
























Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
For centuries, the condition now known as craniosynostosis—defined as the premature fusion of the skull’s sutures—has challenged the understanding of physicians and anatomists. Although historical accounts of this disorder date back hundreds of years, it is only within the past century that significant advancements in surgical techniques and genetic research have transformed its diagnosis and treatment. In this article, the author delves into the early descriptions that first captured the medical community’s attention, explores the molecular mechanisms that have since been uncovered, and traces the remarkable progress in surgical management that continues to shape patient outcomes today.
|
• Craniosynostosis is a condition characterized by the premature fusion of one or more of the cranial sutures. | |
|
• Its prevalence is 1 in 2500 births worldwide. | |
|
• It can be seen in normal individuals or as part of a multisystem syndrome. | |
|
• Genetic factors associated with it include mutations in EFNB1, EFNA4, MSX2, FGFR1-3, SHOC2, TWIST1, POR, RAB23, GLI3 and RECQL4 genes. | |
|
• Surgical release of the affected suture is an effective way of correcting skull deformity and preventing neurocognitive impairment. Endoscopic techniques have been employed. |
Craniosynostosis is the premature fusion or abnormal development of one or more cranial sutures. Although the disorder was recognized in antiquity, some speculate that the Athenian leader Pericles had sagittal synostosis (27). Sommering observed in 1791 that skull growth occurs along cranial sutures, with growth failure at a suture resulting in skull deformity (109). In 1851, Virchow described various types of craniosynostosis and noted that normal growth occurs perpendicular to cranial sutures, whereas compensatory growth in craniosynostosis occurs parallel to the closed suture (121). Craniosynostosis occurring as part of dysgenetic syndromes was first reported by Apert, who described oxycephaly with syndactyly in 1906, and by Crouzon, who described craniofacial dysostosis in 1912. Numerous other syndromes were subsequently identified, though debates over attribution remain ongoing (21). Surgical treatment was pioneered in the 1890s by Lannelongue and Lane, who performed strip craniectomy to remove the fused suture. Initially, little distinction was made between congenital, primary craniosynostosis and microcephaly (32). Early surgeries resulted in high mortality and poor outcomes, and a sharp critique by Jacobi in 1894 led to a decline in popularity (55). The tide began to turn with Mehner’s report of the first successful strip craniectomy in 1921 (18), and by the 1930s, more careful case selection and the more extensive procedures of Bauer (04) and King (63) further solidified the role of surgical intervention.
Nomenclature in craniosynostosis remains complex due to multiple classification systems. One approach divides cases into primary and secondary craniosynostosis. Primary synostosis results from intrinsic disease processes that close sutures before or shortly after birth, leading to abnormal skull growth patterns. In contrast, secondary craniosynostosis occurs when brain growth is arrested or metabolic disorders prevent normal suture patency. Causes of secondary craniosynostosis include microcephaly, shunted hydrocephalus, amniotic band syndrome, positional flattening of the calvarium, and rickets. Some forms of craniosynostosis are syndromic, part of inherited conditions that also involve other congenital anomalies. Over 100 syndromic synostoses have been identified (19), many of which affect multiple sutures, particularly the coronal (Table 1). Nonsyndromic craniosynostosis, by contrast, is an isolated, often sporadic birth defect. Anatomical classification systems further categorize craniosynostosis based on the affected sutures and the resulting head shapes.
|
Name |
Associated findings |
Inheritance |
|
Crouzon disease |
Midface hypoplasia, exorbitism, hypertelorism |
Autosomal dominant |
|
Apert syndrome |
Same as Crouzon disease, plus syndactyly of all four limbs |
Autosomal dominant |
|
Pfeiffer syndrome |
Same as Crouzon disease, plus beaked nose, broad thumbs and great toes, may be syndactyly |
Autosomal dominant |
|
Saethre-Chotzen syndrome |
Facial asymmetry, strabismus, low hairline, variable syndactyly, normal thumbs and great toes |
Autosomal dominant |
|
Carpenter syndrome |
Mental deficiency, short stature, obesity, variable syndactyly, heart and limb defects |
Autosomal recessive |
|
Cloverleaf skull |
Can be associated with above syndromes, thanatophoric dysplasia |
Autosomal dominant |
|
Baller-Gerold syndrome |
Radial, carpal, and digital aplasia |
Autosomal recessive |
|
Antley-Bixler syndrome |
Same as Crouzon disease, plus choanal atresia and stenosis, multiple limb anomalies |
Autosomal recessive |
Characteristic changes in head shape reflect involvement of specific cranial sutures. Normal skull development initiates around day 23 to 26 of gestation. Craniosynostosis can be associated with over 130 different syndromes; however, most commonly present as a single abnormality (09). Over time, a bewildering number of names have been proposed to describe the anatomical consequences of this involvement.
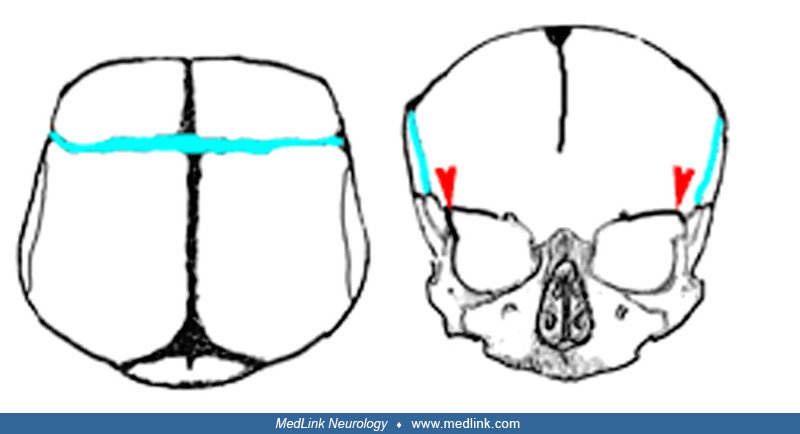
Table 2 describes the common names and the responsible suture(s); the accompanying figures illustrate head shape (somewhat exaggerated).
