

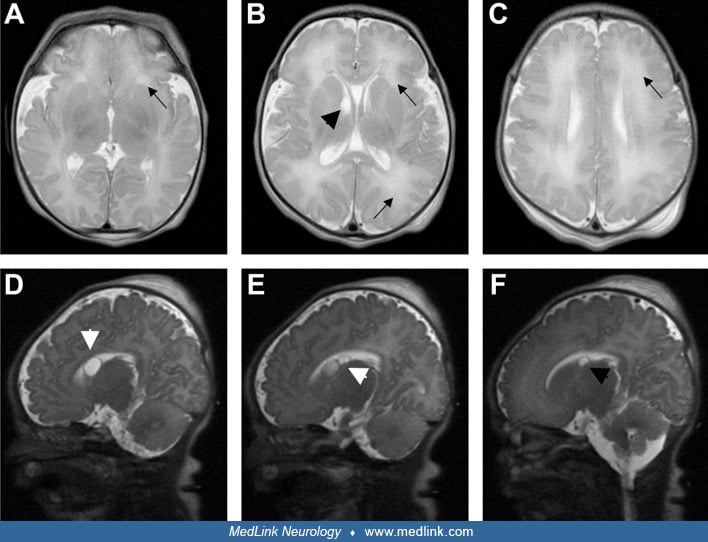

Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Peroxisomal disorders are genetic conditions that are characterized by either a dysfunction of or a total loss of peroxisomes in the cell. These diseases can be divided into biogenesis disorders that are due to a global defect in peroxisome assembly or to single enzyme defects. Peroxisome biogenesis disorders are a heterogeneous group of rare autosomal recessive diseases in which there is a failure to form functional peroxisomes, resulting in deficiencies of multiple enzymes targeted to this organelle and progressive multisystem diseases. This article highlights the molecular and biochemical pathogenesis of these diseases, with a focus on peroxisome assembly defects, and highlights the marked strides in understanding the pathophysiology.
Peroxisomal assembly and biogenesis disorder can present with: | |
• Neurologic abnormalities (ie, neuronal migration defects and germinal cysts, polyneuropathy, epilepsy, ataxia, and leukodystrophy) | |
• Retinal abnormalities (ie, retinal dystrophy) and sensorineural hearing loss | |
• Bone abnormalities | |
• Liver dysfunction and liver fibrosis with bile acid defects leading to cirrhosis | |
• Dysmorphic features (including high forehead, epicanthal folds, micrognathia, very large fontanelles, and shallow supraorbital ridges) | |
• The age of onset often corresponds with severity. |
Peroxisomes are single-layer lipid bilayer organelles that play a role in lipid metabolism, beta oxidation, and reduction of reactive oxygen species.
Peroxisomes were first identified as “microbodies” in mouse kidney in 1958, and purified microbodies from rat liver were then found to be active in peroxide-linked oxidation reactions, leading to the name "peroxisomes" (40). These single-membraned organelles were found in all eukaryotic cells, and their role in fatty acid oxidation, and their absence in patients with Zellweger syndrome (31) stimulated interest in peroxisome biology. A reliable assay for peroxisome dysfunction based on elevated serum very-long-chain fatty acids (with carbon chain lengths of C24 and above) defined a new category of human metabolic disease (47).
The failure to assemble normal peroxisomes and the impaired ability to import peroxisome matrix proteins results in multiple deficiencies of peroxisomal enzymes (peroxisome biogenesis and assembly disorder). Disorders of peroxisome assembly can be divided into two classes: the peroxisome biogenesis disorders–Zellweger syndrome spectrum and rhizomelic chondrodysplasia punctata. The peroxisome biogenesis disorders may affect the brain, retina, craniofacies, kidney, and skeleton (79; 15; 40).
Peroxisome biogenesis disorders-Zellweger spectrum disorders (PBD-ZSD) are defined by a spectrum of disease ranging from mild to severe and encompassing the clinical disorders of Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. These disorders were described before the relationship to peroxisome deficiency was known; thus, the terms do not relate directly to the underlying gene defect. More recently, these conditions have been described as mild, intermediate, or severe peroxisome biogenesis disorders.
