



Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
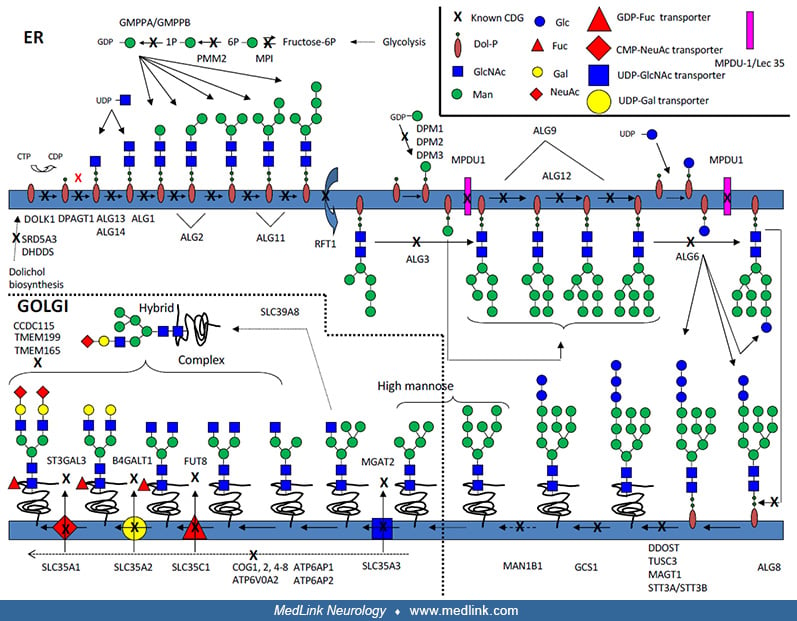
The congenital disorders of glycosylation constitute a large group of inherited disorders with multiorgan expression of symptoms. In most patients, there are signs of nervous system involvement where typical manifestations include structural abnormalities (eg, rapidly progressive cerebellar atrophy), developmental delay, intellectual disability, stroke-like episodes, epileptic seizures, and demyelinating neuropathy. Patient presentations can demonstrate significant differences between and within the different subgroups. In this article, the author explains the basic genetic and biochemical background to these syndromes and describes the most common subtypes in terms of initial clinical signs, diagnostic work-up, prognosis and clinical progression, and, in a few instances, treatment.
• The congenital disorders of glycosylation (CDG) syndromes constitute a group of severe syndromes with a broad phenotypic expression within and between subtypes. | |
• A majority of the patients can be diagnosed using a transferrin glycosylation test. | |
• There is no specific treatment in most subtypes. | |
• CDG should be excluded in all patients with unclear intellectual disability in combination with symptoms of dysfunction in other organ systems. |
Historical note. In 1980, the Belgian professor Jaak Jaeken described the first patients affected with congenital disorders of glycosylation in an abstract in Pediatric Research (37). The patients (2-year-old twin sisters) presented with psychomotor retardation, delayed bone growth, and fluctuating levels of several glycoprotein hormones. Some years later, it was shown that these patients also had deficient glycosylation of transferrin (34), and the term “carbohydrate-deficient glycoprotein syndrome” (CDGS) was eventually suggested (36). More clinical phenotypes emerged, and several subtypes were coined (CDGS-Ia, -Ib, -IIa, etc.). In 1995, the first molecular cause of CDGS was published, deficiency of phosphomannomutase 2 (CDGS-Ia; now PMM2-CDG). PMM2 catalyzes the interconversion of mannose-6-P and mannose-1-P (71). Subsequently, the cause of CDGS-IIa (MGAT2-CDG) as well as the cause and a suggested therapy (alimentary addition of mannose) of CDGS-Ib (MPI-CDG) were published (11; 55).
Nomenclature. In 1999, the first international workshop on CDGS was held, and the participating scientists agreed on a modified nomenclature, including a change in the translation of the acronym CDG to “congenital disorder of glycosylation.” Naming subtypes was based on CDG type (I or II, see below) and chronological order of detection (next available lower-case letter); that is, CDGS-Ia became CDG-Ia, CDGS-IIa became CDG-IIa, and so forth.