|
Involved suture |
Anatomical names (cephal = "head") |
|
Sagittal |
scaphocephaly (boat-shaped), dolichocephaly (long) |
|
Coronal (bilateral) |
brachycephaly (short) |
|
Coronal (unilateral) |
plagiocephaly (diagonal) |
|
Metopic |
trigonocephaly (triangle-shaped) |
|
Lambdoidal (bilateral) |
posterior or occipital brachycephaly |
|
Lambdoidal (unilateral) |
posterior or occipital plagiocephaly |
|
Multiple sutures* |
pansynostosis (all), oxycephaly (conical), acrocephaly (pointed), turricephaly or turmschadel (tower), cloverleaf skull or kleeblattschadel |
|
* = many anatomical variants | |
Because craniofacial bones grow as an integrated unit, most synostoses cause significant change in basicranial and midfacial bones. Metopic and coronal stenoses result in small anterior fossae; coronal stenosis produces shallow orbits; lambdoidal and bilateral coronal stenosis produce forward displacement of the petrous bone. Stenosis of the coronal suture as it continues as the sphenofrontal suture and the sphenoethmoidal suture affect the size of the nasopharynx and apposition of the epiglottis to the soft palate. Isolated sagittal synostosis, the most common, is rarely associated with deformity of the skull base or facial bones.
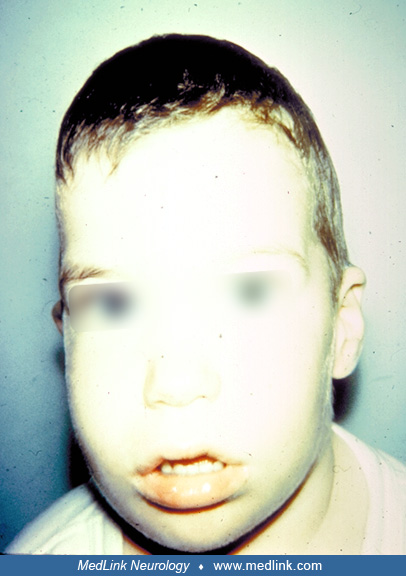
As the extent of craniosynostosis increases, the number of associated problems grows. Ophthalmologic disorders, including amblyopia, strabismus, and refractive error, remain prominent. Although Hertle and colleagues documented the frequent occurrence of amblyopia in syndromic craniosynostosis, more recent studies have further delineated the range and prevalence of visual impairments (48). Airway obstruction and feeding difficulties are also common, especially in syndromic forms (98). Although decreased central visual acuity and optic atrophy are less frequently observed, updated guidelines and modern imaging techniques have improved early detection and management (72). Intracranial hypertension and neurodevelopmental disorders can also be a complication.
Developmental delays are generally uncommon in cases of single-suture craniosynostosis, as previously documented (110; 61), though minor delays can frequently be detected through detailed neurodevelopmental testing (104; 105). Evidence suggests that sagittal nonsyndromic craniosynostosis is associated with a high prevalence—greater than 50%—of executive function deficits, as well as altered neocortical structural and functional connectivity (05). Children with multiple suture closures, especially those with syndromic craniosynostosis, are at increased risk of developmental delays, and the presence of congenital cerebral anomalies within certain syndromes is well documented (99). Furthermore, there is evidence that these children may experience progressive neurologic deterioration, likely driven by restricted cranial growth and subsequent intracranial hypertension (28; 37; 59). Although elevated intracranial pressure is a recognized complication of multiple suture synostosis—occurring in 26% to 54% of cases—it can also be present, though less commonly, in isolated suture closures (93; 17). Hydrocephalus, meanwhile, affects at least 12% of syndromic craniosynostosis cases, but it remains rare in isolated synostosis (36; 41; 17). Another complex feature, hindbrain herniation (acquired Chiari malformation), may occur in syndromic craniosynostosis and cloverleaf skull, sometimes necessitating decompressive surgery. However, the underlying etiology—whether purely due to intracranial crowding or compounded by suboccipital bony deformities—remains uncertain (16; 86).
Outcome should concentrate on two areas, the child's appearance and his or her neurologic development. The craniofacial deformity tends to increase with time and may stigmatize the affected child. Decompressive surgery is effective in limiting deformity (38), especially if done early in life.
Rapid brain growth and the flexible cranium allow the head to assume a more nearly normal shape. There is wide variation of cosmetic outcomes among the different types of synostoses; this appears to be more a matter of the underlying deformities than surgical technique (29).