All these disorders are due to mutations in the same set of peroxisome biogenesis genes, referred to as the PEX genes. Human disease-causing PEX genes are homologous to peroxisome biogenesis machinery in other species, and they were originally determined through a complementation approach in which patient fibroblast cell lines were collected and fused for biochemical complementation. This approach defined at least 13 complementation groups, each found to represent a different gene defect (48). Many PEX genes were subsequently identified by screening human cDNA libraries (23). Thirteen PEX genes are associated with Zellweger spectrum disorders (ranging from mild to severe in phenotype) (65; 66). In contrast, rhizomelic chondrodysplasia type 1 is associated with a defect in the PEX7 gene, or specific mutations in PEX5 (13).
A variety of historic labels have been ascribed to the Zellweger spectrum (cerebrohepatorenal syndrome, hyperpipecolic acidemia, neonatal adrenoleukodystrophy, infantile Refsum disease). Current nomenclature includes peroxisome biogenesis disorders-Zellweger spectrum disorders (PBD-ZSD), peroxisome biogenesis disorders–rhizomelic chondrodysplasia punctata, and peroxisome single enzyme defects, such as X-linked adrenoleukodystrophy (X-ALD).
|
• There are a range of phenotypes from severe to mild, encompassing Zellweger syndrome to rhizomelic chondrodysplasia punctata phenotypes. | |
|
• More severe phenotypes (eg, Zellweger syndrome) present and are identified at earlier ages. |
Peroxisome biogenesis disorders-Zellweger spectrum disorders (PBD-ZSD) presents with a range of severity, including severe, intermediate, and mild. The variation in disease severity is secondary to the effect of the mutation on protein function but not to the specific PEX gene involved. A summary of clinical phenotypes is found in Table 1. Newborn screening for X-linked adrenoleukodystrophy can incidentally identify individuals with PBD-ZSD and a number of single enzyme defects, such as D-bifunctional protein deficiency. Because it has been added to the recommended uniform screening panel in the United States, there may be an increased number of individuals identified in this way, but clinical accuracy is an important issue. Table 2 shows possible results for screening for this differential.
|
Syndrome |
Clinical |
|
Severe PBD-ZSD (previously known as Zellweger syndrome) |
Severe hypotonia |
|
Intermediate PBD-ZSD (previously known as neonatal adrenoleukodystrophy) |
Hypotonia |
|
Mild PBD-ZSD (previously known as infantile Refsum disease) |
Variable cognitive and motor deficiencies |
|
Rhizomelic chondrodysplasia punctata (RCDP) |
Prominent skeletal abnormalities with proximal shortening of long bones and coronal clefts in vertebrae |
|
Disorder |
VLCFA |
Plasmalogens |
Phytanic acid |
Pristanic acid |
Bile acids | |
|
PBD-ZSD |
↑ |
↓ or N |
↑ or N |
↑-N |
↑-N | |
|
Rhizomelic chondrodysplasia punctata |
N |
↓ |
N-↑ |
↓-N |
N | |
|
X-linked adrenoleukodystrophy (X-ALD) |
↑ |
N |
N |
N |
N | |
|
Refsum disease |
N |
N |
↑ |
↓ |
N | |
|
α-Methyl-acyl-CoA racemase deficiency |
N |
N |
(↑) |
↑ |
↑ | |
|
D-bifunctional protein |
↑ |
N |
↑ or N |
↑ or N |
↑ | |
|
Acyl-CoA oxidase (ACOX) |
↑ |
N |
↑ or N |
↑ or N |
↑ | |
|
| ||||||
Peroxisome biogenesis disorders-Zellweger spectrum disorders (PBD-ZSD). The spectrum Zellweger ranges from most severe (Zellweger Syndrome) to mild (infantile Refsum disease). Severe cases present at birth with significant disease (Table 1). Intermediate and mild cases were formerly labeled neonatal adrenoleukodystrophy and infantile Refsum disease, and these can present later in life (several months to a few years of age) and are more variable in terms of the degree of facial dysmorphism and other symptoms (Table 1) (09; 26). Children with these disorders have significant morbidity, including pigmentary retinopathy, hearing loss, liver and adrenal dysfunction, seizures, and intellectual disability.
The clinical severity is determined by the level of facial dysmorphology, degree of hypotonia, level of delay in psychomotor development, timing of presentation, and amount of chondrodysplasia punctata (as compared to individuals with rhizomelic chondrodysplasia punctata).