Biochemically, one can distinguish between 2 pathological patterns of transferrin underglycosylation, coined CDG type I and type II. Type I deficiencies suggest errors in the synthesis or transfer of the dolichol-linked precursor for N-glycosylation (located to the cytosol or the endoplasmic reticulum), whereas type II deficiencies impair trimming or modification of the protein-bound oligosaccharides (late endoplasmic reticulum and the Golgi). Patients with an unknown gene deficiency were referred to as either CDG-Ix or IIx. Due to improved diagnostic techniques, the number of subtypes increased rapidly during the beginning of this century (22).
In 2009, yet another nomenclature was suggested because glycosylation disorders were identified in other glycosylation pathways (such as O-linked glycosylation and lipid glycosylation) in addition to defects of enzymes and transporters in N-linked protein glycosylation. The revised nomenclature uses the official gene symbol followed by “-CDG” as designation of the individual subtypes; hence, CDG-Ia is now referred to as PMM2-CDG, CDG-Ib is MPI-CDG, CDG-IIa is MGAT2-CDG, and so forth (35). In 2022, there is still an ongoing debate as to which genetic disorders should qualify as a CDG syndrome, where the list of CDGs now has surpassed 160 different genetic syndromes with more than 220 different phenotypes (25). A digital discussion involving +40 glyco-scientists and physicians was organized by professors Freeze, Jaeken, and Matthijs to discuss this matter further (25). It was agreed that the classification of a disorder as a ”CDG” requires that there is a measurable effect on any type of glycosylation but that one should be inclusive rather than exclusive. Examples of genetic/biochemical criteria to be classified as a CDG were (example of gene): (1) gene evolved in any glycosylation pathway (ALG1, B3GLCT, B4GALT7, DPM1, EXT1/2, FKRP, MOGS, MPI, OGT, PAPSS2, PGM1, PIGA, PIGL, PMM2, POMT1, SLC35A2, SRD5A3, STT3A); (2) glyco-gene conserved in evolution (ALG13); (3) gene in ER, Golgi, post-Golgi trafficking altering the synthesis of any glycan (COG4, COG7, GET4, SEC23B, TRAPPC11); or (4) gene involved in metal ion or pH homeostasis that alters glycan synthesis (ATP6AP2, ATP6VOA2, SLC39A8, TMEM199). Examples of nonqualifying genes were: part of a glycosylation-altering molecular complex, but lacking evidence of altered glycans (TRAPP6B, TRAPPC12) (25).
Clinical manifestations. The congenital disorders of glycosylation are multi-organ syndromes where the clinical presentations differ widely both within and between the different subtypes (24; 30; 23; 19).
Even siblings with congenital disorders of glycosylation can have different presentations, perhaps indicating a modifying role of the environment and the genetic background. In most known subtypes, fewer than 20 patients have been described. Therefore, subtype-specific features are hard to postulate. Nonetheless, some features, such as developmental delay and hepatic involvement, are present in most known forms of congenital disorders of glycosylation.
PMM2-CDG. The most common and well-described subtype of congenital disorders of glycosylation is PMM2-CDG; data suggest approximately 50% of patients with CDG type I have PMM2-CDG (70). The “classic” phenotype includes muscular hypotonia, failure to thrive, developmental delay, cerebellar atrophy, esotropia, peripheral neuropathy, retinopathy, liver involvement, inverted nipples, and peculiar fat pads (02). However, as screening for CDG has been applied to a broader population, many patients with PMM2-CDG have different phenotypes (28). In classic PMM2-CDG, patients have been described to go through 4 stages: the infantile multisystemic stage, the childhood ataxia or intellectual disability stage, the teenage leg atrophy stage, and the adult hypogonadal stage (36). PMM2-CDG survival into adulthood is estimated at 75% to 80%, and the highest mortality rates are in the early years, particularly in patients with a more pronounced visceral form of disease (13; 02). An international clinical guideline for the diagnosis, treatment, and follow-up of PMM2-CDG has been published (02).