A more serious problem in children with multiple suture craniosynostosis is that of neurodevelopmental impairment, especially in those with syndromic forms. Once considered an integral part of various syndromes, impaired cognitive development is often the result of associated cerebral anomalies (99). However, there is evidence for progressive deterioration, both clinically (93) and on electrophysiological monitoring, in many of these children (28). In a national sample of 82 children with syndromic craniosynostosis, Maliepaard and colleagues found that the children had full scale IQs similar to the normative population but were at increased risk for development of intellectual disability, internalizing, and social and attention problems (70). Additionally, children with Apert syndrome had lower full-scale IQs and children with Muenke syndrome had more social, attention, and inattention problems. There are often symptoms and signs of intracranial hypertension, which may lead to blindness. This suggests that increased intracranial pressure secondary to restricted brain growth and hydrocephalus (or other factors) may be a contributing factor. Indeed, early and aggressive cranial decompression has allowed relatively normal neurodevelopmental progress, even in some children with syndromes thought to be uniformly associated with mental retardation (96). However, restenosis, hydrocephalus, and even hindbrain herniation may occur despite surgical decompression (118). Treatment of hydrocephalus poses a difficult management issue because it can secondarily contribute to premature suture closure and growth arrest (115). Even children with successful surgery for single suture synostosis can develop craniosynostosis of multiple sutures and increased intracranial pressure; postoperative patients must be carefully followed (53). Early surgery does not assure normal cognitive development; children with early repair of sagittal or metopic synostosis do not score as well as controls at school age (122; 104; 105). However, early surgery improves developmental level (06). Additionally, early surgical reconstruction helps limit progressive deformity of the cranial base. Infants retain malleability in the skull leading to easier shaping (09). Early diagnosis and treatment are essential factors in preventing serious complications and improving long-term outcome (37; 59).
The full-term product of a normal pregnancy, labor, and delivery was noted to have a long, narrow head. There was no family history of craniocerebral deformities, and the child was neuroradiologically normal on examination. Skull radiographs and CT scans confirmed isolated synostosis of the sagittal suture. The deformity progressed, and at 6 weeks of age the child was admitted for surgery.
An extensive decompression of the skull was performed, with excision of the stenotic suture and enlargement of adjacent sutures down to the skull base.
Improvement in cranial appearance was rapid and has been maintained despite rapid regrowth of craniectomized bone.
Subsequent appearance and neurologic development are normal.
Primary synostoses are commonly divided into nonsyndromic and syndromic types. Most cases of nonsyndromic craniosynostosis are isolated and sporadic, although a small proportion reflect autosomal dominant inheritance. Twin studies and familial aggregation data suggest both genetic and environmental contributions to nonsyndromic midline synostoses (65). Exome sequencing of 291 parent-offspring trios has revealed heterozygous damaging de novo mutations in genes regulating Wnt, BMP, and Ras/ERK signaling, suggesting novel genetic contributions to nonsyndromic craniosynostosis (119). These findings also demonstrate overlaps in the pathophysiology between nonsyndromic and syndromic forms.
Syndromic craniosynostoses are usually inherited, although sporadic cases occur. The genetic basis of these syndromes is highly heterogeneous, involving mutations in genes, such as FGFR1-3, TWIST1, and EFNB1, among others. These genes encode proteins that regulate intramembranous ossification, highlighting shared molecular pathways across multiple craniosynostosis phenotypes (67; 79). Other contributing factors include chromosomal anomalies, teratogenic exposures (eg, valproic acid, retinoic acid, phenytoin), and environmental influences like fertility treatments, which have been associated with increased craniosynostosis risk (20; 40; 92).
Secondary craniosynostosis arises from premature suture closure due to noninherited factors. It can be seen in conditions, such as microcephaly, hydrocephalus, metabolic disorders (eg, hypophosphatemic rickets, hyperthyroidism), and mucopolysaccharidoses. Hematologic conditions, including sickle cell anemia and the thalassemias, have also been implicated through mechanisms of marrow hyperplasia and osseous overgrowth (19). Additionally, amniotic band syndrome has been linked to craniosynostosis alongside craniofacial clefts and other congenital anomalies.
The historical understanding of cranial morphogenesis provides context for these findings. The 19th-century models proposed that skull growth occurs by apposition at suture margins. Sommering and Virchow noted that premature suture closure impedes perpendicular growth, with compensatory parallel growth influencing skull shape (109; 121). Some aspects of this hypothesis have been enlarged to explain the evolution of abnormal head shape in craniosynostosis. Normal closure of sutures varies between individuals; however, the metopic suture usually closes within the first 2 years of life. The sagittal, coronal, and lambdoid sutures close by 40 years of age, whereas the squamosal, occipitomastoid, and sphenotemporal sutures may remain partly open at age 70 years (25). However, the basic premise that skull growth occurs only at cranial sutures cannot be correct.
Furthermore, the calvarium consists of membranous bone, sandwiched between the outer dural layer (endocranium) and the pericranium. Calvarial growth occurs by a repetitive cycle of suture widening and ossification of enlarging membranous bones (33).
Moss presented an explanation of normal and abnormal skull growth that relied on biomechanical forces originating from the skull base (78). He proposed that dural bands, originating at the skull base, created growth vectors for the calvarium. Cranial sutures formed along these vectors kept patent by biomechanical forces. Interference with these vectors, usually the result of synchondrosis at the skull base, caused the affected suture to close prematurely. Transfer of force to adjacent growth sites completes the deformity.
However, investigations have cast doubt on the Moss hypothesis. Experimental closure of vault sutures can cause craniostenosis and facial deformities typical of the clinical syndromes (90). This reversal of the direction of influence undermines the idea of the skull base as the origin of changes. Local dura, rather than transmitted forces, seems to be most important in determining sutural patency (12) and the timing of suture closure (68). There is accumulating evidence that biomechanical forces do not play a major role in sutural growth and closure; these processes instead respond to complex cell signaling mechanisms (84; 01). It has long been presumed that restricted head growth is responsible for the associated intracranial hypertension and mental retardation. However, computer analysis of intracranial volumes over time suggests that skull constriction effects of synostosis dissipate over the first few months of life (102; 71; 80; 101). Furthermore, impaired venous drainage may play a larger and more durable role than previously suspected (117; 95; 44). Increased intracranial pressure is thought to result from restricted venous outflow, secondary to the obstruction of the skull base venous structures from alterations in the cranial vault (10; 115). A similar role has been implicated for upper airway obstruction and hydrocephalus (44; 116). The latter is an especially common finding in children with syndrome craniosynostosis, and it poses a significant management issue. CSF diversion, although beneficial for the treatment of hydrocephalus, may contribute to further fusion of the sutures and growth arrest for the cranial vault (44; 115).