Individuals with severe Zellweger spectrum are often diagnosed early, and most die within the first year of life secondary to infection, aspiration, progressive liver disease, or underlying neurologic dysfunction.
Patients with mild PBD-ZSD may survive through adolescence and may manifest moderate to severe cognitive impairment, progressive retinopathy, and deafness.
Although these disease names remain useful for teaching purposes, individual patients can exhibit a great deal of variation in terms of severity (09).
General appearance. Patients with severe Zellweger syndrome may have a normal birth weight but often exhibit hypotonia and growth failure. The characteristic facial appearance includes a high forehead, large anterior fontanelle, flat supra-orbital ridges, epicanthal folds, broad nasal bridge, small nose with anteverted nares, and external ear abnormalities (40). The stereotypic Zellweger facies is less apparent in milder phenotypes.
Eye abnormalities. Visual defects include corneal clouding, cataracts, and congenital glaucoma as well as optic atrophy and pigmentary retinopathy. Virtually all patients with infantile Refsum have pigmentary degeneration of the retina, which can be progressive, and a depressed electroretinogram (ERG). Hyperpigmentation, characterized by areas of photoreceptor degeneration with patchy hypertrophy, nodular hyperplasia, and atrophy of the pigment epithelium is observed in neonatal adrenoleukodystrophy patients (42).
Neurologic features. There are three major types of nervous system involvement: neuronal migration abnormalities, white matter abnormalities, and selective neuronal involvement (58). These processes are reflected on examination by profound hypotonia, poor suck, and depressed neonatal and deep tendon reflexes. In severe Zellweger syndrome, seizures are common, and there is minimal psychomotor development. Hearing loss and retinal degeneration occur in all milder patients. Leukodystrophy may develop in longer-surviving patients. The EEG, brainstem auditory evoked responses, and somatosensory evoked responses are typically abnormal in Zellweger syndrome (33). MRI of the brain can show impaired myelination, abnormal cortical gyral patterns, ventricular dilatation, and germinolytic cysts (06).
For the most part, individuals with Zellweger spectrum disorders have intellectual disability, but there are reports of average intellect in some milder patients (60; 64). A caregiver survey reported approximately 63% of individuals with Zellweger syndrome have abnormal EEGs, and about 48% of these patients have clinical seizures (09).
Visceral abnormalities. Hepatocellular disease with liver enlargement is observed in 78% of Zellweger spectrum cases, fibrosis in 76%, micronodular cirrhosis in 37%, and cholestasis in 59%. Liver biopsy may reveal striking iron storage and periportal inflammation, but the iron storage is diminished after the age of 20 weeks. There are renal cortical cysts best visualized by CT scan, with 78 of 80 patients demonstrating renal cysts at autopsy. Renal functional defects include generalized aminoaciduria and proteinuria. Adrenal function may be deficient on provocative testing or during crises, and severe infections are frequent. Cardiac anomalies are also increased, with 32% of patients having ventricular septal defects and 22% having aortic abnormalities (26).
Genital anomalies include cryptorchidism or labial hypoplasia and clitoromegaly (40).
Rhizomelic chondrodysplasia punctata spectrum (RCDP). Around 90% of patients with RCDP have severe disease and rarely survive beyond childhood; 25% die in early infancy. RCDP is distinguished by shortening of the proximal limbs (rhizomelia; humerus more affected than femur), extensive punctate calcifications in cartilage (chondrodysplasia punctata), congenital cataracts, and profound growth and developmental delays. Most children have seizures. Cervical spine stenosis may be underappreciated, and spinal cord compression secondary to the chondrodysplasia is reported (37). Ichthyotic skin changes are noted in less than one third of individuals. Most children will have increased respiratory tract infections, increased aspiration risk, and small chest size.
As in Zellweger spectrum, there is a slightly increased incidence over that in the general population for congenital malformations such as cleft palate and renal and cardiac malformations. Death is often secondary to respiratory complications from chronic aspiration, restricted chest wall movement, and a small pulmonary cage (76; 52). Cranial imaging and brain MR spectroscopy have shown delayed myelination, decreased choline-to-creatine ratios, and increased levels of mobile lipids, thought to reflect the deficiency of plasmalogens, which are substantial components of myelin (01).