Patients with PMM2-CDG are usually born after normal pregnancies with normal birth weights (28; 26). In some instances, hydrops fetalis (46) or pericardial effusions (42) have been noted prenatally or in the neonate, and there are descriptions of cardiomyopathy being present at birth (01; 42). Also, prolonged hypoglycemia has been described as the first symptom of PMM2-CDG (06; 52). During infancy, the predominant neurologic features of PMM2-CDG are hypotonia and developmental delay (particularly gross motor skills), with evolving facial hypotonia, esotropia, and unusual eye movements. Neuroimaging often shows a diminished cerebellar vermis in a process that is now believed to be rapidly progressive atrophy. Further, global atrophy and hydrocephalus with acquired microcephaly are common (24; 23). Other common findings are failure to thrive, inverted nipples, and subcutaneous fat pads. Most patients show some visceral involvement (cardiac, hepatic, renal, or intestinal), which may be catastrophic (36; 28; 24). Infections can precipitate episodes of pericardial effusions with cardiac failure, hepatic failure, coma, and thrombotic or hemorrhagic events, and patients are often susceptible to infections. Muscle reflexes usually diminish early and are most often completely absent by 1 year of age.
Partial seizures and stroke-like episodes often become apparent later in the first decade of life. The etiology of the stroke-like episodes is unclear and possibly multifactorial, but they typically occur during or after infections or head trauma (24). One paper suggests that the cause may be underglycosylation of the voltage-sensitive calcium channel encoded by CACNA1A (33). Patients may present with hemiplegia, unconsciousness, or blindness, and the episodes last from hours to several days but usually resolve completely (in rare instances secondary sequelae may persist). In this phase of the disease, the visceral findings usually regress (24; 23).
In adolescence and adulthood, the patients have fixed neurologic deficits, including severe cerebellar ataxia, peripheral neuropathy with distal wasting, and intellectual disability. Patients often develop signs of retinopathy and fixed strabismus. A large proportion becomes legally blind. Many show kyphoscoliosis. Patients’ communication skills vary, but most often they speak in short sentences in a staccato manner, often incomprehensible to persons without a relation to the patient. Peripheral neuropathy is evident, and the overwhelming majority gradually depends on a wheelchair for locomotion.
Female patients with PMM2-CDG lack pubertal development and show primary ovarian failure with hypergonadotropic hypogonadism (14; 48). Puberty can be hormonally induced; however, the risk of causing thrombotic events should be considered, especially in patients who have had stroke-like episodes in the past. Male patients undergo puberty; however, it may sometimes be delayed. Testosterone levels are in the low to normal range (14; 48).
ALG6-CDG. ALG6-CDG is the second most common N-linked CDG, with more than 100 known patients to date. The phenotype of this disorder is generally milder than that of PMM2-CDG (43); however, several have died due to its complications (50).
The main findings in ALG6-CDG include a moderate degree of intellectual disability, ataxia, hypotonia, esotropia, epilepsy, and proximal muscle weakness (50). Severe protein-losing enteropathy in the context of gastroenteritis has been described (15; 50). The primary findings in the few known adult patients with ALG6-CDG were intellectual disability, deep-vein thrombosis, benign intracranial hypertension, virilization, and skeletal anomalies (67).
ALG1-CDG. This subtype was described in 2004 by 3 independent groups and was initially considered an unusual and very severe subtype of CDG because 4 of 7 described cases died in early childhood (27; 39; 61). With the introduction of next-generation sequencing, however, the number of known patients has increased considerably over the last few years (more than 50 known patients). Of over 40 identified cases diagnosed since 2009, more than 70% survived beyond the first year of life (54). Many affected individuals show a PMM2-CDG-like phenotype with developmental delay/intellectual disability, microcephaly, strabismus, seizures, coagulation abnormalities, and abnormal fat distribution (51; 54). Therefore, ALG1-CDG should be excluded in any case with a “classic” CDG phenotype but normal PMM2-analysis. Interestingly, in all ALG1-CDG patients tested so far, mass spectrometric transferrin analysis shows the presence of a xeno-tetrasaccharide, specific to this subtype, which can allow for a subtype distinction before the genetic analysis is performed (05).
PGM1-CDG. This defect (phosphoglucomutase 1 deficiency) (69) was initially described as a glycogen storage disorder (later named glycogenesis type XIV) (65) and very few patients have been described. However, in 2014, Tegtmeyer and colleagues described 19 more patients from 16 families (68). The subtype (PGM1-CDG) was redefined as a mixed-type congenital disorder of glycosylation (ie, having features of both type 1 and type 2 underglycosylation). Interestingly, they suggested that these patients do not have a neurologic phenotype due to other isoforms of phosphoglucomutase being expressed in the brain. Instead, they suffer from hypoglycemia, liver pathology with hypertransaminasemia, cardiac problems including arrhythmia and dilated cardiomyopathy, muscular weakness and orofacial abnormalities such as a bifid uvula (greater than 80% of patients), and a cleft palate. A more recent paper, however, reports intellectual disability and motor delay in a significant number of the patients (17/41) (57). This is one of few congenital disorders of glycosylation subtypes that seems to respond to the alimentary addition of saccharides; in this case, galactose or lactose (68). A consensus guideline for diagnosis, follow-up, and treatment has been published (03).
