The syndrome is also characterized by autonomic hyperactivity, such as tachycardia, profuse sweating, hypertension, and hyperthermia. Withdrawal symptoms commonly begin 6 to 8 hours after abstinence and are most pronounced 24 to 72 hours after abstinence.
Alcohol withdrawal in a postsurgical setting may cause an increase in surgical complications, a situation demonstrated with elective spinal fusion surgery (122).
Alcohol withdrawal delirium. Alcohol withdrawal leads to a short-lived delirium syndrome and is usually easy to differentiate from the direct effects of alcohol on the brain. However, in some individuals, features of delirium, such as paranoia, auditory hallucinations, and attentional deficits, persist for many months. The etiology of this prolonged and attenuated delirium syndrome is unknown. Benzodiazepines used to treat alcohol withdrawal may also occasionally induce delirium (15).
Alcohol withdrawal seizures. Alcohol withdrawal may lead to tonic-clonic (grand mal) seizures, usually after years of alcohol abuse (337). Convulsions typically occur 6 to 48 hours after the last drink and may occur singly or in a brief cluster. Alcohol withdrawal seizures typically occur as blood alcohol approaches zero, or shortly thereafter, and are rarely seen after more than 2 days of abstinence; however, a relative fall in blood alcohol levels during sustained drinking can also sometimes produce withdrawal seizures while blood alcohol levels are still at levels typically associated with intoxication. Unless an underlying neuropathology exists, the seizures are rarely focal. Generally, electroencephalograms on such patients are mildly abnormal and usually revert to normal within a few days. Status epilepticus is rare.
Seizures after an isolated episode of intoxication or after a short binge should suggest the possibility of an underlying seizure susceptibility (eg, from prior cortical trauma) or other contributing factors (additional toxic exposure, hypoxia, electrolyte abnormality, etc.). Similarly, seizures during active heavy drinking or after more than a week without alcohol raises the possibility of pathogenic mechanisms other than withdrawal per se (129).
Subacute encephalopathy with seizures in alcoholics syndrome. Subacute encephalopathy with seizures in alcoholics syndrome is a well-defined subtype of focal nonconvulsive status epilepticus in patients with chronic alcoholism (33; 96; 97). In a study of 34 cases, all presented with altered mental status, 41% had a history of epileptic seizures associated with acute withdrawal, 71% had bilateral tonic-clonic seizures, 59% had focal motor seizures, 44% had focal impaired awareness seizures, and 24% met criteria for focal nonconvulsive status epilepticus (96; 97). Additionally, 82% had transient neurologic deficits, and persisting cerebral sequelae were mentioned in 26%. Eighty-five percent had lateralized periodic discharges on EEG. Areas of increased T2/FLAIR signal and restricted diffusion were mentioned in 65%. Transfer to the intensive care unit was necessary in 24%. Thirty-eight percent had recurrent episodes. The most used anti-seizure medication was levetiracetam, followed by phenytoin and lacosamide.
Wernicke-Korsakoff syndrome. Wernicke-Korsakoff syndrome is the best example of acquired nutritional deficiency in alcoholism (335). Wernicke encephalopathy and Korsakoff syndrome (or Korsakoff “psychosis”) are successive stages of thiamine (vitamin B1) deficiency. Increased metabolic demands, glucose infusion, and sudden resumption of dietary intake after a period of malnourishment are risk factors in precipitating acute symptoms.
The initial phase in Wernicke encephalopathy is classically characterized by the triad of mental confusion, oculomotor disturbance, and gait ataxia. Oculomotor disturbances can include nystagmus, abducens or conjugate gaze palsies, and ptosis (335; 45; 293). The encephalopathy may progress to hypotension, stupor, coma, hypothermia, and death if the underlying thiamine deficiency is unrecognized or untreated (293).
The classic triad is insensitive for the diagnosis of Wernicke encephalopathy (127; 219). In particular, the incidence of oculomotor findings is low in patients later shown to have had Wernicke encephalopathy. Consequently, modified diagnostic criteria have been developed to address the insensitivity of the classic triad. Modified diagnostic criteria for Wernicke encephalopathy require at least two of the following four signs: (1) dietary deficiencies, (2) oculomotor abnormalities, (3) cerebellar dysfunction, and (4) either an altered mental state or mild memory impairment (45).
Unfortunately, Wernicke encephalopathy is frequently underrecognized by clinicians (127; 280), especially when the disorder occurs in settings other than alcohol abuse (296).
Coronal section of cerebral MRI FLAIR sequence showing hyperintense signal change of the mammary bodies in a 66-year-old man with Wernicke encephalopathy complicating prolonged parenteral nutrition. (Source: Slim S, Ayed K, Tri...
Axial section of cerebral MRI FLAIR sequence showing hyperintense signal change of the mammary bodies and periaqueductal gray matter in a 66-year-old man with Wernicke encephalopathy complicating prolonged parenteral nutrition....
In pathological series, there is a consistently high proportion of cases of Wernicke encephalopathy that never had a clinical diagnosis of that condition during life (127; 219; 316; 45): indeed, less than 20% of cases of Wernicke encephalopathy are diagnosed before death.
Korsakoff syndrome is a memory disorder that typically emerges as the acute symptoms and signs of Wernicke encephalopathy subside. The amnesia is characterized by an inability to recall events for a period of a few years before the onset of illness (retrograde amnesia) and an inability to learn new information (anterograde amnesia, ie, loss of anterograde episodic memory) (45). Almost all patients have limited insight into their memory dysfunction. Other cognitive deficits may manifest themselves but are mild relative to the amnesia (166). Many, but not all, patients confabulate, a phenomenon that may be due to associated frontal lobe dysfunction (30); confabulation may resolve as frontal lobe function improves. Attention, language, and spatial navigation are usually normal. Although usually subacute in onset, Korsakoff syndrome can develop insidiously without evidence of acute encephalopathy, ataxia, or oculomotor findings (331). Approximately 80% of untreated patients with Wernicke encephalopathy subsequently evolve to a Korsakovian state (335).
Alcoholics with Wernicke-Korsakoff syndrome can be differentiated from those with Wernicke encephalopathy (without Korsakoff syndrome), but the severity and stability of memory loss, regardless of other cognitive deficits (45).
Wernicke-Korsakoff syndrome is generally diagnosed in younger patients rather than those diagnosed with alcohol-related dementia (45).
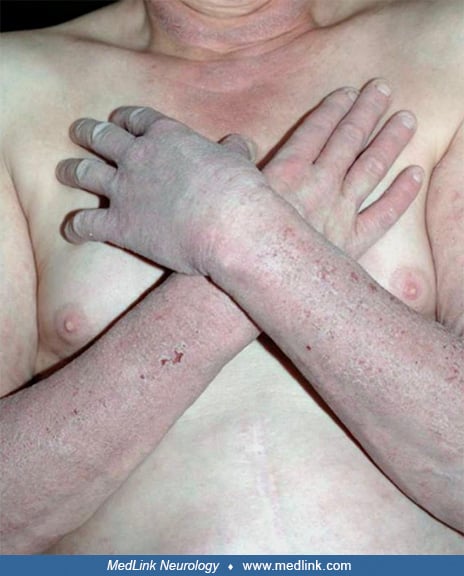
Alcoholic pellagra. Pellagra has been recognized among alcoholics for more than a century (157; 359). The common manifestations of this disorder are encapsulated in “the four Ds”: diarrhea, dermatitis, dementia, and death (177; 179). The typical cutaneous features of pellagra include peeling, redness, scaling, and thickening of sun-exposed areas (177; 179).
However, the dermatitis of pellagra ranges from obvious scaly erythema to more subtle changes that are often mistaken for the photodamage typically seen in the elderly (from a lifetime of cumulative exposure to solar irradiation). Neuropsychiatric features of pellagra include irritability, depressed mood, fatigue, neurasthenia, and poor attention and concentration (177; 179). In more advanced cases, lethargy, confusion, psychotic symptoms, spastic weakness of the limbs, and Babinski signs may be observed. Other findings may include aversion to bright light, glossitis, and dilated cardiomyopathy (130). A range of other less common medical and neurologic manifestations can also be seen as a result of alcoholic pellagra, either due to the pellagra or the chronic effects of alcohol, including, for example, black urine (from urobilinogen) (59), benign symmetrical lipomatosis (98), peripheral neuropathy (124), paratonia, and various forms of myoclonus (282; 284; 242).
Pellagrous encephalopathy in alcoholics is often overlooked, in part because it is frequently mistaken for alcohol withdrawal delirium or Wernicke encephalopathy (282; Oldham and Novic 2012; 196). Also, because the nutritional disorders of alcoholics are often mixed, alcoholic pellagra may be seen in combination with Wernicke encephalopathy or Marchiafava-Bignami disease, a situation likely to foster diagnostic confusion among clinicians (282; 243; 310).
Marchiafava-Bignami syndrome. Marchiafava-Bignami syndrome is a progressive alcoholism-associated neurologic syndrome of corpus callosum demyelination and necrosis and subsequent atrophy (294). The syndrome is thought to be due to a combination of vitamin B deficiency (including thiamine deficiency), malnutrition, and alcohol abuse (particularly large quantities of red wine) (134). The clinical presentation is varied, and premortem diagnosis was almost impossible before the era of modern neuroimaging (331; 289; 117). Some patients present with slowly progressive psychomotor slowing, incontinence, frontal release signs, and wide-based gait. Dysarthria, hemiparesis, apraxia, or aphasia may be present (162; 244). Occasionally, patients may present in stupor or coma. MRI or CT scans may reveal lesions in the corpus callosum, anterior commissure, and, less commonly, in the centrum semiovale (226) and lateral frontal cortex (145).
Alcoholic cognitive impairment (alcohol-related brain damage). Chronic alcohol abuse in the absence of nutritional deficiencies or organ failure has also been associated with changes in cognitive abilities in detoxified chronic alcoholics, with deficits in recent memory, visuospatial ability, abstract reasoning, speed of information processing, and novel problem solving (271; 171; 260). Commonly, neuropsychological testing shows a decline in performance IQ but not verbal IQ. Aphasia, apraxia, and agnosia are uncommon. Typically, the degree of impairment is mild to moderate, with most patients able to carry out daily activities. Widespread cognitive impairment is best encompassed within "alcohol-related brain damage" or "alcoholic cognitive impairment," with Korsakoff syndrome reserved for those with isolated or disproportionate memory impairment (166).
Alcoholic dementia. Alcohol-induced dementia is a syndrome characterized by deficits in memory and intellectual abilities severe enough to interfere with daily functioning. Although no formal diagnostic criteria have been established, Oslin and colleagues proposed that there must be a clinical diagnosis of dementia that remains at least 60 days after the last exposure to alcohol and a history of excessive alcohol consumption for greater than 5 years (237). This syndrome has multiple etiologies and presents with a range of clinical symptoms and abnormalities. Often, dementia attributed to alcoholism is actually dementia due to other etiologies present in an individual who drinks alcohol (248). There is evidence that extreme quantities of alcohol can cause dementia (40) and that low levels may be somewhat protective against dementia (31; 270) and death (84).
Hepatic encephalopathy. Hepatic encephalopathy is a metabolic disturbance associated with liver disease or portosystemic shunting. It is characterized by disturbances of consciousness. Most commonly, hepatic encephalopathy is a chronic disorder that occurs in the setting of alcoholic cirrhosis with portosystemic shunts. There is a wide range of presentations progressing successively through stages: (1) unimpaired cognitive function with intact consciousness; (2) lethargy and confusion/ delirium; (3) somnolence and disorientation; and (4) coma. In the early stages, mood disorders, sleep disturbances, and personality changes can be seen. Asterixis (ie, negative myoclonus, flapping tremor, liver flap) is a typical feature of presomnolent hepatic encephalopathy (02; 03; 04; 05; 286; 222). Various forms of intercurrent illness (eg, infections, gastrointestinal bleeding from esophageal varices, hypoxia, electrolyte disturbances, etc.) and some medications can precipitate abrupt worsening of hepatic encephalopathy.
Several scales have been proposed for assessing or staging hepatic encephalopathy (343). The most commonly used are the West Haven criteria to differentiate between four grades of clinically overt hepatic encephalopathy (340), often augmented by the addition of minimal hepatic encephalopathy (Table 1).
|
Grade |
Manifestations | |
|
Unimpaired |
• No encephalopathy | |
|
Minimal |
• Psychometric or neuropsychological alterations (eg, psychomotor speed, executive function) | |
|
West Haven criteria grade |
I. |
• Trivial lack of awareness |
|
II. |
• Lethargy or apathy | |
|
III. |
• Somnolence to semi-stupor | |
|
IV. |
• Coma | |
Alcoholic cerebellar degeneration. Alcoholic cerebellar degeneration is a disorder of progressive cerebellar degeneration, sometimes seen in patients with severe alcoholism (336). The rate of progression is variable. The anterior and superior vermis are preferentially affected, giving rise to a remarkably stereotypic syndrome of ataxic stance and gait. A wide-based gait and an inability to tandem walk are the most prominent signs. Limb ataxia, if present, occurs primarily in the legs. Arms are involved only to a slight extent, if at all. The gait disturbance usually develops over several weeks. Sometimes, a mild gait instability may be present for some time and then deteriorate suddenly after a bout of binge drinking or an intercurrent illness. The pathogenesis of this disorder is multifactorial and likely includes thiamine deficiency and the toxic effects of alcohol. The disorder predominantly affects middle-aged men (160). In autopsy series of decedents with a history of chronic ethyl alcohol abuse, alcoholic cerebellar degeneration was diagnosed in anywhere from 11% to 27% of cases (318; 352).
Results of brain trauma during alcohol intoxication. Alcoholic patients are prone to traumatic injuries of the brain and the peripheral nerves. Well-recognized central nervous system complications include subdural and epidural hematoma, cerebral contusion, and posttraumatic epilepsy. Compressive neuropathies may appear after a period of prolonged unconsciousness. These may involve the radial nerve at the spiral groove (Saturday night palsy), the peroneal nerve at the fibular head, or the sciatic nerve in the gluteal region.