Management focuses on treating complications and providing supportive care. Cataract removal may help with returning some vision. Physical therapy may help with contractures. Dietary restriction of phytanic acid may have some role in milder cases, though the experience in the field with dietary therapy for this purpose remains anecdotal. Please discuss with your local metabolic physician and their dietitian if you would like more details about this restriction. Poor feeding may require gastrostomy tube placement to maintain nutrition.
Management of respiratory status and prevention of respiratory illnesses is important. Individuals should undergo the usual childhood vaccinations on schedule as well as annual influenza vaccination and respiratory syncytial virus monoclonal antibody.
Docosahexaenoic acid and other bile acid supplementation may be required if deficient. A high index of suspicion if not tolerating feeding or not absorbing feeds should lead to evaluation of bile acid levels.
Single peroxisomal enzyme abnormalities. Sometimes the peroxisome forms and assembles correctly, but some of its functional enzymes are not formed correctly and consequently do not function fully.
Disorders of peroxisomal beta-oxidation. One major role of the peroxisome is beta-oxidation, and enzymes such as D-bifunctional protein (HSD17B4), acyl-CoA oxidase (ACOX), and sterol carrier protein x (one of ketothiolases, SCP2) can be disturbed. Individuals can have physical and biochemical findings similar to Zellweger syndrome but with normal plasmalogens (elevated VLCFA) and increased specific bile acids.
X-linked adrenoleukodystrophy. X-linked adrenoleukodystrophy is one of the more common peroxisomal disorders. Individuals with X-linked adrenoleukodystrophy have a deficiency of ATP-binding cassette transporter for the import of VLCFAs (or their CoA intermediates) into the peroxisome. This is caused by the gene ABCD1 on the X-chromosome. As a result, males are often more severely affected than females. Females can be severely affected depending on their X-chromosome lyonization status.
There is a spectrum of disease ranging from childhood cerebral adrenoleukodystrophy to adrenomyeloneuropathy to Addison disease only, all of which are discussed in this article. X-ALD has been added to the recommended uniform screening panel in the United States. For those with positive newborn screen results and confirmed genetic changes in ABCD1, screening recommendations to identify the childhood cerebral form are early, allowing for interventions with early hematopoietic stem cell transplant.
The childhood cerebral form (usually seen in about 35% of boys 4 to 12 years of age) presents in childhood with school failure, behavioral changes, visual/hearing loss, and intellectual regression (68). For the childhood cerebral form, early hematopoietic stem cell transplantation can be done. It is important that the timing is appropriate and protocols exist for screening and timing of interventions. Elivaldogene autotemcel is an FDA-approved one-time gene therapy for boys between 4 and 17 years of age with early cerebral X-ALD. PPAR-gamma agonists are also being studied in reducing demyelination and inflammation in cerebral X-ALD.
Newborn screening has been initiated for X-linked adrenoleukodystrophy in nearly all the states in the United States. Different states have slightly different approaches, but the screening focuses on measuring the C26:0 by tandem mass spectrometry and then using C26:0-lysophosphatidylcholine (LPC) by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry (71). Positive newborn screens are confirmed typically by very-long chain fatty acid analysis, phytanic acid level, pipecolic acid levels, plasmalogens, and molecular testing for mutations in ABCD1.
If found to have mutation in ABCD1 and positive biochemical testing (or positive biochemical testing consistent with the diagnosis and is awaiting genetic testing), the patient undergoes serial MRIs to look for the development of lesion(s) consistent with X-linked adrenoleukodystrophy. Mallack and colleagues provide recommendations for the timing of these serial MRIs (eg, yearly, etc.); different institutions may choose different intervals (43).
Adrenomyeloneuropathy form presents in early adulthood in males (approximately 65%) and in at least 20% of females at ages greater than 35 years with progressive spastic paraparesis, sphincter problems, impotence, mixed demyelination, and axonal peripheral neuropathy.
Addison disease, due to adrenal insufficiency, can be the only symptom or in conjunction with other symptoms. Patients can have hyperpigmentation, fatigue, muscle weakness, hypoglycemia, salt craving, irritability, depression, hypotension, and bradycardia. In addition, they can develop adrenal crisis when under physical or psychological stress, which can lead to severe hypotension, hypoglycemia/hyperglycemia, and confusion. All individuals with X-linked adrenoleukodystrophy (or if suspected based on newborn screening results before confirmation) should have their adrenocorticotrophic hormone (ACTH) and cortisol levels checked regularly (eg, every 6 months) and abnormal levels followed by endocrinology for adrenal replacement therapy with steroids including stress dosing when under significant physical or psychological stress.