Advances in molecular genetics have significantly enhanced our understanding of the role of fibroblast growth factor receptors in syndromic and nonsyndromic craniosynostosis (125). Fibroblast growth factor receptors are tyrosine kinases that transduce extracellular signals and regulate cranial suture development. Mutations in FGFR2 lead to a spectrum of craniofacial abnormalities, including Crouzon, Pfeiffer, Apert, and Jackson-Weiss syndromes (111; 82; 13; 39). These mutations are associated with increased alkaline phosphatase activity and osteocalcin production, leading to osteoblast proliferation and premature suture fusion (26). The FGFR2 gene is located on chromosome 10q, whereas FGFR1 and FGFR3, which are also implicated in autosomal dominant craniosynostosis, are found on chromosomes 8p and 4p, respectively.
In Apert syndrome, two specific adjacent amino acid substitutions drive the FGFR2 mutation, whereas other syndromic craniosynostoses exhibit mutations involving amino acid substitutions, deletions, insertions, or splice-site changes. FGFR3 mutations have also been implicated in some nonsyndromic craniosynostosis cases (94). Beyond growth factor receptors, craniosynostosis may also arise from mutations in homeobox genes and genes involved in apoptosis, both of which are critical in suture formation and maintenance (49; 11). The observation that identical mutations can produce varied phenotypes suggests that additional, yet unidentified, genetic modifiers contribute to craniosynostosis pathogenesis (64).
Furthermore, several genes have been implicated in nonsyndromic single-suture craniosynostosis. Specifically, TWIST1, a transcriptional regulator critical for mesenchymal cell differentiation, has been linked to single-suture craniosynostosis (67). In Drosophila, homozygous mutations in twist disrupt embryo gastrulation, leading to the failure of mesodermal-derived organ development and complete head eversion. Advances in genome-wide screening and high-resolution genomic resequencing have led to the identification of additional gene variants associated with different types of single-suture craniosynostosis (75; 24). These findings underscore the complex genetic interplay involved in cranial suture biology and highlight the importance of molecular signaling pathways in the pathogenesis of craniosynostosis (124)
The incidence of craniosynostosis is estimated to be 1 in 2000 children (66), although the incidence of any of more than 100 syndromic craniosynostoses varies widely. It is estimated that Crouzon syndrome occurs once in 25,000 births, whereas Apert syndrome occurs once in 100,000 births. A craniosynostosis registry maintained for 3 years in Colorado revealed a birth prevalence of 14.1 per 10,000 live births (02).
The sagittal suture is the most commonly involved suture, constituting more than half of all craniosynostosis. Sagittal synostosis is rarely syndromic and is more frequent in males by a ratio of 3:1 (54). Unilateral or bilateral involvement of the coronal suture is the next most common; however, the male:female ratio is 1:2 (66). Bilateral coronal synostosis is more frequently familial than unilateral synostosis, and it is often associated with involvement of other sutures. Metopic synostosis occurs in less than 10% of nonsyndromic craniosynostosis. However, most series represent patients with clinically significant craniosynostosis. Lesser degrees of metopic stenosis may exhibit a palpable ridge and slight hypotelorism; this is probably the commonest craniosynostosis (106). Examination of portraits and the exhumed skull of the composer Wolfgang Amadeus Mozart confirms he had metopic synostosis (91). Progression of isolated, single suture synostosis to that of multiple sutures is uncommon.
There appears to be an increased risk of craniosynostosis in the offspring of mothers who smoke or live at high altitude (03) and who take certain medications during pregnancy (20). Fetal ultrasound studies have identified syndromic (62; 108) and isolated (113) sutural synostosis early in pregnancy. Apert syndrome has been confirmed by molecular diagnosis of amniotic fluid (34). There is laboratory evidence that opening involved sutures in utero can minimize craniocerebral changes (114).
In the neonate, craniosynostosis must be differentiated from modeling of the calvarium, which may be caused by abnormal intrauterine positioning or vaginal delivery. Premature infants may have dolichocephaly without sagittal synostosis (51). Many cultures practice infant head wrapping and molding, another source of confusion (35). Asymmetric skull growth may be found in children with chronic subdural hematomas, slowly growing brain tumors, and cerebral hemiatrophy (33).
Multiple secondary suture synostosis may occur in disorders with decreased brain volume or microcephaly. These include fetal alcohol syndrome, prenatal infection, hypoxic-ischemic encephalopathy, Chiari malformation, shunted hydrocephalus, and a variety of underlying genetic or metabolic disorders. Appropriate diagnosis of microcephaly may avert unnecessary surgery for secondary craniosynostosis.
The most difficult distinction is in the infant with occipital plagiocephaly. This condition, attributed to unilateral lambdoidal synostosis, was uncommon before the 1990s. A large increase was attributed by Kane and colleagues to the increase in the supine positioning of infants for sleeping (60). In 1992 the American Academy of Pediatrics recommended the “back to sleep” position to prevent sudden infant death syndrome (SIDS). Intrauterine cranial restriction may also play a role (69), and these children usually exhibit some cranial flattening at birth (83). Certainly, there appear clinically to be two syndromes: (1) the rare, true lambdoidal synostosis and (2) the much more common lambdoid positional molding (15). Positional plagiocephaly occurs in the absence of suture synostosis and is due to external forces that deform the shape of the skull (ie, when the child is supine) (107). In this condition there is flattening of one side of the occiput, with anterior displacement of the ipsilateral ear. Repositioning of the child to the nonaffected side is effective for mild cases. In more severe deformities, helmet therapy is employed with good results for children up to 18 months of age (23).