Osmotic demyelination syndrome. Central pontine myelinolysis/extrapontine myelinolysis (CPM/EPM) and its association with alcoholic and malnourished patients was reported by Adams and colleagues in 1959 (06). They found that rapid changes in electrolyte concentration, most commonly of sodium, are associated with CPM/EPM. Today, these entities are also called the osmotic demyelination syndrome (261). Central pontine myelinolysis is a neurobehavioral disorder associated with rapid onset of quadriparesis, pseudobulbar palsy, pupillary abnormalities, behavioral changes, and sometimes coma (71; 314). Not all cases have documented hyponatremia (74).
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). Tobacco-alcohol optic neuropathy--part of the large group of nutritional and toxic optic neuropathies—is a rare disorder of decreased central vision associated with nutritional deficiencies and tobacco smoking (70; 297). Tobacco-alcohol optic neuropathy is characterized by bilateral visual disturbances with grossly diminished visual acuity, papillomacular bundle damage, symmetric scotomas, acquired disturbances of color vision, and mostly normal fundi (276; 174; 104; 28). In a series of 40 cases, central scotomas were present in 80%, whereas centrocecal scotomas (ie, located between the central point of fixation and the blind spot with a roughly horizontal oval shape) prevailed in the rest (174). The acquired disturbances of color vision usually involve the red-green sense (84%) (174). The amplitudes of the visual evoked potentials are typically reduced and deformed (174).
Alcoholic neuropathy. Among the most prevalent neurologic syndromes in alcoholism is a distal, predominantly sensory or sensorimotor polyneuropathy (29; 62; 214). Tingling or burning pain is often the symptom that brings the patient to medical attention. Dysesthesias are most prominent over the soles and toes and may be severe enough to interfere with walking. As the disease progresses, loss of sensation becomes more pronounced, and neuropathic pain often paradoxically lessens in severity. Neurologic examination reveals abnormally elevated sensory thresholds to vibration, temperature, and pinprick. Distal muscle atrophy and mild weakness are sometimes seen. Ankle tendon reflexes are diminished or absent. Romberg sign, gait disturbances, and more widespread areflexia, weakness, and sensory loss may be seen in advanced cases. Autonomic disturbances (eg, impotence, sweating abnormalities, and orthostatic hypotension) are common but often overlooked (214; 44). Rare, neuropathic "Charcot" joints may develop from deafferentation, and hoarseness may develop if the neuropathy involves the recurrent laryngeal nerves.
Pressure-induced focal neuropathies can also be linked to the development of myopathy (be considered as a secondary alcoholic neuropathy). The most common is a radial nerve palsy (ie, the so-called “Saturday night palsy” as it typically followed the carousing of a Saturday night) that resulted from radial nerve compression between the humerus and a hard object (eg, the arm of a park bench) during a drunken stupor.
Alcohol-induced (dry) beriberi. Some nutritionally compromised alcoholics may develop a subacute neuropathy due to thiamine deficiency—dry beriberi. Symptoms may evolve over a period of weeks or months. The most common presentation is flaccid weakness, and at its nadir, many cannot walk independently (218; 56; 136; 274; 76; 308). Other features include numbness/paresthesia, dysautonomia, vocal cord dysfunction, dysphagia, and nystagmus. Some may have concomitant Wernicke-Korsakoff syndrome (79).
Alcoholic myopathy. Alcohol can produce several myopathic disorders, including acute alcoholic myopathy with or without myoglobinuria, hypokalemic myopathy, chronic atrophic myopathy, and cardiomyopathy (245; 203; 332; 176; 182; 323; 94; 161). Acute alcoholic myopathy (also termed “alcoholic rhabdomyolysis and acute alcoholic necrotizing myopathy”) is an uncommon syndrome of abrupt muscle injury that typically occurs in malnourished chronic alcoholics following a binge or in the first days of alcohol withdrawal; experimental studies have demonstrated that both alcohol and nutritional factors are necessary to produce this syndrome (26; 176; 182). Severity ranges from asymptomatic transient elevation of creatine kinase to frank rhabdomyolysis with myoglobinuria. Although in most instances, full recovery occurs within days to weeks, death may occur in the setting of acute renal failure and hyperkalemia. Chronic alcoholic myopathy is a gradually evolving syndrome of proximal weakness, atrophy, and gait disturbance that frequently complicates years of alcohol abuse. Muscle strength correlates with lifetime consumption of ethanol. Recovery occurs if alcohol is avoided, but the timeframe of improvement is weeks to months, in contrast to the rapid recovery typical of acute alcoholic myopathy. Pathogenic mechanisms include impaired gene expression and protein synthesis as well as increased oxidative damage and apoptosis.
Traumatic or pressure-induced rhabdomyolysis resulting from a drunken stupor can also be considered a secondary alcoholic myopathy.
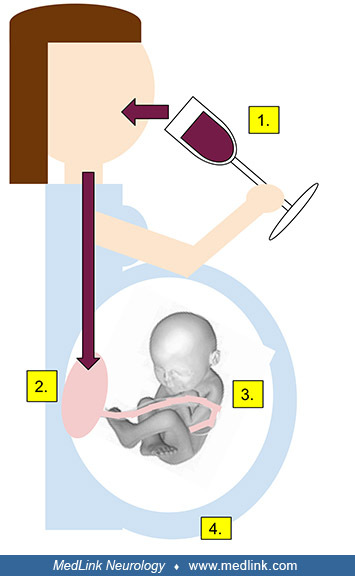
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Prenatal exposure to high levels of alcohol at critical periods of fetal development can impact neural crest development, disrupting the normal processes of facial and brain development, and potentially inducing birth defects that combine morphological stigmata (eg, facial dysmorphism) with neurologic and neuropsychological deficits (304; 224; 39; 175; 186; 191; 273; 299). The range of facial dysmorphism and neurologic and neuropsychologic disorders is called fetal alcohol spectrum disorder, of which fetal alcohol syndrome is the most severe form. The following table details the different fetal alcohol spectrum disorders.
|
Craniofacial dysmorphism | ||
|
• Small head circumference | ||
|
Behavioral problems | ||
|
• Aversion to social/eye contact | ||
|
Cognitive deficits | ||
|
• Mental retardation | ||
|
Neuropathology | ||
|
• Microcephaly | ||
|
| ||
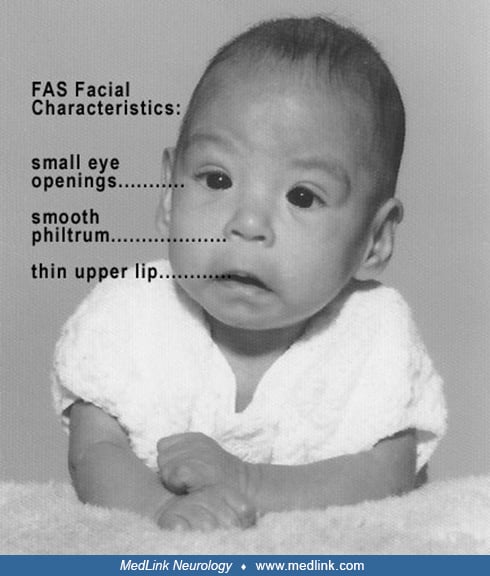
Diagnosis of fetal alcohol syndrome requires identification of a specific pattern of craniofacial dysmorphology (ie, short palpebral fissures; severe midfacial hypoplasia causing a “flat” midface; long flat or “deficient” philtrum; and a thin, elongated upper lip), but most individuals with behavioral and neurologic sequelae of heavy prenatal ethanol exposure do not exhibit defining facial characteristics (191; 299). Minor dysmorphic features can include ptosis, abnormal ear helices, “railroad track” ears, “hockey stick” palmar creases, and a turned-up nose. Short stature and microcephaly are common and may persist into adulthood. Almost half of affected children have significant learning disabilities, and most others have mild intellectual impairment. Speech delay and hyperactivity are common. Nonspecific mild neurologic deficits may include odor and taste abnormalities, fine motor control deficits, and hearing loss.
Brain malformations related to prenatal exposure to ethanol include microcephaly, agenesis/dysgenesis of the corpus callosum, cerebellar hypoplasia, a smaller hippocampus and basal ganglia, neuronal migration errors, holoprosencephaly, myelomeningocele, optic nerve hypoplasia, and hydrocephalus (267; 144; 200).
Children with fetal alcohol syndrome have a higher incidence of vision problems and eye pathology than normal children, including amblyopia, strabismus, hyperopia, anisometropia, and astigmatism (212; 330; 118). Compared with nonaffected children, children with fetal alcohol spectrum disorders showed an amblyopia-like pattern of vision deficit, including deficits in visual acuity, contrast sensitivity, and stereoacuity, in the absence of the optical and oculomotor disruptions of early visual experience that usually precede this condition (330). Evidence from animal models suggests that the deficits in spatial vision may be due to alterations in the functional architecture of the neocortex that occurs following prenatal alcohol exposure (330). Eyelid abnormalities are also more common in children with fetal alcohol syndrome, including blepharophimosis (a congenital anomaly in which the eyelids are underdeveloped such that they cannot open as far as usual and permanently cover part of the eyes), epicanthus (rounded, downward-directed fold of skin covering the caruncular area of the eye), telecanthus (an abnormally increased distance between the medial canthi), and ptosis (212; 118).
Alcohol-related central and peripheral nervous system dysfunction may improve with abstinence from alcohol and correction of malnutrition. The degree of recovery depends on the type of damage as well as the severity and chronicity of the disorder.
Alcohol withdrawal. Alcohol withdrawal is associated with increased cost, longer hospitalizations, and higher risk of medical complications and in-hospital mortality after acute ischemic stroke (07).
Wernicke-Korsakoff syndrome. With early recognition and rapid treatment with intravenous thiamine, Wernicke syndrome may resolve with mild or no sequelae. However, without prompt treatment, many survivors are left with severe amnesia (Korsakoff syndrome) (165). Among those with longstanding or recurrent encephalopathy, persistent cognitive dysfunction or the Korsakoff syndrome eventually emerges. In addition, untreated patients with Wernicke encephalopathy have a significantly increased risk of death, and most patients who succumb are still drinking at the time of death (45).
The prognosis of Korsakoff syndrome (and more specifically of Korsakoff disease) is generally bleak, and most patients are left with a permanent and devastating memory disorder, even if other neurologic features (eg, nystagmus, ataxia, and neuropathy) may improve markedly or even recover completely (93; 311; 198). Some limited recovery of the amnestic disorder may occur within 1 to 3 months and may continue for up to 1 year or more (311). Only about 20% of patients make a substantial recovery (335). Patients' lack of insight further complicates their behavioral management. Most patients with Korsakoff syndrome require institutionalization in some form because of the profound memory impairment (45). Patients may survive for many years, and their death is typically unrelated to the original neurologic disease. Cognitive test performance of detoxified alcoholic Korsakoff patients remains stable over at least 2 years; neither accelerated cognitive decline nor onset of dementia-like symptoms is observed (106).
Alcoholic pellagra. Alcoholic pellagra has a good prognosis for short-term recovery if promptly recognized and treated, but the issues that led to development of the condition in the first place (ie, severe alcoholism and malnutrition) are associated with significant long-term morbidity and mortality.
Marchiafava-Bignami syndrome. Marchiafava-Bignami syndrome has a high mortality rate, and survivors frequently have significant disability, though limited improvement is possible (134). A longer duration of alcohol consumption, inadequate thiamine supplementation, altered consciousness, pyramidal signs, and pneumonia are suggested predictors of a poor prognosis in patients with alcoholic Marchiafava-Bignami syndrome (350).
Alcoholic cognitive impairment. Perhaps the best prognosis is associated with the milder cognitive changes seen in chronic alcoholics with good nutrition. Several studies have shown that with months and years of abstinence, improvements in visuospatial ability and memory can occur, although older alcoholics are less likely to reach the performance levels of their nonalcoholic age cohort. Partial or complete reversal of the brain shrinkage seen on neuroimaging may also occur.
Alcoholic dementia. With alcohol-related dementias, prognosis varies according to the extent to which permanent brain damage has occurred.
Hepatic encephalopathy. The overall prognosis for recovery is poor in patients with alcoholic hepatic encephalopathy. Mortality is commonly due to sepsis, gastrointestinal hemorrhage, or raised intracranial pressure (45).
Alcoholic cerebellar degeneration. Alcoholic cerebellar degeneration causes irreversible damage, mostly involving the vermis, but also in other areas of the cerebellum (17).
Osmotic demyelination syndrome. The outcome in osmotic demyelination syndrome (CPM/EPM) is variable, and some cases may be asymptomatic: in an autopsy series of 220 consecutive decedents with chronic liver disease, 21 had CPM that was not clinically evident before death (295).
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). The outcome is variable and unpredictable.
Alcoholic neuropathy. In patients with peripheral neuropathy, dysesthesia often persists years after the initial manifestation, even with alcohol cessation. Unfortunately, tobacco-alcohol optic neuropathy is often underdiagnosed or only detected at a stage when full vision recovery is not possible (60).
Alcohol-induced (dry) beriberi. The prognosis of alcohol-induced dry beriberi is variable. Some recover promptly with thiamine (56), whereas others, with more advanced disease, may recover slowly and incompletely over a period of months to years (218; 136; 79; 76). Deaths may occur from acute heart failure (Shoshin beriberi) or pulmonary embolism (274).
Alcoholic myopathy. In acute alcoholic myopathy, massive muscle injury triggered by acute alcohol abuse may result in multiple organ failure and death. However, alcoholic rhabdomyolysis generally has a good prognosis if renal failure is avoided or is aggressively treated (18). Muscle has a remarkable capacity to regenerate, and most patients with alcoholic rhabdomyolysis recover full muscle function. Even patients who have had multiple episodes of myoglobinuria may have no lasting skeletal muscle effects. In patients with chronic alcoholic myopathy, improvement is also usual when ethanol is avoided.
Individuals with acute alcoholic muscular syndrome with muscle cramps recover within 2 to 4 weeks if they remain abstinent (246).
A 5-year study of the natural history of chronic alcoholic myopathy showed that only half of the sober patients recovered to normal strength, indicating that chronic alcoholic myopathy is only partially reversible. In some alcoholics, even a substantial reduction in alcohol consumption may be as effective as complete abstinence in improving muscle strength or preventing its deterioration (91).