Refsum disease (not to be confused with infantile Refsum disease). Refsum disease presents in school age and is due to abnormal phytanoyl-CoA hydroxylase (PHYH) 90% of the time, which is imported by peroxisomal targeting signal receptor 2 (PTS2) (encoded by PEX7 and accounting for approximately 10% of cases).
Individuals often present with retinitis pigmentosa and then develop other symptoms such as anosmia, polyneuropathy, cerebellar ataxia, and deafness (77). Skin ichthyosis is common and often starts in adolescence. Approximately 30% of individuals will have short metatarsals (especially the fourth). Most individuals with Refsum disease have average intellectual ability. Cardiac arrhythmias and cardiomyopathy can be life-limiting.
Dietary limitation of phytanic acid and a high-calorie diet help treat the ichthyosis, sensory neuropathy, and ataxia. In some acute cases, plasmapheresis or lipid apheresis are used in life-threatening arrhythmias or extreme weakness.
Alpha-methyl-acyl-CoA racemase deficiency. Alpha-methyl-acyl-CoA racemase deficiency presents in adulthood with sensorimotor neuropathy or as a neonate with hepatopathy and encephalopathy. It is a deficiency of the isomerization of pristanic acid and bile acid intermediates needed for beta-oxidation (AMACR gene). Individuals have increased pristanic and possible phytanic acid. Therapy focuses on replacement of bile acids in infancy because this enzyme isomerizes pristanic acid and bile acid intermediates for beta-oxidation, and its loss leads to deficiency in bile acids.
ACOX1: Peroxisomal acyl-CoA oxidase deficiency and Mitchell syndrome. ACOX1 encodes the peroxisomal acyl-CoA oxidase. Peroxisomal beta-oxidation of fatty acids involves three key components: the acyl-coA oxidase, the D-bifunctional protein encoded by HSD17B4, and 3-oxoacyl-coA thiolase (ACAA1). Autosomal recessive loss of function of ACOX1 leads to a neonatal and infantile disorder with deficiency of peroxisomal acyl-CoA oxidase (56). This condition, which was originally called “pseudoneonatal adrenoleukodystrophy” leads to defects in beta oxidation and overlapping features with PBD-ZSD (30). Clinical features include neonatal hypotonia, epilepsy, leukodystrophy, neurologic regression, hepatic steatosis, hearing loss, nystagmus, strabismus, optic atrophy, and retinal degeneration.
More recently, a de novo heterozygous missense variant in ACOX1 (N237S) was identified in patients with peripheral neuropathy, ataxia, leukoencephalopathy, and seizures with age of presentation in adolescence or adulthood (17). These patients exhibit a unique neuropathology distinct from peroxisomal acyl-CoA oxidase deficiency (35).
Genetic testing for confirmation. Clinical presentation and biochemical findings are sufficient to identify particular phenotypes, and traditionally this approach followed by molecular testing with gene panels was the typical diagnostic approach. However, with the use of X-ALD newborn screening and more widespread testing with whole exome sequencing and whole genome sequencing earlier in life, many patients are identified early in life with specific genetic variants. Biochemical testing is essential to validate variants of uncertain significance and to gauge the severity and extent of the peroxisomal defect.
The average age at death for severe Zellweger syndrome patients was around 12 weeks, with liver failure and respiratory infections as common precipitating factors (78); currently, it has only increased to a mean of about 6 months, with most affected individuals dying by 12 months (26). Patients with neonatal adrenoleukodystrophy and infantile Refsum disease vary in prognosis; although they have multiple disabilities, they survive to a later age. Complications include refractory seizures, liver disease with gastrointestinal bleeding and coagulopathy, failure to thrive, adrenal insufficiency, susceptibility to routine infection, and osteopenia secondary to hepatorenal disease. Surviving patients usually have progressive visual and hearing loss. Nearly all patients are mentally disabled. A study has suggested that in combination, the activities of dihydroxyacetone phosphate: acyltransferase and C26:0 acid oxidation correlate with life expectancy (32), but this study requires corroboration. Homozygosity for PEX1-I700fs and PEX1-G843D are associated with severe and mild disease, respectively (55; 65).