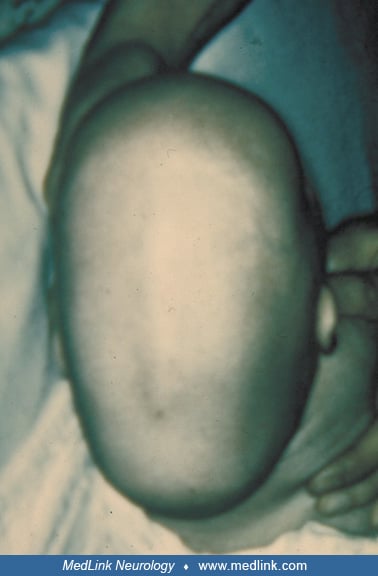
Because craniosynostosis often starts in utero, the condition should be diagnosable at or shortly after birth. Careful examination of head shape and facial appearance is essential to identify both syndromic and nonsyndromic craniosynostoses, as is limb and general examination for the former. Examination of parents and review of their childhood photographs is important. Anthropometric measurements of the affected child's skull are typically abnormal. In addition to measuring the occipitofrontal circumference, it is helpful to measure the cephalic index, which is the ratio of biparietal diameter to anteroposterior diameter x 100. The normal cephalic index ranges from 78 to 84; scaphocephaly is associated with a decreased value and brachycephaly with an increased value.
A diagnosis of craniosynostosis is usually made by clinical examination; findings include a head shape consistent with the palpably closed suture(s), often characteristic orbital, maxillary, and limb changes. A palpable ridge may replace the stenosed suture.
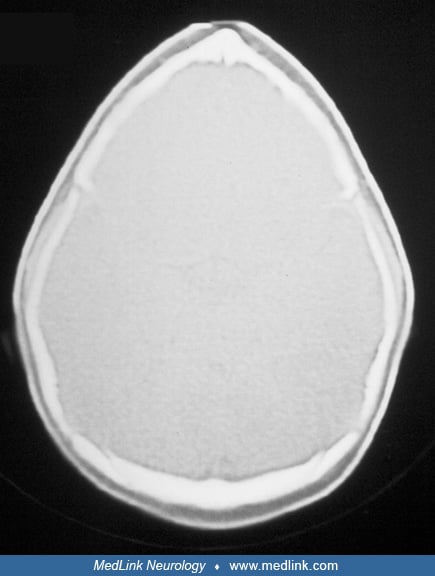
Radiologic evaluation offers confirmation and provides supplemental information that may be useful for management. Classic findings on routine x-ray include sclerosis or indistinctness along the suture and ridging of bone at the suture. Skull radiographs, however, may show synostosis along a few millimeters of suture, despite clinical evidence of inadequate skull growth along the length of the suture. CT scanning has been considered the best modality for diagnosing craniosynostosis, although problems with volume averaging occasionally interfere with demonstration of small areas of synostosis.
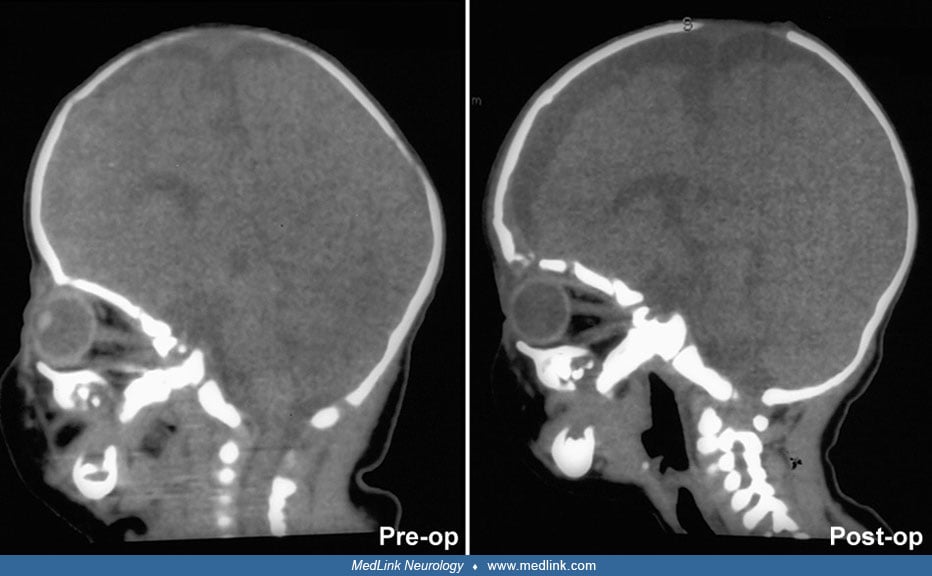
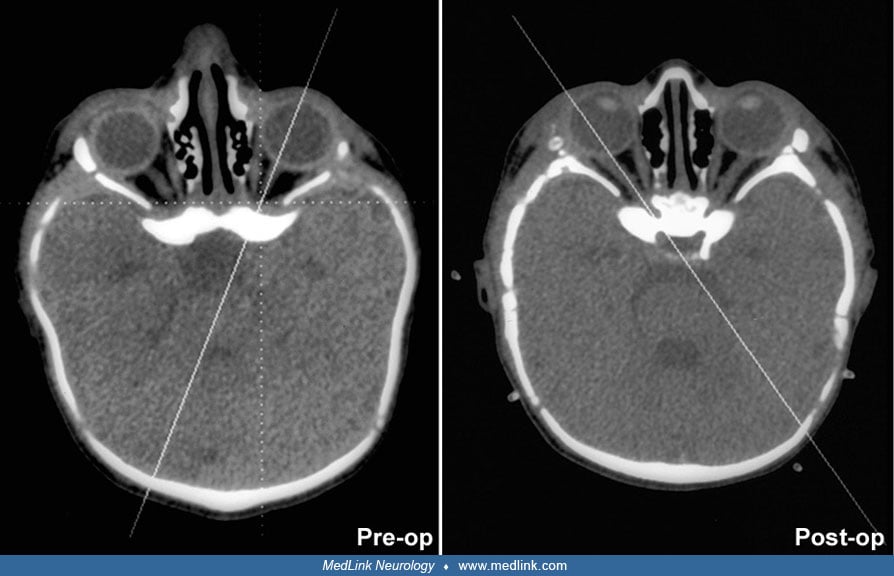
Thin-section, 3-dimensional CT enhances specificity of diagnosis and provides additional information about associated abnormalities of the basicranial and facial bones (07; 74; 79). Neuroimaging also permits evaluation of the brain, which may be abnormal in children with syndromic synostoses. Like bicoronal synostosis, metopic synostosis is associated with other abnormalities, such as cleft palate and holoprosencephaly. MRI offers additional information about neuronal migration abnormalities, hindbrain herniation and brainstem compression, delayed myelination, and oropharyngeal obstruction (46). Rozovsky and associates showed that cranial ultrasound is useful as a first-line imaging examination for diagnosis of craniosynostosis as it provided substantial accuracy for detection with 100% sensitivity and 98% specificity (97). Additionally, the use of cranial ultrasound removes the harmful effects of ionizing radiation, which can be particularly deleterious in young infants (97). Also, it can be diagnosed intrauterine by a variety of neuroimaging techniques that can improve outcome in these children (47). Neuroimaging may not be essential unless there is some question regarding the diagnosis or where help is needed in surgical planning (14).