Loss of paraspinal muscle mass is a male gender-specific consequence of cirrhosis that predicts complications and death (88). Loss of paraspinal muscle mass was an independent predictor of the occurrence of bacterial infections, spontaneous bacterial peritonitis, hepatic encephalopathy, and hepatorenal syndrome (88).
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Secondary disabilities (eg, school dropouts, criminal behavior, and substance/alcohol abuse problems) are common in young adults with fetal alcohol spectrum disorders (215).
Case 1. Wernicke-Korsakoff syndrome. A 48-year-old man with chronic alcoholism was brought to the emergency room after being found at home in a confused state. He was awake but disoriented. Examination showed a partial right abducens palsy and nystagmus in all directions of gaze. Tendon reflexes were normal in both arms and at both knees but absent at the ankles. Babinski sign was absent bilaterally. He was unable to walk independently and fell easily to either side. Brain CT showed mild atrophy of the cerebellar and cerebral hemispheres. Routine blood studies were notable for a serum sodium of 130 mg/dL. He was treated with intravenous fluids, thiamine, and multivitamins.
Repeat examination 2 weeks later showed an alert and coherent patient; however, he thought the year was 1978 and the president was Ronald Reagan. He could not recall events of the last several days but seemed undisturbed by his difficulties. His eye movements were full, with prominent end-gaze nystagmus. He had normal strength in the limbs. Finger-to-nose testing was performed normally, but there was mild dysmetria with heel-to-shin testing. He walked with a wide-based gait, and tandem gait was impossible. Sensory examination revealed severe impairment of vibratory sensation in the toes, normal joint position sensation, and mild dysesthesias to light touch distal to both ankles. Ankle jerks continued to be absent.
With abstinence from alcohol and resumption of a normal diet, his gait improved slowly over a year. He continued to have significant memory deficits and remained unable to function independently. He also complained of burning and tingling pain in the distal lower extremities and received only partial relief from amitriptyline.
Case 2. Cognitive and behavioral changes accompanied by a history of alcohol abuse. A 76-year-old right-handed man presented with a 4-year history of erratic behavior and memory difficulties. Before our evaluation, he had a 15-year history of heavy alcohol abuse that persisted after inpatient rehabilitation and participation in Alcoholics Anonymous. Approximately 1 year before the examination, he began to have difficulty learning his new area code and dialing phone numbers. Both his drive and interest in life were diminished. Although previously easygoing, he became explosive and irritable, often frightening his wife. Unexpectedly, he converted from a liberal Democrat to a fanatical follower of a right-wing political sect and spent much of his time associating with followers of this group. A few months before the evaluation, he had been caught having an extramarital affair. There was no history of stroke, head injury, or psychiatric disorder. His medications were nifedipine and enalapril.
On examination, the patient was noted to be bright and alert but showed no insight into his problems and blamed his wife for scheduling the doctor’s appointment. He was irritable, disinhibited, and somewhat explosive. Mini-Mental State Exam score was 23/30. On memory testing, he had a slow acquisition rate and a rapid rate of forgetfulness. Remote memory was good with cues. He interpreted proverbs concretely and made several perseverations when asked to perform frontal executive tasks. He had normal language and perfect naming but impaired word-list generation. There were mild visuospatial difficulties. Cranial nerves II through XII were normal, as were motor bulk, power, and tone. Sensory examination showed severe loss of position and vibration sense distal to the elbows and knees. Reflexes were decreased. Babinski signs were present bilaterally. Gait was normal. MRI scan of the brain showed mild to moderate ventricular dilatation and severe cortical atrophy without acute hemorrhage, midline shift, or focal lesions. SPECT scan demonstrated extensive bilateral frontal hypoperfusion and bilateral temporal hypoperfusion.
The patient’s memory loss was relatively mild but was consistent with early Alzheimer disease. Yet, his disinhibition and profound difficulty on frontal systems tasks, along with a SPECT scan with marked frontal involvement, were atypical for early Alzheimer disease and brought up the possibility of either an alcohol-induced frontal syndrome or a degenerative frontotemporal dementia.
Subsequently, the patient’s course was remarkable for progressive cognitive and behavioral difficulties and unrelenting alcohol consumption. Eventually, he became incontinent and required placement in a nursing home where he died. Autopsy revealed severe frontal and hippocampal atrophy and moderate temporal atrophy in the neocortex. On microscopic examination, moderate numbers of neuritic and diffuse plaques in the neocortex and moderate numbers of plaques in the hippocampus and entorhinal cortex, primarily on the right, were found. There was a marked loss of neurons in the CA-1 and subiculum of the hippocampus but no neurofibrillary tangles and no Pick bodies. Cerebellar structures were normal. Unfortunately, the mammillary bodies were not available for analysis. The patient’s plaque score was sufficient for a diagnosis of probable Alzheimer disease using CERAD criteria, despite atypical features like the absence of neurofibrillary tangles in the cortex and severe neuron loss in frontal cortex.
In retrospect, this gentleman suffered from a mixed disorder with Alzheimer disease leading to progressive memory loss and dementia. The alcohol abuse seems to have exacerbated the memory disturbance. The profound neuronal loss in the frontal lobes was not consistent with Alzheimer disease and may have been secondary to this patient’s chronic alcoholism (125). Hence, the behavioral deficits exhibited by this gentleman may truly represent the ongoing effects of alcohol abuse.
|
• In alcohol intoxication, ethanol fleetingly and weakly binds or interacts with receptor sites on various proteins in a manner similar to that of inhaled anesthetic agents, and it can also change or stabilize ion channels. | |
|
• Alcohol ingestion impairs cellular and molecular processes, causing neurodegenerative change through excitotoxicity, free radical formation, and neuroinflammatory damage. | |
|
• Alcohol dependence is a complex brain disorder, and its etiology encompasses a vast array of physiologic, immunologic, genetic, hereditary, social, and behavioral factors. | |
|
• Alcohol use disorder is a psychiatric diagnosis comprising both dependence and abuse; over 50% of patients treated for alcohol use disorders carry one or more additional psychiatric disorders. | |
|
• Tolerance develops with frequent or prolonged alcohol intoxication through both metabolic and functional mechanisms: metabolic tolerance refers to changes in the efficiency or capacity to metabolize ethanol, whereas functional tolerance refers to a lessened response to alcohol independent of the rate of alcohol metabolism. | |
|
• In contrast to prior notions that ethanol and other alcohols exerted their effects on the CNS by nonselectively disrupting the lipid bilayers of neurons, modern evidence has demonstrated that ligand-gated ion channels are primarily responsible for mediating the effects of ethanol. | |
|
• Wernicke-Korsakoff syndrome is due to thiamine deficiency. | |
|
• Korsakoff disease is most commonly associated with chronic alcohol abuse, in which case low dietary intake of thiamine is compounded by alcohol-induced impairments in thiamine absorption and metabolism. | |
|
• At autopsy, the major gross pathologic changes in Wernicke encephalopathy include petechial hemorrhages, grayish and reddish discoloration, and slight softening of the tissues in structures surrounding the third and fourth ventricles. | |
|
• The neuropathologic changes in Korsakoff syndrome are identical in distribution and histologic character to those of Wernicke encephalopathy, showing only the more chronic findings of the earlier pathologic processes. | |
|
• Alcoholic pellagra is caused by a deficiency of either nicotinic acid (ie, niacin/vitamin B3) or tryptophan, usually in combination with a lack of other amino acids and micronutrients. | |
|
• Chronic alcoholism is a risk factor for liver disease, particularly alcoholic cirrhosis with portosystemic shunting, which can result in hepatic encephalopathy. | |
|
• Various pathogenic mechanisms have been proposed for hepatic encephalopathy, including those that involve hyperammonemia, neuroinflammation, false neurotransmitters, and increased GABA neurotransmission. | |
|
• Malnutrition is probably not a significant factor in alcoholic cerebellar degeneration; instead, direct neurotoxicity of ethanol is suspected to be the cause of this disorder. | |
|
• Acute alcoholic myopathy is caused by severe alcoholic binges, usually in drinkers of long duration; in contrast, chronic alcoholic myopathy is caused by prolonged, consistent alcohol abuse rather than binge drinking. |
Alcohol intoxication. Ethanol fleetingly and weakly binds or interacts with receptor sites on various proteins in a manner similar to that of inhaled anesthetic agents, and it can also change or stabilize ion channels.
Inhibitory GABA and glycine receptor subunits are promising candidate genes associated with alcohol dependence, and these subunits, along with a group of ligand-gated ion channels, may be the target site responsible for acute intoxication. Over time, with chronic intake, various physiologic adaptations occur (128; 321).
Excessive ethanol is toxic to the nervous system (58; 77). Moderate alcohol exposure can induce angiogenesis through induction of vascular endothelial growth factor (116). This may explain the cardiovascular-protective effects of modest alcohol consumption. Ethanol, under experimental conditions, has wide-ranging effects on gene expression (229) and various neuronal constituents, including lipid membranes, receptors for GABA N-methyl-D-aspartate (290; 351) and 5-hydroxytryptamine (355), ion channels, G-proteins (53; 291), and second messengers.
Alcohol ingestion impairs cellular and molecular processes, causing neurodegenerative change through excitotoxicity, free radical formation, and neuroinflammatory damage. These neurodegenerative changes may involve membrane proteins, neurotransmitters, ion channels, and signaling pathways (08; 65). Alcohol-induced dysfunction or dysregulation of microglia (which play a major role in immune responses to cerebral insults) may induce or exacerbate neurotoxicity (131).
Thirty ounces of an 86-proof alcoholic beverage contain 2250 calories, or approximately 100% of the daily caloric requirement. These are empty calories, as a typical alcoholic beverage contains a negligible amount of protein, vitamins, and minerals; therefore, serious malnutrition is prevalent in people with alcoholism. On the other hand, adequate diet or even nutritional supplements do not prevent many neurologic complications of alcoholism.
Except for acute alcohol intoxication and alcohol-withdrawal syndrome, neurologic complications of alcoholism are likely due to either nutritional deficiency, a direct toxic effect of ethanol, or secondary effects from prior metabolic derangement or cumulative neurologic trauma acquired during bouts of alcohol intoxication (58; 77).
Most ingested ethyl alcohol is oxidized to acetaldehyde in the liver. Acetaldehyde is further metabolized to acetic acid, subsequently forming acetyl coenzyme A. During this process, via the cytosolic oxidative pathway of ethyl alcohol metabolism, NAD is reduced to NADH via alcohol dehydrogenase. NADH, the reduced form of NAD, accumulates in mitochondria, hindering mitochondrial metabolism.
A fraction of ingested alcohol is metabolized before reaching the bloodstream, a phenomenon known as first-pass metabolism (283). The elimination of first-pass metabolism following gastrectomy and gastric bypass increases blood alcohol concentrations, the bioavailability of alcohol, and the risk of alcohol-related diseases (283). Compared with a control group of participants who did not undergo surgery and had equivalent age, body max index, and alcohol drinking patterns, women with sleeve gastrectomy had a shorter Tmax, higher peak blood alcohol concentration, and greater area under the curve but a similar systemic alcohol elimination rate.
(A) The sleeve gastrectomy (SG) group included 12 women, and the control group included nine women. (B and D) Area under the curve (AUC) for concentration-time curves for both groups. Whiskers indicate the standard error of the...
In the subset of participants who were matched for Tmax to control for gastric emptying rate, the area under the curve was increased by 34% in the sleeve gastrectomy group. These findings suggest that alcohol first-pass metabolism occurs in the stomach in women and provide a plausible mechanism for the observed increase in alcohol-related disease after bariatric surgery (156; 209).
As alcohol is metabolized, a "hangover" may develop with characteristically unpleasant symptoms, including headache, nausea, fatigue, and drowsiness. Fast elimination of ethanol is associated with experiencing less severe hangovers (197). The mechanism of hangover headache (veisalgia cephalgia) is unknown (205) but has long been attributed to the formation of acetaldehyde during the metabolism of ethanol. However, (1) there is a direct relationship between blood ethanol concentration and hangover severity, whereas this association is not significant for acetaldehyde (197); and (2) direct administration of acetate in rats increased nociceptive behaviors, suggesting that acetate, not acetaldehyde, accumulation results in hangover-like hypersensitivity (205). In addition, an inflammatory process may be involved in the pathogenesis of hangover headaches: hangover severity is significantly and positively correlated with blood concentrations of biomarkers of the inflammatory response to alcohol [interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP)] (325). Four hours after alcohol consumption, blood ethanol concentration, but not acetaldehyde, is significantly and positively associated with elevated levels of IL-6, suggesting a direct inflammatory effect of ethanol. In addition, biomarkers of oxidative stress (ie, malondialdehyde and 8-isoprostrane) are significantly correlated with hangover severity, suggesting that oxidative stress also contributes to the inflammatory response. Regardless of the exact metabolites and processes involved, a hangover may be alleviated by altering the rate of alcohol metabolism and facilitating elimination of metabolites by affecting the activity of alcohol dehydrogenase (ADH) or aldehyde dehydrogenase (ALDH) enzymes. For example, in a rat model, rosiglitazone alleviated the symptoms of ethanol-induced hangover by inducing ALD2 expression (149).
Alcohol dependence. Alcohol dependence is a complex brain disorder, and its etiology encompasses a vast array of physiologic, immunologic, genetic, hereditary, social, and behavioral factors. A spectrum of use and abuse exists, and progression to more severe levels does not always occur. Habitual intake can lead to abuse, resulting in conflicts in a person’s work and social environments. Abuse can wax and wane over time. Chronic severe abuse can lead to health, legal, and financial problems. Addiction is the end result.
Alcohol use disorder. Alcohol use disorder is a psychiatric diagnosis comprising both dependence and abuse; over 50% of patients treated for alcohol use disorders carry one or more additional psychiatric disorders (303). Two of the genes most strongly suspected of engendering a decreased risk of abusing alcohol are alcohol dehydrogenase types 2 and 1B (82). Thus far, genome-wide association studies have yielded inconsistent results, reinforcing the genetic complexity of alcohol use disorder. A 2015 meta-analysis of twin and adoption studies reported a heritability of approximately 50% (329).