Defects of the conserved oligomeric Golgi (COG) complex. In 2004, it was shown that mutations in COG subunit 7 (COG7) cause a new CDG subgroup, the COG-CDGs (78). Biochemically they had a combined N- and O-linked deficiency, with hypoglycosylation of both transferrin and apolipoprotein C-III, one of the very few serum proteins that show exclusive O-linked glycosylation. Since then, deficiencies in COG 1, 2, 4, 5, 6, and 8 have been described (58). There is a considerable variation in which organs are involved and the severity of the phenotype in those affected, but there is always a degree of developmental delay/intellectual disability (mild to severe), microcephaly, and hypotonia; often there is also epilepsy. Other findings include facial dysmorphias, skeletal dysplasia, failure to thrive, hepatomegaly, and cardiac involvement (heart failure, hypertrophy, congenital heart defects). Brain imaging can be normal, but some patients showed cerebral and cerebellar atrophy (24). A particularly interesting phenotype (Saul-Wilson syndrome) that includes skeletal dysplasia, short stature, and developmental delay was reported in 14 individuals and was caused by the same heterozygous mutation in COG4 (20), implying a yet unknown function for this particular COG subunit.
Cutis laxa syndromes. Cutis laxa is a dermatological condition with inelastic, loose, and sagging skin. Some patients with autosomal recessive cutis laxa II and wrinkled skin syndrome have mutations in ATP6V0A2, encoding the alpha2 subunit of vacuolar-type proton-ATPase. Interestingly, these patients show a combined N- and O-linked glycosylation deficiency, which merits its classification as a CDG (ATP6V0A2-CDG) (38).
The more than 20 described patients with ATP6V0A2-CDG will always have wrinkly skin and typically show a large fontanelle with delayed closure, down-slanting palpebral fissures, joint laxity, muscular hypotonia, and eye anomalies (strabismus and myopia). Microcephaly is common with variable intellectual disability and developmental delay. Half of the patients show brain abnormalities, such as partial pachygyria or cobblestone-like lissencephaly. Seizures are uncommon but can occur in older children. The early feeding difficulties, growth retardation, and skin condition usually resolves with age (49; 31).
Congenital myasthenia syndromes. Congenital myasthenic syndromes primarily present clinically by fatigable muscle weakness. They are due to impaired signal transmission at the neuromuscular synapse (18). Over the last 5 years, genes that have previously been connected to a more CDG-like phenotype have been found to cause a separate phenotype with congenital myasthenia as the major symptom. These genes include DPAGT1, GFPT1, ALG2, and ALG14 (62; 04; 12). The pathogenesis probably involves defective glycosylation of the acetylcholine receptor subunits (04). In total, more than 50 families have been identified.
SLC35A2-CDG (galactose transporter deficiency). This is the most prevalent X-linked congenital disorders of glycosylation, with about one hundred patients known (oral communication, Bobby Ng, Sanford-Burnham-Prebys Medical Discovery Institute, La Jolla, CA). Most of the patients are girls; however, boys with mosaic mutations also exist. The original description was of 3 unrelated patients with mainly neurologic and skeletal problems, including epileptic encephalopathy, developmental delay, ocular manifestations (ocular flutter, nystagmus, and retinitis pigmentosa), facial dysmorphisms, and brain malformations including cerebellar hypoplasia (53). There seems to be three distinct phenotypic groups, including either solely neurologic features, neurologic and skeletal features combined, or an exclusively skeletal phenotype (unpublished). Galactose therapy has been tried in some patients with partial response (75). Interestingly, somatic mosaicism of the SLC35A2 gene in the brain has been shown to cause focal intractable seizures in mild malformation of optical development with oligodendroglial hyperplasia in epilepsy (63; 73; 07).