Secondary radiologic signs of craniosynostosis are caused by the deforming pressure of growing brain after suture closure. Elevated intracranial pressure in children with multiple suture stenosis may lead to increased convolutional markings, erosion of the floor of the sella turcica, and flattening of the anterior and posterior clinoids.
Cervical spine radiographs should be considered to identify craniovertebral abnormalities or instability that may lead to spinal cord injury during prone positioning for surgery.
Neuropsychological testing should be obtained to provide a useful baseline and serial monitor of developmental progress in children with craniosynostosis. With younger children, in whom such testing is difficult and not always reliable, serial evoked responses can be used to monitor neurologic development (28).
Treatment approaches depend on the type of craniosynostosis and the age of the child. Knowledge of the child's developmental potential and parental expectations may also guide selection of management options. There is no clear-cut evidence that positional or deformational occipital plagiocephaly requires surgical correction or orthotic devices. There is also less enthusiasm for operation in milder forms of metopic synostosis, as untreated cases may not progress.
Surgery for craniosynostosis is guided largely by the sutures involved and the patient’s age and appearance.
Simple linear craniectomy rarely produces satisfactory results; more complex procedures are typically required for removal of abnormal sutures and reconstruction of the skull. Blood loss and transfusion needs can be minimized by technical adaptations (112). Some surgeons recommend a staged craniectomy, whereas others recommend a morcellation procedure or "expanding cranioplasty," allowing immediate, partial reshaping of the calvarium (77; 52). This procedure involves separating the calvarium into several segments of bone that can be repositioned, thereby allowing future growth and remodeling. Hashim and colleagues compared neuropsychological outcomes following treatment with whole-vault cranioplasty or endoscopic strip craniectomy for patients with sagittal craniosynostosis and reported that patients undergoing early whole-vault cranioplasty attained higher intelligence quotient and achievement scores relative to those undergoing strip craniectomy (43).
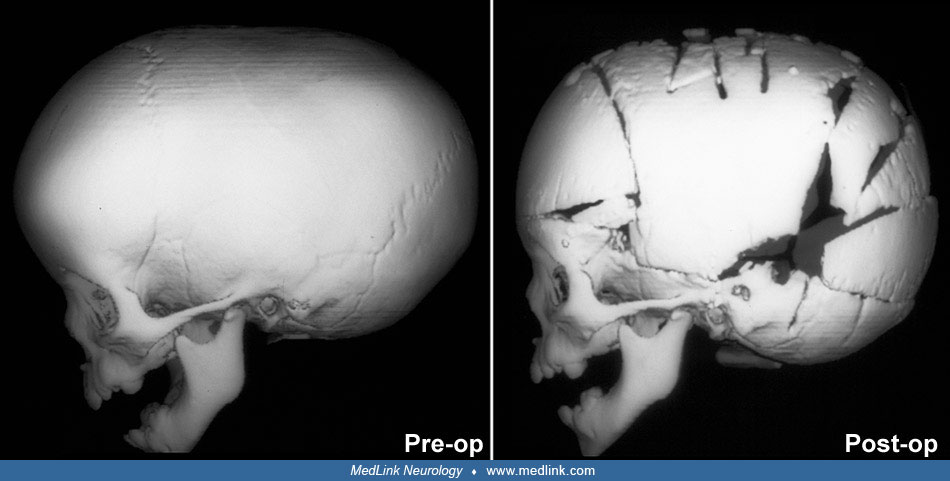
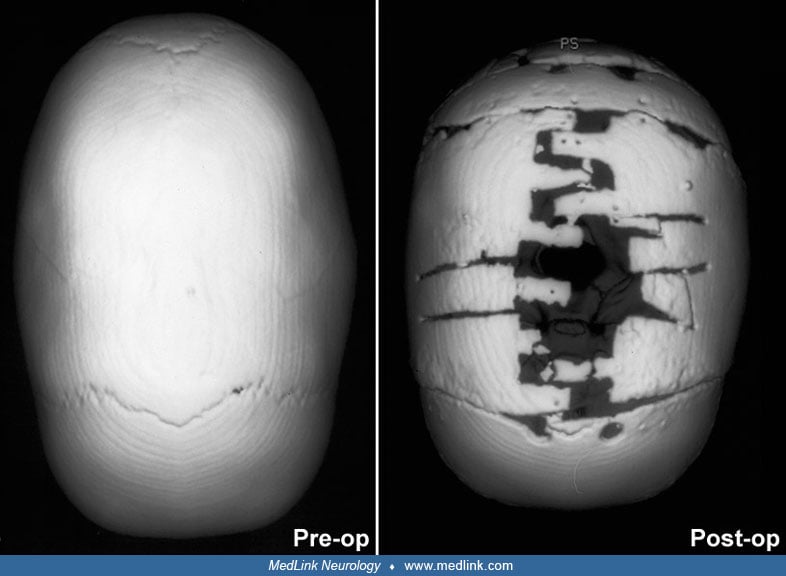
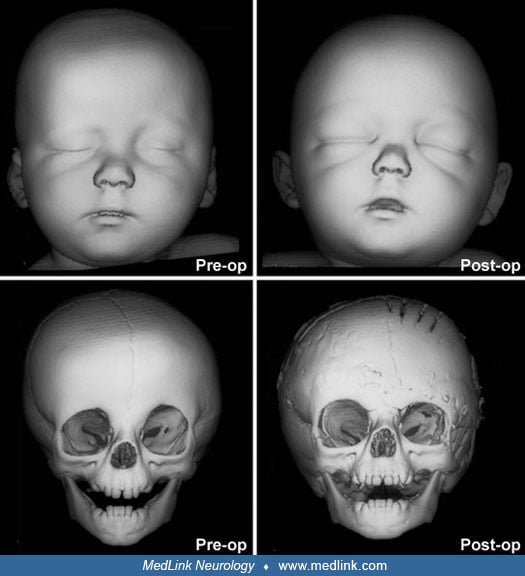
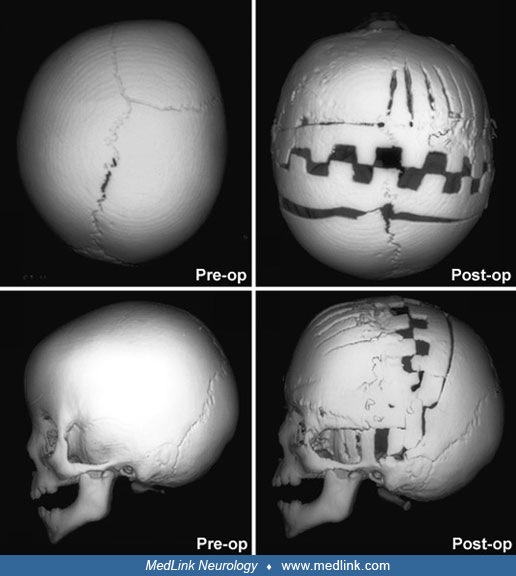
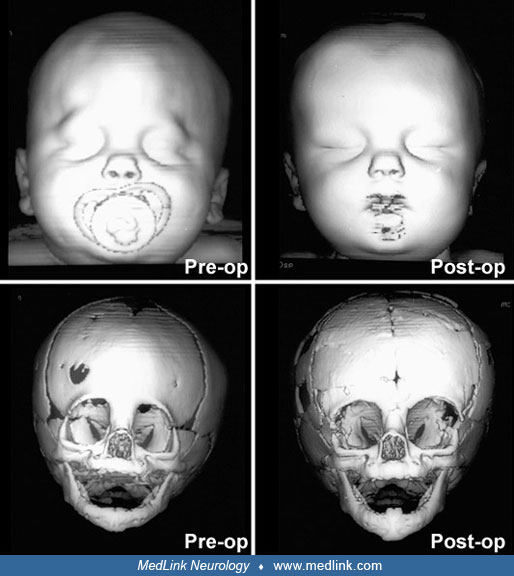
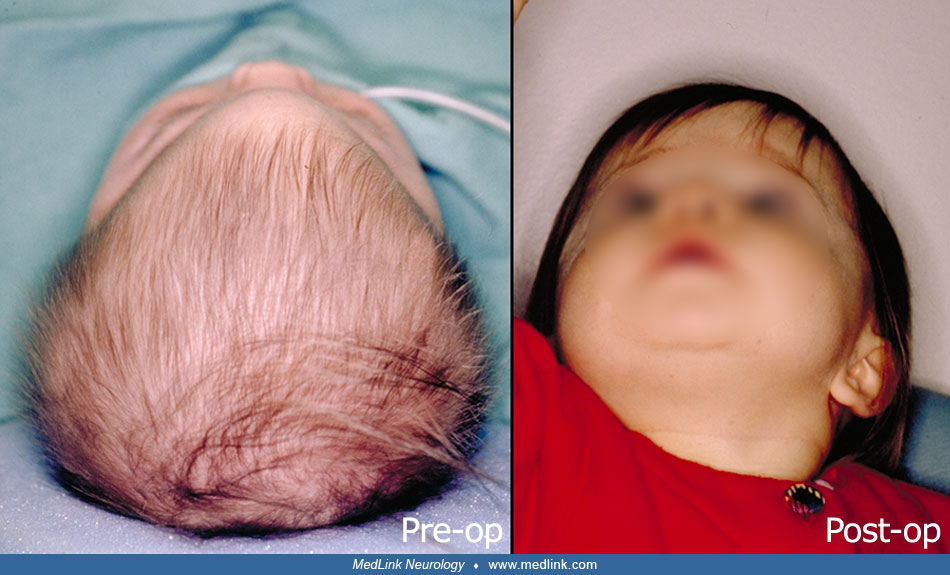