Most patients with rhizomelic chondrodysplasia punctata patients show early demise. Of those that survive infancy, 50% are alive at age 5 years, but only a fraction are alive at 10 years. The majority of children do not progress in their development beyond the 3-month level (76). Due to this grave prognosis, historically families have chosen to severely curtail aggressive interventions and have chosen comfort care only. Seizures and respiratory complications are the typical causes of death. Homozygosity for PEX7-L292X predicts a severe course (12).
A newborn female was referred for evaluation of severe hypotonia, poor feeding, large anterior fontanelle, bilateral talipes equinovarus, bitemporal hollowing, broad nasal root, and down-turned corners of the mouth.
The family and gestational histories were normal. Ophthalmology examination revealed retinitis pigmentosa, and there was hepatomegaly (liver edge 4 cm below the right costal margin) with elevated liver enzyme levels including serum alanine aminotransferase (237 U/mL), alkaline phosphatase (1380 U/mL), and lactic dehydrogenase (610 U/mL). A skeletal survey revealed large patellae and punctate calcification around the hips and delayed bone age. A normal karyotype, elevated urinary pipecolic acid, decreased red blood cell plasmalogens, and increased serum very-long-chain fatty acids were consistent with the diagnosis of Zellweger syndrome (47). The child exhibited minimal developmental progress and died at 6 months of age and disseminated intravascular coagulation and hepatic failure.
Peroxisomes are necessary for beta-oxidation for very-long-chain fatty acids, pristanic acid, and intermediates of bile acids. They also play a role in alpha-oxidation of fatty acids (eg, phytanic acid), biosynthesis of bioethers (eg, plasmalogens), and glycoxylate detoxification. Peroxisomes also provide a protected environment for elimination by catalase of reactive oxygen species.
Disorders of peroxisome assembly are autosomal recessive conditions. About 85% of patients with a Zellweger spectrum phenotype and elevated plasma very-long-chain fatty acids have a defect in peroxisome assembly. The remaining patients with Zellweger phenotype have single enzyme defects in one of two enzymes required for peroxisomal fatty acid metabolism: Acyl-CoA oxidase or D-bifunctional protein (73). Rarely, exceptional cases in which only one allele is expressed can appear to mimic autosomal dominant inheritance (27).
About 95% of patients with rhizomelic chondrodysplasia punctata have a defect in PEX7, referred to as type 1 (13; 50). The remaining patients have single enzyme defects in one of the first two peroxisomal steps of plasmalogen synthesis: dihydroxyacetone phosphate: acyltransferase (type 2) and alkyl-dihydroxyacetone phosphate synthase (type 3) (20). These are all inherited in an autosomal recessive manner.
The Zellweger spectrum is associated with defects in at least 13 different PEX genes that encode proteins required for peroxisome biogenesis and maintenance. It is not possible to predict the PEX gene defect of a Zellweger spectrum patient based on phenotype alone. In contrast, biochemical testing distinguishes the three types of rhizomelic chondrodysplasia punctata. The estimated frequency of each gene defect in patients with Zellweger spectrum is shown in Table 3. PEX1 is by far the most common cause of Zellweger spectrum defects, and approximately 96% of these patients have a defect in one of six genes: PEX1, PEX2, PEX6, PEX10, PEX12, or PEX26 (65; 66).