Clinical vignette. A 7-month-old boy was seen by a child neurologist and was recognized to be hypotonic with poor head control. The boy did not reach for objects, sit, or roll over, and reflexes were decreased. A nerve biopsy showed chronic axonal neuropathy, and a brain MRI showed ventriculomegaly ex vacuo with atrophy. Sonar spectroscopy revealed high levels of lactate in the gray matter. At age 18 to 24 months, he developed myoclonic jerks with electroencephalographic slow waves consistent with seizures. At age 8.5 years, the patient had severe intellectual disability, no speech, and no bowel and bladder control. Biochemically he showed signs of liver pathology with increased serum ALT. A neurometabolic investigation (mitochondrial disease, lysosomal disease, peroxisomal disease, aciduria) was normal, as was his karyotype. Due to signs and symptoms from dysfunction of several organ systems, including the brain, glycosylation of transferrin was checked by mass spectrometry. On this analysis, the patient was found to have a type II pattern with normal chain occupancy but lacking sialic acids. Analysis of the COG complex revealed a lack of COG8, consistent with the diagnosis COG8-CDG (CDG-IIh) (40).
In classical PMM2-CDG, approximately 80% of patients survive into adulthood. The diseases generally enter a rather stable state after puberty. In other subtypes, survival into adulthood is much reduced, where the presence of severe early-onset neurologic symptoms as epileptic encephalopathy constitute strong risk factors.
Glycosylation is the collective name for the processes in which carbohydrates are added to proteins and lipids, thereby modifying their properties (16; 24; 23). Almost all glycosylation takes place in the endoplasmic reticulum and the Golgi. Hence, all proteins that are translated into the endoplasmic reticulum will face the glycosylation machinery and are possible candidates of becoming glycoproteins if they have consensus sequences for glycosylation. In asparagine (N)-linked glycosylation, a precursor oligosaccharide containing 14 monosaccharides is preformed on a lipid anchor in the endoplasmic reticulum then transferred en bloc to the growing polypeptide.
This structure is then trimmed by glycosidases, a process vital for correct folding of many glycoproteins. The oligosaccharides are then further rebuilt in the Golgi, which is important for several aspects of the “life” of the proteins, including their initial folding and trafficking to the proper functioning and regulation of their degradation.
All eukaryotic cells contain N-linked glycoproteins. Therefore, errors in these glycosylation processes can cause signs and symptoms in many organ systems in affected patients.
Addition of carbohydrates to proteins requires a complex machinery and demands a coordinated expression of hundreds of genes. In fact, more than 1% of the expressed human genome is predicted to encode proteins involved in the glycosylation process (21). Though all syndromes due to pathologic mutations in a gene involved in glycosylation could be referred to as a congenital disorder of glycosylation, many diseases that fit this criterion have already been included in other classification systems (eg, congenital muscular dystrophies with deficiencies in O-linked mannosylation, such as muscle-eye-brain disease or Walker-Warburg syndrome). Therefore, this article focuses on disorders with defective N-linked glycosylation.
The congenital disorders of glycosylation are multifaceted, and patients are often suspected to have other disorders (eg, mitochondrial disease) before correct diagnosis is reached (08). The diagnosis of classical PMM2-CDG is, however, rather straightforward and seldom missed. In general, congenital disorders of glycosylation should be excluded, at least by analysis of transferrin glycosylation status, in any patient with intellectual disability/developmental delay in combination with symptoms from at least one other organ system (such as liver, endocrine, or coagulation) (17). Most patients in the last 10 years diagnosed with a CDG syndrome were found via clinical sequencing programs, and CDG was not the primary diagnosis suspected.
The initial testing for congenital disorders of glycosylation should include analysis of N-glycosylation of serum transferrin. Traditionally, this is done by isoelectric focusing (IEF), a method still widely used (64). However, in some diagnostic laboratories, the method of choice is affinity chromatography followed by mass spectrometry (41). Other methods, including HPLC separation of the differently glycanated species (29) and capillary zone electrophoresis (09), have been used. When pathological, these methods will suggest either a type I, a type II, or a mixed pattern. If the pattern is suggestive of a type II or mixed pattern (ie, suggestive of a Golgi pathology), analysis of underglycosylation of apo-lipoprotein CIII (suggestive of O-linked glycosylation errors) may be deployed to further deepen the analysis (77). There are several potential problems with the transferrin analyses. A positive test can occur in uncontrolled hereditary fructose intolerance, galactosemia, certain hepatic pathologies, alcoholism, and rare mutations at the transferrin glycosylation site (56). Also, preterm infants with congenital disorders of glycosylation have normal transferrin patterns; normal transferrin has been reported in patients who were later proven to have congenital disorders of glycosylation (72); and confirmed CDG cases sometimes normalize transferrin glycosylation. This is suggestive of an optimal window for transferrin diagnosis, and unpublished results from thousands of transferrin tests at the Mayo Clinic clearly show an optimal period between 6 and 18 months of age (Raymond, K, personal communication).