Alcohol withdrawal. Tolerance develops with frequent or prolonged alcohol intoxication through both metabolic or functional mechanisms; metabolic tolerance refers to changes in the efficiency or capacity to metabolize ethanol, whereas functional tolerance refers to a lessened response to alcohol independent of the rate of alcohol metabolism.
In contrast to the prior notions that ethanol and other alcohols exerted their effects on the CNS by nonselectively disrupting the lipid bilayers of neurons, modern evidence has demonstrated that ligand-gated ion channels are primarily responsible for mediating the effects of ethanol (72).
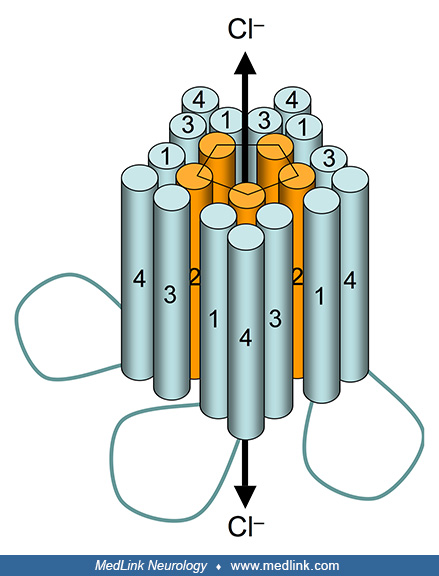
In particular, gamma-aminobutyric acid type A (GABAA) receptors occupy a central role in mediating the effects of ethanol on the CNS. GABA is the primary inhibitory neurotransmitter in the mammalian CNS; consequently, activation of GABAA receptors by GABA decreases neuronal excitability. Ethanol produces both short- and long-term modulation of GABAA receptors (72). Not only does ethanol enhance inhibitory GABAergic neurotransmission (194), but it also suppresses excitatory glutamate release by inhibiting NMDA receptors (87; 48).
Chronic alcohol ingestion leads to increased release of endogenous opiates, activation of GABAA receptors, up-regulation of NMDA-type glutamate receptors (141; 320), increased dopaminergic transmission, and increased serotonin release. When chronic consumption suddenly ceases, GABAA inhibitory actions are decreased, and NMDA excitatory actions are increased, resulting in increased neurotransmitter excitotoxicity effects that can result in hallucinations, tremor, seizures, delirium, and increased body temperature, heart rate, and blood pressure.
In addition to alcohol (ethanol), other positive allosteric modulators of GABAA receptors include benzodiazepines, barbiturates, meprobamate, methaqualone, zolpidem, and propofol. In particular, the ionotropic GABAA receptor protein complex is also the molecular target of the benzodiazepine class of tranquilizer drugs.
Because they act through similar mechanisms, alcohol (ethanol), benzodiazepines, barbiturates, and other CNS “depressants” can produce cross-tolerance. Withdrawal from these substances can produce similar withdrawal syndromes.
Wernicke-Korsakoff syndrome. Wernicke-Korsakoff syndrome is due to thiamine deficiency. Korsakoff disease is most commonly associated with chronic alcohol abuse, in which case low dietary intake of thiamine is compounded by alcohol-induced impairments in thiamine absorption and metabolism (301; 281; 356). There may also be a contribution from an age-related vulnerability to diencephalic amnesia produced by thiamine deficiency (251).
At autopsy, the major gross pathologic changes in Wernicke encephalopathy include petechial hemorrhages, grayish and reddish discoloration, and slight softening of the tissues with neuronal loss, gliosis, and vascular damage in structures surrounding the third and fourth ventricles and the cerebral aqueduct (318; 172). This anatomically localized capillary dysfunction is due to thiamine deficiency and is not a direct toxic effect of alcohol (17). Regions most prominently affected are the mammillary bodies, dorsal medial nucleus of the thalamus, periaqueductal region, and the tegmentum of the pons. The encephalopathic component of Wernicke encephalopathy is associated with leakage of capillaries around the third ventricle, whereas the ophthalmoplegia and ataxia are secondary to hemorrhages around the aqueduct of Sylvius in the midbrain and the fourth ventricle in the medulla. Cortical changes are frequently, but not universally, reported and are seen as ventricular enlargement and sulci widening, especially in the frontal lobe (331). These pathological changes may occur rapidly, with the onset of clinically apparent Wernicke encephalopathy, or insidiously, without clinically apparent signs of Wernicke encephalopathy.
The neuropathologic changes in Korsakoff syndrome are identical in distribution and histologic character to those of Wernicke encephalopathy, showing only the more chronic findings of the earlier pathologic processes. Bilateral, symmetrically placed, punctate lesions in the area of the third ventricle, fourth ventricle, and aqueduct are the hallmarks of Korsakoff disease. Lesions in the midbrain and cerebellum are responsible for the neurologic symptoms of the Wernicke stage, whereas lesions in the diencephalon (including the mammillothalamic tract) are critical for the amnesia that characterizes Korsakoff disease (353). The characteristic neuropathology of Korsakoff disease includes neuronal loss, microhemorrhages, and gliosis in the paraventricular and periaqueductal grey matter. Interactions involving the thalamus, mammillary bodies, hippocampus, frontal lobes, and cerebellum are important for new memory formation and executive function; the impairment of these circuits significantly contribute to the cognitive defects found in Korsakoff disease (150; 250). However, the minimal lesion(s) necessary for severe amnesia has not been resolved. The mammillary bodies are affected in approximately 80% of cases; however, shrinkage of the mammillary bodies and frontal lobes is visible on MRI for both patients with Korsakoff disease and non-Korsakoff alcoholics (143; 86), even if it tends to be more severe in those with Korsakoff disease. Victor and colleagues suggested that damage to the dorsomedial nucleus of the thalamus is essential in the causation of memory deficits of Korsakoff disease (335), whereas others have implicated both the mammillary bodies and other thalamic nuclei in the anterior and midline areas, including the preretinal nucleus (199; 43; 123; 326; 165; 173). Neurodegeneration of the anterior thalamic nuclei is apparently a characteristic and specific feature in alcoholics with Korsakoff syndrome (123; 326).
Alcoholic pellagra. Alcoholic pellagra is caused by a deficiency of either nicotinic acid (ie, niacin/vitamin B3) or tryptophan, usually in combination with a lack of other amino acids and micronutrients (282; 331; 177; 179). Inadequate intake of either niacin or tryptophan was historically most common in areas where corn or millet (with a high leucine content) was the primary foodstuff, but endemic pellagra is now rare because cereals and bread are supplemented with niacin. Secondary deficiency may occur due to certain medications, diarrhea, alcoholism, cirrhosis, or some combination of these. Alcoholics with pellagra have a higher prevalence of protein malnutrition than nonpellagrous alcoholics, as reflected in greater frequencies of anemia and hypoalbuminemia and in lower serum potassium levels (68). Depression in pellagrins may be due to a serotonin deficiency caused by decreased tryptophan availability to the brain (22). Alcoholism can induce or aggravate pellagra by (1) inducing malnutrition, gastrointestinal disturbances, and B vitamin deficiencies; (2) inhibiting the conversion of tryptophan to niacin; and (3) promoting the accumulation of the heme precursor 5-aminolaevulinic acid and porphyrins (154; 22).
Marchiafava-Bignami disease. In Marchiafava-Bignami disease, there is demyelination of the corpus callosum. The pathogenesis of Marchiafava-Bignami disease is unknown, but the underlying pathologic changes in the corpus callosum resemble those of central pontine myelinolysis with selective destruction of white matter; in addition, Marchiafava-Bignami disease has rarely been seen in reported alcohol abstainers, suggesting that other factors (eg, nutritional) may play a role. A link between abuse of red wine and the disorder has been suggested (220).
Alcoholic dementia. Chronic and excessive alcohol use has both direct and indirect effects on central nervous system function. A small study suggested a possible association between reduced brain glucose metabolism and cortical thickness in alcohol use disorder (315). Whether or not the direct effects of alcohol abuse can independently produce a dementia syndrome continues to be debated (192). If an alcohol dementia truly exists, it is often mild enough that the ability to perform activities of daily living is retained. There is a much stronger consensus that indirect effects of alcoholism produce dementia. These indirect etiologies include metabolic thiamine or niacin deficiency. Marchiafava-Bignami disease, which is associated with the degeneration of the corpus callosum, is also considered a complication of alcohol, possibly red wine.
The biological bases for alcohol-induced dementia vary. Alcohol quickly crosses the blood-brain barrier, and both alcohol and its major metabolite, acetaldehyde, can be neurotoxic. Prolonged use seems to lead to neuronal loss, neuroglial proliferation, dendritic simplification, and reduction in synaptic complexity. The frontal and parietal lobes may be particularly sensitive to the effects of alcohol, although this is far from proven. Studies using well-nourished animals have shown ethanol-induced reductions in choline acetyltransferase, cell loss in the nucleus basalis, and altered dendritic spines in the hippocampus (57). In humans, these effects are observable on CT and MRI as enlargement of the lateral and third ventricles and mild to moderate sulcal widening. The fact that the atrophy is reversible after sustained periods of abstinence argues that some of these changes are due to shrinkage rather than outright cell death. Cerebellar degeneration, particularly in the region of the anterior and superior vermis, is also commonly seen in chronic alcoholics (317).
Hepatic encephalopathy. Chronic alcoholism is a risk factor for liver disease and particularly alcoholic cirrhosis with portosystemic shunting, which can result in hepatic encephalopathy. Various pathogenic mechanisms have been proposed for hepatic encephalopathy, including those that involve hyperammonemia, neuroinflammation, false neurotransmitters, and increased GABA neurotransmission. Arterial blood ammonia concentrations are typically elevated in hepatic encephalopathy, with fluctuating ammonia levels roughly correlating with the severity of clinical disease, although a small proportion of patients develop symptoms with normal blood ammonia levels. Moreover, measures that act to reduce ammonia levels are helpful in treating hepatic encephalopathy.
There are no gross abnormalities in the brains of patients who die due to hepatic coma. Microscopic examination shows diffuse hyperplasia of Alzheimer type 2 astrocytes of the cerebral cortex and subcortical grey matter structures (eg, basal ganglia, thalamus, dentate nuclei, and brain stem nuclei).
These pathologic cells have a large vacuolated nucleus with chromatin displaced to one side.
Alcoholic cerebellar degeneration. Malnutrition is probably not a significant factor in alcoholic cerebellar degeneration; instead, direct neurotoxicity of ethanol is suspected to be the cause of this disorder. Pathologic changes in alcoholic cortical cerebellar degeneration involve particularly the anterior and superior portions of the cerebellar vermis and hemispheres.
Osmotic demyelination syndrome. Central and extrapontine myelinolysis, disorders of the osmotic demyelination syndrome, are uncommon demyelinating disorders most frequently involving the pons, midbrain, cerebellum, and lateral geniculate body. CPM/EPM is almost always associated with comorbid chronic conditions, including chronic alcoholism, various causes of hypo- and hypernatremia, liver transplantation, lung infections, malignancies, hemodialysis, and central nervous system dysfunction (159). The underlying etiology of these disorders is complex and not yet clearly delineated; however, there is a strong association with osmotic stress, with the most common predisposing factor being hyponatremia in 78% of cases (261; 138; 292). The aquaporin water channels have also been implicated (252).
Neuroimaging most commonly reveals symmetric demyelinating lesions in the base of the pons, midbrain, cerebellum, lateral geniculate nucleus, deep gray nuclei, spinal cord, and cerebral cortex (322). Neuropathologic features include relatively well-preserved cell bodies with demyelinated axons in the pons and occasionally lesions in the striatum, thalamus, cerebellum, and cerebral white matter (57). Rojiani and colleagues described the following types of lesions: spongy (early demyelination), vacuolar (reactive), and hypercellular (265).
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). Tobacco-alcohol optic neuropathy typically occurs in nutritionally deficient patients who abuse both tobacco and alcohol (28; 113; 23). The pathogenesis of tobacco-alcohol amblyopia has been controversial but may involve alterations of methionine and S-adenosyl-L-methionine metabolism, with contributions from cyanide in tobacco smoke, vitamin deficiencies (eg, folate, B12, thiamine), and other nutritional deficiencies (70; 114; 115).
Many have subsumed the outdated term tobacco-alcohol optic neuropathy under the larger group of nutritional amblyopia (297), and more recently, the improved terms nutritional optic neuropathy (113; 309; 114) or toxic-nutritional optic neuropathy (28; 23). Indeed, some have argued that tobacco-alcohol is a misnomer because the condition is primarily mediated by nutritional deficits and not ethanol toxicity per se (52; 113), even though the same argument could be leveled at other nutritional neurologic disorders resulting from alcohol abuse combined with a nutritionally deficient diet (eg, Wernicke-Korsakoff syndrome and alcoholic pellagra). Identified nutritional defects in patients with tobacco-alcohol optic neuropathy include deficiencies of folate (vitamin B9), cobalamin (vitamin B12), and thiamine (vitamin B1) (103; 342; 70; 104). The role of cyanide in tobacco smoke is unclear because serum thiocyanate levels in "tobacco amblyopes" are relatively reduced compared with healthy smokers (102); therefore, if cyanide derived from tobacco smoke is indeed a causative factor in the development of tobacco-alcohol optic neuropathy, the biochemical defect may be a failure of conversion of cyanide to thiocyanate, its chief detoxification product (298; 102).
Although optic neuropathy is generally assumed in such cases, there is little evidence to suggest that the locus of pathology is restricted to the optic nerve. MRI of the optic nerve is typically normal (153), and electrophysiological evidence indicates that some affected people likely have retinal dysfunction involving the macula (28). Moreover, histopathological studies in animal models showed lesions in the retina, optic nerve and tract, and the maculopapillary bundle (263; 297).
Alcoholic neuropathy. The etiology of alcoholic polyneuropathy is unresolved. In the first half of the 20th century, many authors suggested that alcoholic polyneuropathy resulted from vitamin deficiency and, eventually, by the late 1930s (following the synthesis of thiamine in 1926) that it resulted from thiamine deficiency specifically (285; 213; Jolliffe and Colbert 1936). One study of 37 alcoholic patients with neuropathy found that the underlying pathology by electrophysiological studies and nerve biopsy was typically due to axonal degeneration, whereas cases of nutritional neuropathy showed evidence of segmental demyelination (29). More than 60% of these showed no clinical evidence of malnutrition, but the pattern of abnormalities was the same in nerves from alcoholic patients with nutritional deficiency as in those without nutritional deficiency. However, the clinical-pathologic similarity to beriberi (neuropathy due to thiamine deficiency) suggests that nutritional factors, and particularly thiamine deficiency, play a role in some cases (69; 232). The difference may simply depend on the degree of malnutrition and the amount of thiamine supplementation received by individual patients.