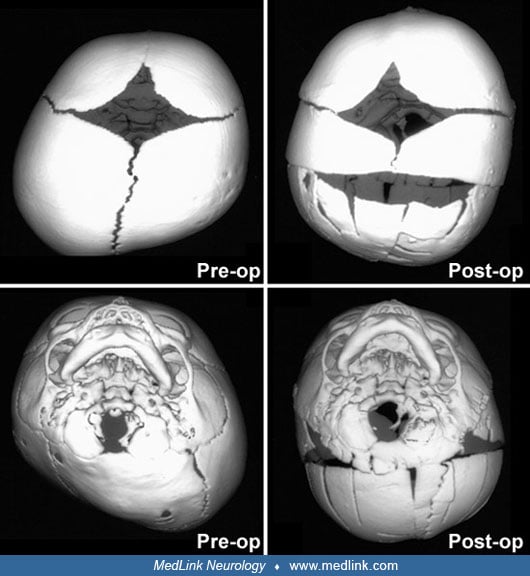
Timing of surgical intervention is important, as it affects the capability of the craniofacial bones to achieve a more nearly normal pattern of growth (87; 89; 103). Because the rate of brain growth decreases in older children, those with craniosynostosis may require more extensive surgery to achieve results similar to those achieved in younger children with limited surgery. Sagittal synostosis is best treated as early as 6 weeks of age. Surgery by 3 to 6 months of age permits restoration of near-normal skull contour. Surgery for children under 1 year of age requires loose fixation of bone segments to allow for brain growth, whereas those over 3 years of age require rigid fixation (88). Because metallic hardware often erodes through thin scalps, absorbable cranial fixation devices have been developed (30). Although early surgery is optimal for most calvarial abnormalities associated with craniosynostosis, surgical treatment of associated facial abnormalities should be delayed. Premature treatment of midface hypoplasia impairs growth potential and ultimately causes greater facial deformity. Changes of the cranial base that accompany synostosis of individual cranial sutures develop slowly. Based on their studies of orbital development, Bentley and colleagues recommend that frontoorbital advancement surgery be delayed until late in the first year of life (08). Distraction methods have high success rates in older children (81). Illustrations of surgical technique and results include skin incision, sagittal synostosis, and synostoses.
A minimally invasive surgical approach has surfaced with the introduction and widespread use of endoscopic suturectomy (76). This is based on the principle that if timely intervention occurs, the rapidly growing brain will cause expansion of the skull into the normal shape. The procedure involves the insertion of the endoscope through small scalp incisions and the subsequent removal of a strip of bone including the fused suture (76). No internal fixation is used. It is generally recommended for younger children and involves the use of a postoperative cranial molding helmet. Surgeons have been able to demonstrate minimal blood loss, short operative times, early hospital discharge, and excellent functional and cosmetic results for these procedures (56; 50). The efficacy of this technique has been established in the treatment of isolated sagittal synostosis (56). Several groups have expanded the use of the endoscope for other forms of single suture synostosis (57; 58; 42). Additionally, cost analysis has shown that endoscope-assisted craniectomy plus helmet therapy is a less costly surgical option for patients with sagittal craniosynostosis than open cranial vault repair (123).