If the patient has a classic type I pattern of transferrin underglycosylation, PMM2-CDG (CDG-Ia) should be ruled out or confirmed as it is the commonest subtype. Some laboratories offer enzymatic testing for PMM2 activity in white blood cells or fibroblasts, whereas others employ genetic testing of the PMM2 gene immediately. If the patient presents without or with only mild neurologic symptoms, MPI-CDG should be excluded as this is a treatable, but potentially fatal, condition. This is done using an enzyme assay on white blood cells or fibroblasts, but it is generally not available at clinical laboratories as a routine (CDG laboratories in the United States and Europe should be contacted for more information).
If the subtype remains obscure after this initial screen, targeted whole exome or genome analysis should be performed (22) even though some laboratories still perform lipid-linked oligosaccharide analysis as part of the investigative path.
In most congenital disorders of glycosylation subtypes there is no specific treatment available, and symptomatic treatment should be employed for those conditions that are associated with the CDG syndromes, including epilepsy, heart failure, and reflux. Active physiotherapy is critical for maintaining muscular function.
The aldose reductase inhibitor epalrestat, used for treating diabetic neuropathy, was found as a potential therapy for PMM2-CDG in a multispecies drug-repurposing screen (32), and a few patients have been treated with this drug with promising results (nonpublished data). A placebo-controlled study has been designed (ClinicalTrials.gov NCT04925960) and will soon start to recruit patients. Further, use of acetazolamide has shown promising results in treating ataxia in PMM2-CDG (45), and a placebo-controlled study (ClinicalTrials.gov NCT04679389) is soon to be completed.
Some patients with PMM2-CDG with recurrent stroke-like episodes are kept on acetylsalicylic acid. Osteoporosis and fractures are common in older patients, and puberty can be induced in adolescent girls to prevent this. The risk of thrombotic events should be considered, and some patients are kept on warfarin, low molecular weight heparin, or oral factor Xa inhibitors for several years after the induction. Bisphosphonates are indicated for patients who have had several fractures.
Frequent check-ups during the first years of life are vital and should include liver enzymes analysis, cardiac ultrasound, and coagulation investigations. Ophthalmologic investigation, including ERG, should be employed bi- to tri-annually.
Only a few other subtypes are currently treatable. MPI-CDG, the only subtype without neurologic deficits, shows the best results. Here, supplementing the food with mannose has a dramatic effect on coagulopathy, protein-losing enteropathy, and hypoglycemic episodes present in this subtype. In one case, when mannose was given intravenously due to fasting before surgery, the patient went into unconsciousness and experienced seizures, possibly due to the accumulation of Man-6P and intracellular depletion of ATP (60). Thus, caution must be taken in similar situations, and intravenous glucose administration may be used to reverse the effect. A clinical management guide has been published (10).
Fucose has been tried in SLC35C1-CDG (CDG-IIc), but appreciable effects have been absent in most patients (44; 66).
Galactose and lactose are currently used in PGM1-CDG and there seems to be some stabilizing effects with normalization of transaminases, coagulations factors, and hormones. Furthermore, there also seem to be beneficial effects on the episodic rhabdomyolysis and hypoglycemia (74; 76).
The 2 ultra-rare subtypes, TMEM165-CDG and SLC39A8-CDG, also seem to benefit from galactose treatment. In SLC39A8-CDG, addition of manganese is also helpful (74).
Most patients with congenital disorders of glycosylation are infertile. Successful pregnancy with normal outcome has occurred in a patient with MPI-CDG (not published). There is no information available concerning specific management in the event of a pregnancy.
There are very few papers describing the use of anesthesia in congenital disorders of glycosylation (47; 59); however, coagulation issues are discussed in a patient with ALG6-CDG (47).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Erik A Eklund MD PhD
Dr. Eklund of Lund University has no relevant financial relationships to disclose.
See Profile
Deepa S Rajan MD
Dr. Rajan of UPMC Children's Hospital of Pittsburgh has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026