Alcohol-induced (dry) beriberi. Alcohol-induced dry beriberi is due to thiamine deficiency, resulting from nutritional compromise in an alcoholic patient.
Alcoholic myopathy. Acute alcoholic myopathy is caused by severe alcoholic binges, usually in drinkers of long duration. Chronic alcoholic myopathy is caused by prolonged, consistent alcohol abuse rather than binge drinking. Although myopathy and neuropathy are frequently both present in chronic alcoholics, the pathogenetic mechanisms involved in each of these forms of tissue damage are presumably not identical because histologic and clinical evidence of myopathy may occur in the absence of symptoms or electrophysiologic signs of neuropathy or malnutrition (203; 323). Nutritional factors may contribute to the pathogenesis of chronic alcoholic myopathy because muscle weakness and histologic myopathy among alcoholics are both more severe in the presence of malnutrition (227).
The pathogenesis of alcoholic myopathy involves multiple interrelated pathways (254; 253; 255). First, impaired gene expression and protein synthesis, as well as increased oxidative damage, act together to reduce the formation of myofibrillar proteins (254; 255). Second, impaired gene expression, increased oxidative damage, and the pro-apoptotic properties of alcohol act to increase apoptosis. Third, additional muscle damage comes from alterations in ion channels and cell membrane permeability, impaired energy metabolism, protein adduct formation, and the toxicity of fatty acid ethyl esters. All of these processes contribute to the death and loss of myocytes and, hence, to progressive myopathy (95).
Fetal alcohol spectrum disorder. Structural alterations associated with prenatal alcohol exposure include decrements in global and regional brain volume, isocortical volume, isocortical thickness, and isocortical surface area with prenatal alcohol exposure, along with alterations in brain shape and symmetry, and disruption of interhemispheric functional connectivity (206; 300; 348; 186; 268; 187). Prenatal alcohol exposure can also produce structural brain abnormalities that involve specific structures in the cerebellum, basal ganglia, and midline corpus callosum (262; 38; 206). Prenatal alcohol exposure is also a risk factor for developing more severe brain abnormalities, including holoprosencephaly (36; 267; 64; 327; 135). Malnutrition is probably not a significant factor in fetal alcohol spectrum disorder; instead, direct neurotoxicity of ethanol is the suspected cause of this disorder.
The developmental neurotoxicity of ethanol may be due to the disruption of L1 cell adhesion molecule signal cascades. Cell adhesion molecules (CAMs) are critical for guiding neural development. Most cell adhesion molecules have been grouped into three families, all of which participate in differential adhesion, signal transduction, and physical/mechanical effects: (1) cadherins; (2) integrins; and (3) members of the immunoglobulin superfamily, including the cell surface transmembrane glycoprotein L1 (L1CAM) (27; 357). L1 mediates adhesion, neurite extension, neuronal migration, and fasciculations that are necessary for proper development of synaptic connections (221). Mutations in the L1 gene on the X chromosome are responsible for multiple allelic X-linked neurologic conditions: (1) X-linked hydrocephalus (hydrocephalus due to stenosis of aqueduct of Sylvius or HSAS) (240); (2) MASA syndrome (The acronym "MASA" stands for four of the signs and symptoms associated with the syndrome: (a) mental retardation, (b) "aphasia" [speech delay], (c) shuffling gait, and (d) adducted thumbs [with cleft palate, microcephaly, dysmyelination, and possibly hydrocephalus]) (100); (3) complicated spastic paraplegia type 1 (SP-1); (4) X-linked agenesis of the corpus callosum; and (5) "CRASH syndrome": corpus callosum hypoplasia, mental retardation, adducted thumbs (or "aphasia" [speech delay]), spastic paraparesis, and hydrocephalus (221; 240; 357).
The observation that patients with fetal alcohol syndrome share similar features to patients with the CRASH syndrome led to the investigation of the effects of ethanol on L1, and ultimately to recognition that alcohol inhibits cell-cell adhesion mediated by L1 (258; 27; 278). For example, concentrations of ethanol achieved in blood and brain after ingesting a single alcoholic beverage are sufficient to inhibit L1-mediated neurite outgrowth in cerebellar granule neurons (258). Such inhibition of L1-mediated neurite outgrowth may result from decreased expression, altered cell surface distribution, impaired signal transduction, or impaired interaction with the cytoskeleton (27).
|
• A genetic predisposition for alcohol abuse has been demonstrated by adoption studies and studies of identical twins and families of individuals with early-onset alcoholism. | |
|
• The low concordance between clinical and pathological diagnosis of Wernicke-Korsakoff syndrome may result from a combination of factors: (1) under-recognition of the disorder by clinicians; (2) pathological changes occurring with subclinical disease. | |
|
• Approximately half of chronic alcoholics have a loss of lean body mass due to loss of muscle tissue, decreased muscle strength, and associated gait abnormalities. | |
|
• Between one third and two thirds of chronic alcoholics have skeletal muscle myopathies, making alcoholic myopathies the most prevalent type of myopathy. | |
|
• Alcoholic rhabdomyolysis predominates in men by a greater than 4-to-1 ratio, whereas in chronic alcoholic myopathy, males and females are equally affected. | |
|
• After exposure to heavy alcohol consumption during pregnancy, 80% of children exhibit abnormalities associated with alcohol exposure. |
About 15 to 19 million Americans may be problem drinkers, and approximately 88,000 adults die yearly due to alcohol-related causes (01). An estimated one in eight total deaths among adults in the U.S. aged 20 to 64 years were attributable to excessive alcohol use, including one in five deaths among adults aged 20 to 49 years (89). The average annual number of deaths from excessive alcohol use increases with age and is greater for men than women (90).
Estimated percentage of total deaths attributable to excessive alcohol use among adults aged 20 to 49 years, United States, 2015 to 2019. (Source: Esser MB, Leung G, Sherk A, et al. Estimated deaths attributable to excessive al...
Average annual number of deaths from excessive alcohol use by age group and period among males (A) and females (B). Deaths from excessive alcohol use includes all decedents whose deaths were attributed to conditions that were f...
A genetic predisposition has been demonstrated by adoption studies, studies of identical twins, and families of individuals with early-onset alcoholism (338). First-degree relatives of alcoholics are seven times more likely to develop alcoholism than the general population.
Alcoholism affects all socioeconomic levels of society and appears to have a strong genetic predisposition (256), although environmental and cultural influences also clearly play a role (338). Adolescents are at increased risk for alcohol abuse disorders and other neuropsychiatric disorders during the adult years when they participate in binge drinking (67).
In a study of 110 participants with a history of alcohol consumption for at least 5 years and clinical evidence of chronic alcoholic liver disease, patients with neurologic manifestations had consumed alcohol for a significantly longer duration (mean 13.9 ± 6.1 years) compared to those without (mean 9.6 ± 4.6 years) (287). Peripheral neuropathy was the most prevalent neurologic manifestation, affecting 85% of participants.
In a 9-year study of 750 children admitted with coma, the etiology of toxic coma was dominated by alcohol and other abused substances (302).
Alcoholism is an independent risk factor for recurrent hypertensive intracerebral hemorrhage (358).
Alcohol-related seizures. Alcohol withdrawal is the most common cause of acute symptomatic seizures seen in an emergency room setting, accounting for about three quarters of such cases (259). Chronic alcoholism and alcohol consumption are risk factors for developing a first generalized tonic-clonic seizure (188). The risk of seizures increases with increasing (current) alcohol use, independent of alcohol withdrawal (225); for unprovoked seizures, the risk of seizures increases in a dose-dependent manner with daily alcohol intake (225).
Wernicke-Korsakoff disease. Alcoholism and alcohol-related nutritional deficiency account for approximately 80% of the cases of Wernicke encephalopathy seen in a hospital setting (211). The incidence of Korsakoff disease has been estimated at approximately 10 per million first psychiatric admissions (55). However, several large autopsy studies have found neuropathological changes consistent with Wernicke-Korsakoff syndrome in 2% to 3% of postmortem examinations, suggesting a much higher prevalence of this syndrome (317; 127; 238). Most of these cases had never received a clinical diagnosis of Wernicke-Korsakoff syndrome. The low concordance between clinical and pathological diagnosis of Wernicke-Korsakoff syndrome may result from a combination of factors: (1) underrecognition of the disorder by clinicians; (2) pathological changes occurring with subclinical disease (328); and (3) Wernicke-Korsakoff encephalopathy superimposed on a more general dementia picture.
Alcoholic pellagra. Alcoholism is a risk factor for nutritional disorders, including pellagra and Wernicke encephalopathy (41). Contributing factors to the development of pellagra in alcoholics include homelessness, failure to eat regularly, and some medications (eg, isoniazid for tuberculosis) (154; 249; 312; 190).
Alcohol-related dementia. In alcohol-related dementia, the frequency of alcohol-related problems varies according to how the problem is defined and which group is being studied. For the elderly, the group most likely to develop a dementia, the prevalence of alcohol problems ranges between 1% and 6% of all community residents, between 10% and 15% of elderly seeking medical attention, and between 28% and 44% of elderly admitted to psychiatric units (99). However, the incidence and prevalence of alcohol-related dementia syndromes have not been clearly established. Heavy alcohol use is reported in 21% to 24% of patients with dementia, but in many of these cases, the primary diagnosis is Alzheimer disease, and the role of alcohol abuse is equivocal. However, alcohol use may increase the risk or severity of dementia, as is noted in the clinical vignette (Case 2) above and in the study of Saunders and colleagues (277). Within an elderly population, moderate drinkers have an odds ratio of 1.8 for incident dementia and 2.8 for an Alzheimer diagnosis relative to nondrinkers (236). A lifetime history of heavy drinking is associated with a 4-fold increase in dementia diagnoses (237), although moderate drinking may diminish one’s chances of cognitive decline in late life (108).
Alcohol-induced (dry) beriberi. Alcohol abuse is a significant risk factor for developing dry beriberi (56; 228).
Alcoholic myopathy. Approximately half of chronic alcoholics have loss of lean body mass due to loss of muscle tissue, decreased muscle strength, and associated gait abnormalities (266). Between one- and two-thirds of chronic alcoholics have skeletal muscle myopathies, making alcoholic myopathies the most prevalent type of myopathy (255).
Heavy alcohol consumption and sedentary lifestyle are independently associated with reduced skeletal mass index in cirrhotic patients (63). The skeletal mass index is commonly used for diagnostic purposes because it includes central skeletal muscles whose mass is independent of activity and water retention, and it corresponds best to the patient’s total muscle mass (63; 266). The skeletal mass index, or more precisely the L3 skeletal muscle index, is determined with the use of abdominal CT scans. On these, the transverse area of the abdominal and paraspinal wall muscles (psoas, erector spinae, quadratus lumborum, transversus abdominis, internal and external oblique muscles and rectus abdominis) is measured at the level of the third lumbar vertebra; this measurement in square centimeters is then divided by the patient’s height squared to obtain the index result.
Alcohol is a common cause of nontraumatic myoglobinuria. Alcoholic rhabdomyolysis predominates in men by a greater than 4-to-1 ratio and usually occurs in the fourth to sixth decades of life. Recurrent episodes are reported in about 30% of cases following an initial episode of rhabdomyolysis. The incidence and prevalence of alcoholic rhabdomyolysis have varied considerably in different studies, probably at least in part due to variations in the intensity and chronicity of drinking and coexistent nutritional disorders among the subjects studied. Most studies suggest that alcoholic rhabdomyolysis is infrequent in alcoholics, occurring in less than 5% of chronic alcoholics. Nevertheless, the rhabdomyolytic variant of acute alcoholic myopathy represents the most common nontraumatic cause of rhabdomyolysis in hospitalized patients.
In chronic alcoholic myopathy, males and females are equally affected. Quantitative strength testing and histologic studies indicate that muscle abnormalities consistent with chronic alcoholic myopathy are common in habitual drinkers, affecting 40% to 60% of patients who chronically abuse alcohol (223).
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Assessment of the effects attributable to prenatal exposure to alcohol is complicated by the difficulties in verifying and quantitating exposure from a group of mothers who almost universally smoked tobacco, often abused other substances, and had poor or nonexistent prenatal care (144); thus, fetal alcohol spectrum disorder in humans is not a monotoxic disorder (144).
After exposure to heavy alcohol consumption during pregnancy, 80% of children exhibit abnormalities associated with alcohol exposure (175); maternal binge drinking and high total weekly intake of alcohol pose the greatest risk of adverse fetal outcomes (175).
|
• Abstinence from alcohol or treatment of alcoholism are the only effective preventive measures for the neurologic complications of alcohol abuse. | |
|
• The means by which alcohol abstinence is achieved in the alcoholic population includes medications (eg, disulfiram, calcium carbimide, naltrexone), support groups (eg, "Alcoholics Anonymous"), specialized addiction treatment programs, and more controversial programs stressing moderation versus total abstinence. | |
|
• Thiamine supplementation of some staple food products (eg, flour) is an easy and safe measure that can potentially improve the thiamine reserve of different human groups and function as a means of primary prevention of thiamine-deficiency disorders. | |
|
• In patients with Wernicke encephalopathy, rapid and aggressive treatment with parenteral thiamine is critical to prevent progression to Korsakoff disease and also to prevent fatal outcomes; this should be done before administration of glucose-enriched fluids. | |
|
• Niacin supplementation of some staple food products (eg, flour, cereals) is an easy and safe measure that can potentially improve the niacin reserve of different human groups and function as a means of primary prevention of niacin-deficiency disorders, including alcoholic pellagra. | |
|
• Total abstinence from alcohol during pregnancy is recommended because of the potential of alcohol-related teratogenic effects (fetal alcohol spectrum disorder and fetal alcohol syndrome). |
Abstinence from alcohol or treatment of alcoholism is the only effective preventive measure for the neurologic complications of alcohol abuse. The means by which abstinence is achieved in the alcoholic population includes medications (eg, disulfiram, calcium carbimide, naltrexone), support groups (eg, "Alcoholics Anonymous"), specialized addiction treatment programs, and more controversial programs stressing moderation versus total abstinence (230).