Children with extensive craniosynostosis require a multidisciplinary approach to optimize diagnosis, treatment, and long-term outcomes. A dedicated craniosynostosis team should include specialists in craniofacial surgery, neurosurgery, developmental pediatrics, genetics, neuropsychiatry, anesthesiology, ophthalmology, and physical therapy, as well as ophthalmologists, geneticists, psychologists, orthodontists, and pediatricians. Comprehensive ophthalmologic evaluation is essential, as visual impairment and intracranial hypertension are significant concerns. Corrected visual acuity is the best measure of central afferent function, whereas fundoscopic and visual field examinations can help assess increased intracranial pressure. Cycloplegic refraction and assessment of individual and conjugate eye movements should be included. Due to the shallow orbits often seen in craniosynostosis, particularly in syndromic cases (31), children are at increased risk for exposure keratopathy, necessitating careful monitoring and intervention when needed.
According to Persing, although the majority of patients with single-suture nonsyndromic craniosynostosis do not experience intracranial hypertension, at least 15% have elevated intracranial pressure, and symptoms may be subtle and difficult to assess in young children. Additionally, strabismus is reported in up to 90% of children with unilateral coronal synostosis, and many of these cases require ophthalmologic intervention (86).
Associated hydrocephalus, which occurs in a subset of craniosynostosis cases, rarely resolves following synostosis surgery and typically requires ventriculoperitoneal shunting (22). Genetic counseling is crucial, especially in syndromic cases where mosaicism, variable penetrance, and expression of dominant mutations can influence recurrence risks and family planning. Pediatricians play a key role in managing hearing, feeding difficulties, and failure to thrive, whereas neuropsychologists are essential for evaluating and addressing developmental and cognitive concerns. Given the psychosocial impact on families, family counseling and support services should be an integral part of care (100).
There is a higher incidence of perinatal complications associated with prenatal craniosynostosis (47).
Increased intracranial pressure, airway impairment, and associated anomalies present special anesthetic risks in these children, requiring careful planning (120; 73). Perioperative airway management is required in a large proportion of children with craniofacial anomalies (85; 73).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Brittany Poinson MD MSEd
Dr. Poinson of Tulane University has no relevant financial relationships to disclose.
See Profile
Alcy R Torres MD FAAP
Dr. Torres of Boston Medical Center and Boston University Chobanian and Avedisian School of Medicine has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026