Wernicke-Korsakoff syndrome. Thiamine supplementation of some staple food products (eg, flour) is an easy and safe measure that can potentially improve the thiamine reserve of different human groups and function as a means of primary prevention of thiamine-deficiency disorders (126).
Because alcohol abuse is the primary cause of Korsakoff disease, moderation of or abstinence from alcohol is an important preventative measure. However, because alcoholics often have multiple presentations with thiamine-deficient states and because they often present after significant neurologic damage has occurred, prevention opportunities are constrained even with optimal recognition and medical management. The possibility of thiamine deficiency should be considered carefully in all patients with a history of alcoholism, malnutrition, or protracted vomiting, with an emphasis on low-threshold diagnosis and prompt treatment (354).
In patients with Wernicke encephalopathy, rapid and aggressive treatment with parenteral thiamine is critical to prevent progression to Korsakoff disease and also to prevent fatal outcomes. This should be done before administration of glucose-enriched fluids. Given proper vitamin supplementation, the confusional state seen in Wernicke encephalopathy typically clears, with marked improvement or resolution of the oculomotor manifestations, ataxia, and peripheral neuropathy (beriberi) (288). Effective treatment and prophylaxis may only be achieved by using parenteral vitamin supplements because oral supplements may not be adequately absorbed (66).
Alcoholic pellagra. Niacin supplementation of some staple food products (eg, flour, cereals) is an easy and safe measure that can potentially improve the niacin reserve of different human groups and function as a means of primary prevention of niacin-deficiency disorders, including alcoholic pellagra.
Alcoholic dementia. Nontraumatic alcohol-related dementia syndromes can be prevented with adequate nutrition, including vitamin supplementation, and limited alcohol consumption.
Alcohol-induced (dry) beriberi. The preventive measures for Wernicke-Korsakoff encephalopathy also apply to alcohol-induced (dry) beriberi.
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Total abstinence from alcohol during pregnancy is recommended because of the potential of alcohol-related teratogenic effects (fetal alcohol spectrum disorder and fetal alcohol syndrome).
(1) Alcohol consumed, (2) alcohol crosses into the placenta, (3) alcohol metabolizes, (4) byproducts of alcohol metabolism are detected in meconium. When alcohol crosses through the placenta, it is circulated throughout the fet...
Alcohol abuse and encephalopathy. A careful history of alcohol consumption and nutritional status are of primary importance in the differential diagnosis. The differential diagnosis of alcohol abuse and encephalopathy should include alcohol withdrawal, alcohol withdrawal delirium, delirium tremens, Wernicke-Korsakoff syndrome, head trauma, postictal confusion, hepatic encephalopathy (decompensated alcoholic liver disease), and various causes of ischemic, structural, metabolic, infectious, or toxic encephalopathy. Because patients under the influence of alcohol are prone to head injury from falls and other accidents, it is important to rule out a traumatic etiology.
Alcohol withdrawal seizures. Differential diagnoses include epilepsy, psychogenic nonepileptic seizures, convulsive syncope, and nonconvulsive syncope.
Wernicke-Korsakoff syndrome. Persistent global amnesia can occur as a consequence of a number of neurologic disorders, including cerebrovascular accidents, rupture and surgical repair of anterior communicating artery aneurysms, encephalitis, and anoxia. The presentation of the amnesic syndrome may be strikingly similar regardless of etiology; therefore, these neurologic conditions should all be considered in the differential diagnosis. Traumatic brain injury may occasionally result in a selective memory disorder as well.
A history of Wernicke encephalopathy facilitates a confident diagnosis of Korsakoff disease as a chronic sequela; however, Korsakoff disease can occasionally occur without signs of a preceding Wernicke encephalopathy. Attention to the course of illness is critical because the memory disorder seen in Alzheimer disease is progressive, whereas it is stable in established Korsakoff disease.
MRI can be useful in the diagnosis of the acute stages of Wernicke encephalopathy, demonstrating hyperintense signals in the mammillary bodies, colliculi, periventricular gray matter, fornix, and thalamus via fluid-attenuated inversion recovery (FLAIR) (307). Neuroimaging findings suggestive of lesions around the third and fourth ventricles support a diagnosis of Korsakoff syndrome, although the absence of such findings does not rule out the possibility that the underlying neuropathology is associated with thiamine deficiency.
Neuropsychological testing is often useful in providing evidence in support of Korsakoff disease. Patients with Korsakoff syndrome often have memory impairment that is disproportionately severe and, unlike Alzheimer disease, performance on IQ tests can fall in the average range. Failure to release from proactive interference is a characteristic feature of the Korsakoff memory deficit, although this deficit may also be observed in patients with frontal lobe pathology.
Alcoholic pellagra. Pellagra should be considered in the differential diagnosis of patients with chronic alcoholism, malnutrition, and amino acid imbalance, especially in those with dermatitis on sun-exposed skin surfaces, chronic diarrhea of unknown etiology (47), or encephalopathy. Pellagrous encephalopathy in alcoholics is often overlooked, in part because it is frequently mistaken for alcohol withdrawal delirium or Wernicke encephalopathy (282; Oldham and Novic 2012; 196).
Marchiafava-Bignami syndrome. Marchiafava-Bignami disease may be incorrectly diagnosed as a decompensation of a psychiatric disorder (eg, schizophrenia) (42). MRI is helpful for diagnosing Marchiafava-Bignami syndrome if demyelination or necrosis of the corpus callosum is observed. Gadolinium enhancement is suggested to be useful in the determination of severe disease (341). Additional clinical symptoms can also include bilateral frontal lobe symptoms, language deficits, gait disturbance, incontinence, and hallucinations. Lesions involving the corpus callosum can also be seen in high-altitude cerebral edema, antiepileptic drug withdrawal, hypoglycemia, hyperglycemia, and infection (109).
Alcoholic dementia. Severe memory disorders can be seen in the context of selective amnestic disorders like Wernicke-Korsakoff syndrome or as part of more global intellectual deterioration. A diagnosis of alcoholic dementia is warranted if patients demonstrate, in addition to amnesia, severe deficits in conceptual and problem-solving abilities as well as marked impairments on visuospatial and visuo-constructive tasks (275; 260). In contrast to the acute onset of Korsakoff disease, the global cognitive deficits seen in alcoholic dementia usually develop more gradually. Differential diagnosis is more difficult when the memory impairment has progressed gradually without a clear-cut history of encephalopathy.
The co-occurrence of dementia and alcohol abuse requires a comprehensive assessment to establish a proper diagnosis. In cases of dementia, the possibility of a primary dementia like Alzheimer disease must be considered. Like many patients with alcohol-induced dementias, patients with Alzheimer disease present with insidious onset, gradual progression, prominent memory loss, and deficits in naming, word generation, visuospatial ability, abstract reasoning, and mental flexibility. It is difficult to definitively rule out the possibility of Alzheimer disease in patients with the above presentation, regardless of the severity of the alcohol history. With sustained abstinence from alcohol, however, significant improvement in cognition and functional ability and the absence of any further deterioration argues against Alzheimer disease pathology.
Frontotemporal dementia should also be considered, especially if disinhibition, impaired social behavior, poor insight, or impaired executive function are early symptoms. Because many patients with frontotemporal dementia develop a craving for carbohydrates, increased alcohol abuse may be secondary to, rather than a cause of, the dementia. As with Alzheimer disease, the differential diagnosis may depend on the patient’s clinical course once abstinence from alcohol has been achieved.
Because alcoholics place themselves at risk for a host of dementia-causing disorders, these too must be considered in the differential diagnosis. For example, alcoholics are at increased risk for cerebral vascular disease. Although hemorrhagic stroke and large vessel infarcts will have a rapid onset, subcortical ischemic vascular disease may also develop gradually and insidiously without clear clinical signs of stroke. Subdural hematomas can also cause dementia, and in the case of alcoholics, a clear history of head injury may not be obtainable. Subdural hematomas typically progress more rapidly than the primary progressive dementias. Electrolyte abnormalities, most notably hyponatremia, are common in the alcoholic population. Alcoholism sometimes coexists with the substance abuse of other drugs. The alcoholic patient population is also at an increased risk of trauma and infection.
As with any dementia workup, it is important to rule out delirium. This is particularly important in patients with a history of alcohol abuse because they are at risk not only for metabolic encephalopathies but also for alcohol intoxication delirium, alcohol withdrawal delirium, alcoholic hallucinosis, and alcohol-related seizures.
Hepatic encephalopathy. Other encephalopathic conditions related to acute and chronic alcoholism can mimic hepatic encephalopathy, including alcohol intoxication, alcohol withdrawal/delirium tremens, Wernicke-Korsakoff syndrome, and pellagra encephalopathy. Other toxic and metabolic encephalopathies should also be considered because alcoholics are at an increased risk of multiorgan dysfunction, electrolyte disorders, sepsis, and nonalcohol toxin exposures.
Alcoholic cerebellar degeneration. Alcoholic cerebellar degeneration is recognized by its subacute course and its propensity to affect the gait with little or no limb involvement. These clinical features, together with radiological and laboratory studies, help to distinguish it from other causes of cerebellar dysfunction, such as intoxication by various drugs, cerebellar ischemia or hemorrhage, viral cerebellitis, posterior fossa neoplasms or abscesses, hypothyroidism, and paraneoplastic cerebellar degeneration. It should also be noted that Torvik and colleagues found evidence of age-dependent cerebellar atrophy in their study of 67 nonalcoholic male patients. They recommended that the diagnosis of alcoholic cerebellar degeneration in patients older than 70 years be “made with caution” (319).
Osmotic demyelination syndrome. Central pontine myelinolysis and Marchiafava-Bignami are relatively rare disorders but should be considered in the differential diagnosis of dementia. Both have a more rapid onset and course than a primary dementia and cause more physical symptoms. In central pontine myelinolysis, symptoms evolve over days to weeks, and patients may have progressive quadriparesis, pseudobulbar palsy, behavioral changes, conjugate gaze palsies, and impaired tongue movements. Cerebrospinal fluid may show elevated pressure and increased protein (77). Demyelination in the base of the pons should be observable on MRI. However, it is noteworthy that several studies revealed that MRI could be misleading in the case of pontine lesions. Some of these lesions were ultimately found to be due to a deep interpeduncular cistern or ischemic changes. Multiple sclerosis and metastases should also be considered (158).
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). Because tobacco-alcohol optic neuropathy and Leber hereditary optic neuropathy (LHON) have a similar phenotype, known Leber hereditary optic neuropathy-associated mutations should be analyzed before establishing a tobacco-alcohol amblyopia diagnosis (167; 309).
Alcoholic neuropathy. Alcoholic polyneuropathy presents as a distal, symmetrical, sensorimotor polyneuropathy. As such, it is indistinguishable from the neuropathies associated with a wide range of common systemic disorders, such as diabetes mellitus, uremia, drug usage, hypothyroidism, and AIDS. Therefore, diagnosing alcoholic polyneuropathy depends on the exclusion of other causes and on the documentation of improvement with abstinence. Disulfiram, a drug widely used in the rehabilitation of alcoholism, also causes a peripheral neuropathy at a dose greater than 125 mg per day (239).
Alcohol-induced (dry) beriberi. Alcohol-induced (dry) beriberi is often confused with alcoholic neuropathy due to direct toxicity of alcohol and Guillain-Barre syndrome (218; 210; 274; 76; 308).
Alcoholic myopathy. In patients with symptoms suggesting acute alcoholic myopathy, the possibility of other acquired causes of rhabdomyolysis should be considered, such as drug abuse (eg, heroin, cocaine, amphetamines), trauma or crush injury, and depletion of phosphate or potassium. In patients with recurrent episodes of rhabdomyolysis, the possibility of an underlying metabolic defect in carbohydrate or lipid metabolism should be considered.
In patients with symptoms suggesting chronic alcoholic myopathy, care must be taken to exclude other causes of chronic myopathic weakness, such as inflammatory myopathies, acquired metabolic myopathies, and limb-girdle dystrophy. The muscle biopsy finding of type II muscle fiber atrophy is also a feature of steroid myopathy, hypophosphatemia, and muscle disuse.
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Because of the potentially serious implications for fetal development (fetal alcohol spectrum disorder and fetal alcohol syndrome), pregnant women should abstain entirely from alcohol.
|
• A complete history should be taken with particular emphasis on alcohol use and nutritional status, protracted vomiting or diarrhea, evidence of an acute episode of Wernicke encephalopathy, and any associated features. | |
|
• The prompt diagnosis of Wernicke encephalopathy is critical, as delayed treatment may lead to irreversible neurologic deficits. | |
|
• Wernicke encephalopathy is an acute neurologic condition classically characterized by the clinical triad of (1) nystagmus with ophthalmoparesis, (2) ataxia, and (3) confusion. Unfortunately, this classic triad is insensitive for the diagnosis of Wernicke encephalopathy. | |
|
• Modified diagnostic criteria for Wernicke encephalopathy require at least two of the following four signs: (1) dietary deficiencies (undernutrition, vitamin deficiency); (2) oculomotor abnormalities (ophthalmoplegia, nystagmus, gaze palsy); (3) cerebellar dysfunction; and (4) either an altered mental state or mild-to-moderate memory impairment or confabulation. | |
|
• In Marchiafava-Bignami disease, MRI shows demyelination and possible areas of focal necrosis in the corpus callosum. | |
|
• No liver function test abnormalities are diagnostic of or specific for hepatic encephalopathy, although elevated blood ammonia levels suggest a diagnosis of hepatic encephalopathy in the proper clinical setting. | |
|
• EEG abnormalities in hepatic encephalopathy include bilateral synchronous delta waves and triphasic waves, particularly in the frontal regions, but these findings may also be seen in other toxic-metabolic encephalopathies, and there is only a rough correlation between the degree of EEG abnormality and the severity (or stage) of hepatic encephalopathy. | |
|
• In alcoholic cerebellar degeneration, brain imaging shows atrophy of the anterior lobes of the cerebellum and the superior vermis. | |
|
• In acute alcoholic myopathy, creatine kinase is moderately or severely increased in serum, whereas in chronic alcoholic myopathy, creatine kinase is normal. | |
|
• Diagnosis of fetal alcohol syndrome requires identification of a specific pattern of craniofacial dysmorphology (ie, severe midfacial hypoplasia, shortening of the palpebral fissures, an elongated upper lip, and deficient philtrum), but most individuals with behavioral and neurologic sequelae of heavy prenatal ethanol exposure do not exhibit defining facial characteristics. |
A complete history should be taken with particular emphasis on alcohol use and nutritional status, protracted vomiting or diarrhea, evidence of an acute episode of Wernicke encephalopathy, and any associated features. Information should also be obtained regarding other medical conditions that may be part of the differential diagnosis (eg, encephalitis, anterior communicating artery aneurysm, head injury, tumor) or that may be precipitating conditions (ie, contributing to the development of thiamine deficiency). A complete physical and neurologic examination is necessary.
Initial laboratory evaluation should also include complete blood count, blood coagulation times, serum electrolytes, liver function tests, blood urea nitrogen, creatinine, bilirubin, serum osmolality, serum alcohol, and urine toxicology screen. Serum osmolality is correlated with alcohol concentration because 100 mg/dL of blood alcohol raises serum osmolality by 22 mOsm/L. Urine toxicology screening is prudent because alcoholics frequently abuse multiple substances. Other laboratory studies (eg, arterial blood gases, vitamin B12 level, thyroid function tests, creatine kinase, etc.) are necessary only if indicated by the clinical condition.
Not infrequently, serious alcoholics ingest other toxic neuroactive substances. Alcoholics sometimes ingest methanol, or ethylene glycol instead of ethanol, or "moonshine" tainted with methanol or heavy metals (eg, lead or arsenic) (233). Ethylene glycol and methanol cause a severe metabolic acidosis with increased anion and osmolal gaps. Ketoacidosis is not a feature of ethylene glycol or methanol poisoning (208).
Focal neurologic findings or unexplained encephalopathy should prompt imaging studies of the brain and, where appropriate, examination of cerebrospinal fluid for evidence of infection or subarachnoid blood.
Wernicke-Korsakoff syndrome. The prompt diagnosis of Wernicke encephalopathy is critical, as delayed treatment may lead to irreversible neurologic deficits. Wernicke encephalopathy is an acute neurologic condition classically characterized by the clinical triad of (1) nystagmus with ophthalmoparesis, (2) ataxia, and (3) confusion. Unfortunately, the classic triad is insensitive for diagnosis of Wernicke encephalopathy (127; 219). In particular, the incidence of oculomotor findings is low in patients later shown to have had Wernicke encephalopathy. In pathological series, there is a consistently high proportion of cases of Wernicke encephalopathy that never had a clinical diagnosis of that condition during life (127; 219; 45).
To address the poor sensitivity of the classic triad, modified diagnostic criteria have been developed. Modified diagnostic criteria for Wernicke encephalopathy require at least two of the following four signs; (1) dietary deficiencies (undernutrition, vitamin deficiency); (2) oculomotor abnormalities (ophthalmoplegia, nystagmus, gaze palsy); (3) cerebellar dysfunction; and (4) either an altered mental state or mild-to-moderate memory impairment/confabulation (45). In a clinicopathologic study of 28 patients, the modified diagnostic criteria improved diagnostic sensitivity from 31% based on the classic triad to 100% compared with pathological diagnoses (45).
|
Requires two of four signs in chronic alcoholics | ||
|
• Dietary deficiencies (eg, malnutrition, vitamin deficiency, low body mass index) | ||
|
• Oculomotor abnormalities (eg, ophthalmoparesis, nystagmus, gaze palsy) | ||
|
• Cerebellar dysfunction (eg, ataxia, balance deficits, dysmetria) | ||
|
• Altered metal status or impaired memory (eg, disorientation, coma, confusion, memory problems, confabulation) | ||
|
| ||
MRI is helpful in diagnosis of Wernicke encephalopathy (140; 132; 16). MRI studies may show symmetric hyperintense signal in paraventricular regions of the thalamus, hypothalamus, mamillary bodies, periaqueductal region, and floor of the fourth ventricle on T2-weighted imaging, FLAIR, and diffusion-weighted imaging (DWI) (140; 132).
Korsakoff disease is a clinical diagnosis based on identifying the characteristic amnesic syndrome, usually following an episode or episodes of Wernicke encephalopathy and occurring in the context of a history of alcohol abuse, malnutrition, or protracted vomiting. A diagnosis of Korsakoff disease can be made only when the confusional state associated with Wernicke encephalopathy has sufficiently cleared. All cognitive functions should be thoroughly evaluated to characterize the pattern and severity of the memory disorder and to screen for the possibility of a general intellectual decline. A diagnosis of Korsakoff syndrome (or more specifically of Korsakoff disease) is warranted only when the memory deficits are much more prominent than other cognitive disorders in the domains of language, visuoperceptual functioning, problem-solving, and judgment (45).
Laboratory tests showing elevation of blood pyruvate levels and reduced transketolase activity in erythrocytes support a diagnosis of Wernicke-Korsakoff syndrome.
Alcoholic pellagra. Alcoholic pellagra is underrecognized. Because endemic pellagra has been eradicated in Western countries, a diagnosis of pellagra is rarely considered, even with risk factors for malnutrition such as chronic alcohol intake, homelessness, or AIDS (234). In a Japanese study, 20 cases of neuropathologically diagnosed pellagra were identified among 74 necropsy cases of chronic alcoholism (142). A therapeutic response to niacin in a patient with the typical symptoms and signs of pellagra establishes the diagnosis.
Marchiafava-Bignami disease. In Marchiafava-Bignami disease, MRI shows demyelination and possibly areas of focal necrosis in the corpus callosum.
Alcohol-related dementia. Diagnostic evaluation for dementia in an alcoholic should include neuroimaging, laboratory tests, and possibly neuropsychological evaluation and EEG. Brain imaging in alcoholic dementia typically shows diffuse cortical atrophy with enlargement of the lateral ventricles; these findings are not specific to alcoholic dementia, however. Brain imaging also helps to exclude alcohol-related traumatic brain injuries, such as subdural hematomas and cerebral contusions. Neuropsychological evaluation should be conducted to measure the degree of cognitive and intellectual impairment and the extent to which it interferes with daily functioning. EEG shows nonspecific loss of background activity with increased theta and delta activity, particularly in the temporal leads bilaterally.
Hepatic encephalopathy. No liver function test abnormalities are diagnostic of or specific for hepatic encephalopathy, although elevated blood ammonia levels suggest diagnosis of hepatic encephalopathy in the proper clinical setting.
Some patients with cirrhosis of the liver, who appear to be otherwise normal on general clinical examination, show impairment on neuropsychological tests of psychomotor speed and executive function—a condition referred to as “minimal hepatic encephalopathy” (24). Reitan's trail-making test is simple and enables serial assessment of the mental state.
EEG abnormalities in hepatic encephalopathy include bilateral synchronous delta waves and triphasic waves, particularly in the frontal regions, but these findings may also be seen in other toxic-metabolic encephalopathies, and there is only a rough correlation between the degree of EEG abnormality and the severity (or stage) of hepatic encephalopathy.
The primary utility of brain imaging in patients with suspected hepatic encephalopathy is excluding other relevant intracranial pathology. Brain imaging may also show cerebral edema in the acute stages of fulminant hepatic encephalopathy and may show cerebral atrophy in chronic cases.
Alcoholic cerebellar degeneration. In alcoholic cerebellar degeneration, brain imaging shows atrophy of the anterior lobes of the cerebellum and the superior vermis. Thyroid function tests should also be considered for individuals with cerebellar ataxia.
Osmotic demyelination syndrome. In central pontine myelinolysis, MRI shows demyelination in the basis pontis.
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). Tobacco-alcohol optic neuropathy is often underdiagnosed or only detected at a stage when the full recovery of vision is not possible (60). Workup of suspected nutritional optic neuropathy in an alcoholic should include MRI of the visual pathways, vitamin levels (B12, folate, and thiamine), and cyanide levels (309). In addition, because tobacco-alcohol optic neuropathy and Leber hereditary optic neuropathy (LHON) have a similar phenotype, known LHON-associated mutations should be analyzed before establishing a tobacco-alcohol amblyopia diagnosis (167; 309). Visual loss may precede optic disc changes detected by optical coherence tomography (152).
Alcoholic neuromuscular disease. Electromyography and nerve conduction studies help to characterize neuropathy or myopathy. Serum vitamin B12 and thyroid function tests are useful for evaluating neuropathy, and serum creatine kinase tests are useful for assessing myopathy. Thyroid function tests should also be considered for individuals with chronic myopathy.
In acute alcoholic myopathy, creatine kinase is moderately or severely increased in serum. The workup should include a careful history and a toxicology screen to identify the possible contribution of other drugs or toxins known to produce myoglobinuria. Screening for metabolic derangements that can cause myoglobinuria, particularly hypokalemia and hypophosphatemia, should be performed. Other causes of pigmenturia (eg, hematuria, hemoglobinuria, and porphyria) should be excluded. In an individual with recurrent rhabdomyolysis or evidence of muscle injury that has occurred apart from alcohol abuse, perform an evaluation for a possible inborn error of muscle metabolism. EMG shows myopathic potentials (short duration, small-amplitude units with normal recruitment) and a high frequency of spontaneous discharges (fibrillations and positive sharp waves) attributed to hyperirritability of muscle fibers associated with active muscle fiber necrosis (231). Muscle biopsy, if performed, shows muscle fiber necrosis (247).
In chronic alcoholic myopathy, creatine kinase is normal. In patients with symptoms suggesting chronic alcoholic myopathy, a family history of muscle disease should be excluded, and laboratory screening should determine whether an underlying endocrine or electrolyte disorder exists. Electromyography and muscle biopsy are necessary to exclude other causes of chronic proximal weakness and atrophy. EMG often shows mixed myopathic and neuropathic features with variable spontaneous discharges (231). The most frequent histological findings are myocytolysis, fiber-size variability, and type IIB-fiber atrophy (247). Cardiomyopathy and hepatic cirrhosis are more frequent in patients with chronic alcoholic myopathy and should be checked for in chronic alcoholics with skeletal myopathy (272).
In alcohol-induced dry beriberi, nerve conduction studies may show an acute motor axonopathy superimposed on a background polyneuropathy or myopathy (218; 136). Clinical features favoring dry beriberi over Guillain-Barre syndrome include the following: (1) 3 or more weeks of symptoms; (2) associated confusion, nystagmus, or vocal cord dysfunction; (3) volume overload; (4) low serum thiamine level; (5) elevated lactate; (6) normal spinal fluid (including normal CSF protein); and (7) absence of a sural-sparing pattern in nerve conduction studies (218; 136; 274). Sural sparing--defined as absent/abnormal median sensory nerve action potential (SNAP) amplitude or absent/abnormal ulnar SNAP amplitude with a normal sural SNAP amplitude--is thought to be a marker for inflammatory demyelinating polyneuropathy.
Nerve conduction studies in patients with alcoholic myopathies often show coincident neuropathic abnormalities (ie, borderline or slowed conduction velocities and absent sensory potentials) because distal neuropathies often coexist with myopathy.
Fetal alcohol spectrum disorder and fetal alcohol syndrome. Fetal alcohol spectrum disorder and fetal alcohol syndrome are frequently underdiagnosed, due to an absence or subtlety of classical facial features, mild neurodevelopmental sequelae, societal attitudes, and suboptimal performance characteristics of current diagnostic criteria. Diagnosis of fetal alcohol syndrome requires identification of a specific pattern of craniofacial dysmorphology (ie, severe midfacial hypoplasia, shortening of the palpebral fissures, an elongated upper lip, and deficient philtrum), but most individuals with behavioral and neurologic sequelae of heavy prenatal ethanol exposure do not exhibit defining facial characteristics (191; 299). Variations of diagnostic criteria exist, contributing to diagnostic confusion (349). Consequently, efforts are ongoing to develop better diagnostic criteria to capture the full spectrum of dysmorphology associated with fetal alcohol spectrum disorders.
Four diagnostic systems for fetal alcohol syndrome and other fetal alcohol spectrum disorders have been developed in North America: (1) The Institute of Medicine (IOM) guidelines (306); (2) The University of Washington diagnostic scheme, "The 4-Digit Diagnostic Code," which ranks the four key features of fetal alcohol spectrum disorders on a 4-level Likert scale and yields 256 descriptive codes that can be categorized into 22 distinct clinical categories, ranging from fetal alcohol syndrome to no findings (20; 21; 19); (3) The Centers for Disease Control guidelines, "Fetal Alcohol Syndrome: Guidelines for Referral and Diagnosis" (32); and (4) the Canadian guidelines for fetal alcohol spectrum diagnoses, which harmonized most differences between The Institute of Medicine and University of Washington's systems (61).
|
• Three drugs are approved by the U.S. Food and Drug Administration for the treatment of alcohol abuse and alcohol dependence: disulfiram, acamprosate, and naltrexone. | |
|
• Benzodiazepines are recommended as a first-line medication for the management of alcohol withdrawal to alleviate discomfort and agitation and to prevent seizures and delirium. | |
|
• All alcoholic patients should be supplemented with parenteral thiamine and other vitamins. | |
|
• Withdrawal seizures are typically self-limited. | |
|
• Benzodiazepines, and not nonbenzodiazepine anticonvulsants, should be used following an alcohol withdrawal seizure to prevent further alcohol withdrawal seizures. | |
|
• Withdrawal seizures do not represent "latent" epilepsy; therefore, treatment with nonbenzodiazepine anticonvulsants is not recommended. If all convulsions are clearly linked to withdrawal, chronic anticonvulsant therapy is not indicated. | |
|
• Chronic management of alcoholic patients prone to recurrent seizures is complicated by the difficulty in separating alcohol-related convulsions and preexisting epileptic conditions. | |
|
• Patients at high risk of Wernicke encephalopathy (eg, malnourished, severe withdrawal) should be given 3 to 5 days of parental thiamine. | |
|
• In patients with suspected Wernicke encephalopathy, parenteral thiamine should be administered twice daily for 5 days. | |
|
• When pellagra is suspected, treatment with oral nicotinamide is an inexpensive, safe, and potentially life-saving intervention. | |
|
• Management of acute exacerbations of chronic hepatic encephalopathy includes identification and correction of precipitating factors, such as correction of diuretic-induced hypokalemia and reduction of the nitrogenous load in the intestine (eg, reducing protein content in the diet, halting gastrointestinal bleeding from esophageal varices, clearing blood and other nitrogenous substances from the colon, etc.). | |
|
• Acute rhabdomyolysis with myoglobinuria requires urgent inpatient interventions to monitor and maintain renal function (with hemodialysis if necessary) and to avoid or correct hyperkalemia. |
Chronic alcoholism. Three drugs are approved by the U.S. Food and Drug Administration for the treatment of alcohol abuse and alcohol dependence: disulfiram, acamprosate, and naltrexone (54; 155; 269). Disulfiram (Antabuse), the first medicine approved for the treatment of alcohol abuse and alcohol dependence, works by causing a severe adverse reaction: when someone taking the medication consumes alcohol, they get nauseated and vomit, creating a deterrent to drinking (54). A meta-analysis of acamprosate and naltrexone supports their efficacy in relapse reduction (147). Nalmefene, an opioid receptor antagonist similar in chemical composition to naltrexone, has been approved by the European Medicine Agency; it is not available in the United States to treat alcohol dependence (264). Gabapentin, a GABA analogue used for treating epilepsy and nerve pain, is being considered as another possible candidate (204). Other nonbenzodiazepine anticonvulsants such as carbamazepine, valproic acid, and topiramate may also eventually prove helpful both in the treatment of alcohol withdrawal and the prevention of alcohol relapse (37).
Promising developments in the treatment of chronic alcoholism include preliminary studies demonstrating the relationship between the administration of exogenous ghrelin and a decrease in serum leptin levels, which may play a role in decreasing alcohol cravings in humans (119). Another double-blind, placebo-controlled human study of 46 patients found evidence suggesting a role of the ghrelin system in connection to alcohol craving and seeking in alcohol use disorder (120).
The single most important therapeutic factor in treating neurologic complications of alcohol abuse in the long term is abstinence from alcohol. Therefore, an alcohol treatment program is a critical component of treatment. Besides alcoholism rehabilitation and nutritional supplementation, treatment of persistent neurologic abnormalities due to chronic ethanol abuse is largely symptomatic.
Alcohol intoxication and withdrawal. Acute management of alcohol intoxication and delirium tremens is primarily supportive. Cardiovascular collapse and respiratory failure occur with a sufficiently high level of intoxication. In delirium tremens, prolonged fever and sweating may also result in fluid loss and secondary hypotension.
Agitation and autonomic disturbances may be treated with benzodiazepines (eg, oral or intravenous chlordiazepoxide, 25 to 100 mg, with a maximum of 300 mg in the first 24 hours). Recommendations from the World Health Organization advocate supported withdrawal from alcohol in patients with alcohol dependence (346). Mild withdrawal symptoms can be treated with carbamazepine or gabapentin on an outpatient basis (313). Benzodiazepines are recommended as a first-line medication in a hospital setting for the management of alcohol withdrawal with moderate-to-severe symptoms to alleviate discomfort and agitation and to prevent seizures and delirium. Long-acting benzodiazepines (eg, chlordiazepoxide or diazepam) are preferred over shorter-acting ones, except in cases of impaired hepatic metabolism (eg, liver failure, elderly). The dose and duration should be individually determined according to the severity of withdrawal and the presence of other medical disorders. In general, benzodiazepine treatment should be limited to the first 3 to 7 days after the cessation of alcohol. Phenobarbital is an alternative to benzodiazepines that is not more likely to lead to oversedation compared with benzodiazepines (75).
All alcoholic patients should be supplemented with parenteral thiamine and other vitamins.
Alcohol withdrawal seizures. Withdrawal seizures are typically self-limited. Benzodiazepines, and not nonbenzodiazepine anticonvulsants, should be used following an alcohol withdrawal seizure to prevent further alcohol withdrawal seizures (346). There is no clear difference in hospital admission, seizures occurring in the emergency department after benzodiazepine administration, or 1-week return visits for discharged patients for patients treated with either diazepam or lorazepam (279). Withdrawal seizures do not represent "latent" epilepsy; therefore, treatment with nonbenzodiazepine anticonvulsants is not recommended (12). If all convulsions are clearly linked to withdrawal, chronic anticonvulsant therapy is not indicated.
Status epilepticus, though rare, needs aggressive intervention with benzodiazepines, anticonvulsants, and treatment of systemic complications.
Chronic management of alcoholic patients prone to recurrent seizures is complicated by the difficulty in separating alcohol-related convulsions and preexisting epileptic conditions.
In patients with epilepsy unrelated to alcohol, drinking in moderation probably has little effect on seizure control or anticonvulsant level (129), whereas heavy or frequent drinking is linked to an increase in seizure frequency (129; 121). Treatment of alcoholism in such individuals is the most effective means of seizure control.
Wernicke-Korsakoff syndrome. As part of withdrawal management, all hospitalized alcoholic patients should be given oral or parenteral thiamine. Patients at high risk of Wernicke encephalopathy (eg, malnourished, severe withdrawal) should be given 3 to 5 days of parental thiamine (346).
In patients with suspected Wernicke encephalopathy, parenteral thiamine should be administered twice daily for 5 days.
Effective treatment and prophylaxis may only be achieved by using parenteral vitamin supplements because oral supplements may not be adequately absorbed (66). Unfortunately, evidence is still insufficient to decide the exact dose, frequency, or duration of thiamine treatment for prevention or treatment of Wernicke-Korsakoff syndrome associated with alcohol abuse (73; 139; 80). Thiamine repletion with at least 100 to 200 mg/day intravenously or intramuscularly is recommended (133), and some advocate for much more aggressive initial treatment regimens of 1000 mg or more daily, particularly for the first 3 to 5 days and as long as 2 months (34; 139).
A double-blind, randomized controlled trial of 127 symptomatic subjects found no significant differences in outcomes for any of the dosage conditions: 100 mg three times daily, 300 mg three times daily, or 500 mg three times daily for 5 days (80). This study found no clear short-term benefit of high-dose thiamine over intermediate or lower doses of thiamine for treating cognitive and neurologic abnormalities related to Wernicke-Korsakoff syndrome. The authors concluded that "the absence of conclusive evidence for the superiority of high-dose thiamine supports a recommendation for patient-specific treatment, while ensuring that the potential impact of other biochemical factors (eg, magnesium and other B vitamin deficiencies) are considered and corrected if necessary" (80).
Alcoholic pellagra. When pellagra is suspected, treatment with oral nicotinamide (100 mg three times daily for 3 to 4 weeks) is an inexpensive, safe, and potentially lifesaving intervention (234; 22). Affected patients should also be treated with other B vitamins and adequate protein nutrition (22).
Marchiafava-Bignami syndrome. Management of this condition is primarily supportive.
Alcoholic dementia. Treatment for alcohol-induced dementias should include abstinence from alcohol and an adequate diet with vitamin supplements. Patients should be given thiamine hydrochloride 100 to 200 mg intravenously, followed by 100 mg intramuscularly every 12 hours. Electrolyte imbalance should also be corrected (110). Withdrawal from alcohol may require inpatient treatment followed by outpatient therapy or support groups such as Alcoholics Anonymous. To the degree that the patients’ cognitive deficits interfere with daily functioning, they may require caregiver support in daily living tasks and may be prohibited from driving. Mental status may improve after patients abstain from alcohol for a sustained period.
Social support is necessary for patients with significant cognitive deficits, as many of them have little insight into their impairment. Gait ataxia requires physical therapy and assistive devices. The dysesthesia of peripheral neuropathy responds partially to tricyclic antidepressants, such as desipramine or amitriptyline at 50 to 150 mg at bedtime. Carbamazepine at 400 to 1200 mg per day or gabapentin at 900 to 1800 mg per day have a similar palliative effect.
Hepatic encephalopathy. Management of acute exacerbations of chronic hepatic encephalopathy includes identification and correction of precipitating factors, such as correction of diuretic-induced hypokalemia and reduction of the nitrogenous load in the intestine (eg, reducing protein content in the diet, halting gastrointestinal bleeding from esophageal varices, clearing blood and other nitrogenous substances from the colon, etc.).
Lactulose (1,4 beta galactoside-fructose) is a nonabsorbable synthetic disaccharide composed of galactose and fructose. Because the small intestine lacks enzymes that can split this synthetic disaccharide, it reaches the large intestine unchanged. In the colon, anaerobic bacteria metabolize lactulose, initially to monosaccharides and then to volatile fatty acids, hydrogen, and methane. Effects of this include increased gas formation and osmolality, as well as acidification (lower pH), within the colon. The increased intraluminal gas and higher osmolality produce a combined volume and osmotic laxative effect, which helps to clear nitrogenous substances from the colon. The reduced intestinal pH shifts ammonia (NH3) produced by gut bacteria to the ammonium ion (NH4+), a charged form that cannot cross biological membranes, including the gut lining. Some colonic bacteria utilize the trapped ammonia as a nitrogen source for protein synthesis, which helps sequester ammonia further and prevent its absorption. Finally, the acidic pH destroys urease-producing bacteria in the colon, decreasing the colonic ammonia derived from bacteria.
In patients with hepatic encephalopathy, lactulose is typically given enterally as a syrup. For acute hepatic encephalopathy, a bolus of 45 ml (30 gm) can be administered and repeated hourly until the first bowel movement. Once the episode of encephalopathy has subsided, the dose can be titrated to achieve two to three soft bowel movements daily (typically 15 to 30 ml, two to four times daily). Lactulose can also be given as a rectal enema with the patient in a left lateral decubitus position.
A meta-analysis of randomized controlled clinical trials showed that lactulose has significant beneficial effects for patients with minimal hepatic encephalopathy compared with placebo or no intervention (195). In particular, lactulose reduced blood ammonia levels, prevented the progression to overt hepatic, and improved health-related quality of life. However, lactulose significantly increased the incidence of diarrhea and did not significantly lower mortality. Other systematic reviews of randomized controlled trials have also suggested that nonabsorbable disaccharides are associated with a beneficial effect on clinically relevant outcomes compared with placebo or no intervention (112).
In the HELP (Hepatic Encephalopathy: Lactulose vs Polyethylene Glycol 3350-Electrolyte Solution) study, a randomized clinical trial in an academic tertiary hospital of 50 patients with cirrhosis admitted for hepatic encephalopathy, polyethylene glycol 3350-electrolyte solution led to more rapid resolution of hepatic encephalopathy than standard therapy with lactulose (257). This suggests that polyethylene glycol 3350-electrolyte solution may be superior to standard lactulose therapy in patients with cirrhosis hospitalized for acute hepatic encephalopathy.
Broad-spectrum nonabsorbable antibiotics, such as rifaximin, are quite effective and safe for treating hepatic encephalopathy. In one systematic review of randomized clinical trials, antibiotics were superior to nonabsorbable disaccharides in improving hepatic encephalopathy, but it is unclear whether this apparent difference is clinically important (09; 10). A later meta-analysis of clinical trials found that rifaximin is at least as effective as other conventional oral agents for treating hepatic encephalopathy and has a better safety profile (85).
In any case, adding rifaximin to lactulose significantly reduces the risk of recurrence and related hospitalization compared with lactulose therapy alone (137). Rifaximin's clinical effect is likely due to its impact on the metabolic function of the gut microbiota rather than a change in the relative bacterial abundance (25).
Surgical treatments for hepatic encephalopathy include procedures to prevent variceal (re)bleeding, procedures to occlude portacaval shunts (using a balloon or open surgical technique), and liver transplantation (potentially definitive in suitable candidates).
Because Wernicke encephalopathy neuropathology is identified in a substantial portion of patients with hepatic encephalopathy, patients who develop hepatic encephalopathy should also be treated presumptively with thiamine (45).
Alcoholic cerebellar degeneration. Management of this condition is primarily supportive.
Osmotic demyelination syndrome. Management of this condition is primarily supportive.
Tobacco-alcohol amblyopia (tobacco-alcohol optic neuropathy). In a study, the visual abnormalities improved in approximately half of the patients with alcohol and tobacco cessation combined with vitamin replacement and then supplementation, but complete recovery was never reached (174). Most cases improve when smoking is stopped or even reduced (104).
Alcoholic neuropathy. A trial of oral thiamine and multivitamins is recommended. Gabapentin and other medications may be needed for symptomatic relief of dysesthesias. Management of this condition is otherwise primarily supportive.
Alcohol-induced (dry) beriberi. The appropriate therapy is thiamine repletion and then supplementation. Depending on severity, thiamine may initially be administered orally if mild or parenterally if moderate or severe.
Alcoholic myopathy. Acute rhabdomyolysis with myoglobinuria requires urgent inpatient interventions to monitor and maintain renal function and to avoid or correct hyperkalemia. Hemodialysis may be needed (18; 241). Blood levels of potassium, phosphate, and magnesium should be determined, and any identified deficiencies should be corrected. In chronic alcoholic myopathy, associated nutritional deficiencies must be corrected, and a diet with adequate protein and carbohydrates ensured.
Fetal alcohol spectrum disorder (and fetal alcohol syndrome). Management of this condition is primarily supportive.
Reduction in tobacco consumption may be associated with better outcomes in the treatment of alcoholism, although alcoholics who abstain from drinking often replace their addiction to alcohol with an inordinate amount of smoking and caffeine use (105).
Prenatal exposure to excessive alcohol can cause fetal alcohol syndrome; hence, the use of any alcohol during pregnancy is contraindicated.
Patients at risk of developing perioperative alcohol withdrawal symptoms can be identified by laboratory evaluation, patient interviews, and questionnaires. Rapid-sequence intubation is recommended (324).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurobehavioral & Cognitive Disorders
Jun. 17, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neuro-Oncology
May. 27, 2026
Neurobehavioral & Cognitive Disorders
May. 20, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Infectious Disorders
May. 01, 2026
Neurogenetic Disorders
Apr. 30, 2026