|
Role in peroxisome assembly |
PEX gene |
Estimated frequency in spectrum |
|
Membrane synthesis |
PEX16 PEX19 PEX3 |
Approx 1% <1% <1% |
|
Peroxisome targeting signal-1 receptor |
PEX5 |
2% and <1% of rhizomelic chondrodysplasia punctata |
|
Docking site for matrix protein import |
PEX13 PEX14 |
1.5% <1% |
|
RING domain proteins, translocation function |
PEX2 PEX10 PEX12 |
3% 3% 7.6% |
|
Recycling of targeting receptors |
PEX26 PEX6 PEX1 |
4% 14% 60% |
|
Peroxisome targeting signal-2 |
PEX7 |
95% of rhizomelic chondrodysplasia punctata |
|
Peroxisome division and proliferation |
PEX11B |
<1% |
General model of peroxisome assembly and matrix import. The cellular origin of peroxisomes has been controversial. Previously it was believed that peroxisomes arose from fission of pre-existing peroxisomes (39), but evidence has arisen suggesting that peroxisomes can also arise de novo from endoplasmic reticulum membranes (38). Peroxisomes do divide and proliferate (24). The key steps of peroxisome matrix protein import are listed in Table 3. The import process begins when matrix proteins are synthesized on free cytosolic ribosomes. PEX5 and PEX7 encode the peroxisome targeting signal receptors and escort matrix proteins bearing the signals PTS1 (C-terminal-SKL) and PTS2 (N-terminal-R/KX5Q/HL-) to the peroxisome membrane. PEX13 and PEX14 encode peroxisome membrane proteins that are the putative docking site for the receptor-matrix protein complex. Peroxisome membrane proteins encoded by the genes PEX10, PEX12, and PEX2 provide the site of matrix protein transport across the membrane. PEX1, PEX6, and PEX26 play an important role in recycling PEX5 and PEX7 for another round of import (74).
Cellular and molecular pathogenesis. PEX gene defects can be characterized based on their impact on peroxisome assembly at the cellular level. For example, patients with PEX3, PEX16, and PEX19 defects appear to have no residual peroxisome structures in cell culture. In contrast, PEX7 defects do not affect peroxisome morphology but fail only to import PTS2 matrix proteins. The remaining PEX gene defects perturb peroxisome metabolism globally but retain residual peroxisome structures, termed ghosts (62).
Most of what is known about peroxisome morphology comes from the study of liver biopsies or cultured skin fibroblasts. Interestingly, there is not always concordance of peroxisome morphology between the same patient’s liver biopsy and cultured skin fibroblasts, with the liver usually displaying more severe abnormalities. In most samples from Zellweger patients, hepatic peroxisomes were not detectable or were present in small numbers (21). This is different from cultured fibroblasts, in which most Zellweger cells have peroxisomes that are reduced in number but enlarged in size as compared to normal controls (62). In rhizomelic chondrodysplasia punctata hepatic peroxisomes appear enlarged but have normal morphology in fibroblasts (21).
Furthermore, the assembly defect is strongly influenced by the class of gene mutation. PEX mutations predicted to result in null alleles are associated with Zellweger syndrome, and missense mutations are more often associated with neonatal adrenoleukodystrophy or infantile Refsum disease. Around 80% of patients with PEX1 defects have at least one of two common mutations, I700fs and G843D. Homozygosity for the nonsense allele I700fs is associated more often with Zellweger syndrome, whereas homozygosity for the missense allele G843D is associated with infantile Refsum disease (55). PEX7-L292X homozygosity is also severe.
Histological abnormalities. In the Zellweger spectrum, neuropathological studies have demonstrated a defect in neuronal migration that begins about the third month of gestation. Macroscopically brains frequently have polymicrogyria and pachygyria, hypoplasia of the corpus callosum, and deficient olfactory lobes (78). Microscopically detectable changes include heterotopias of the cerebral neurons and Purkinje cells of the cerebellum, dysplasia of the olivary complex, and hypomyelination. Demyelination is a frequent complication of older patients. Trilamellar inclusions can be detected in the brain, adrenal gland, and kidney.
In rhizomelic chondrodysplasia punctata, routine brain imaging is normal or has shown delayed myelination with cerebral and cerebellar atrophy. Lens pathology has not been reported. In the CNS, there is a general decrease in neuron number and white matter, dysplastic olives, and progressive cerebellar degeneration (57). In epiphyseal bone, the most marked abnormalities are in the resting cartilage, which shows focal areas of degenerating chondrocytes, calcification, cyst formation, and vascularization. The zone of hypertrophic chondrocytes is diminished and disorganized (22).
Animal models. Null mouse models have been developed for Pex2, Pex5, Pex7, Pex11beta, and Pex13. These mice have severe phenotypes, as expected.
Pex2 Zellweger mice have been useful for defining the neuropathology that is observed in these disorders (28; 29). Biochemical analysis of Pex2 mutant mice shows the characteristic accumulation of very-long-chain fatty acids and deficient plasmalogens in a wide variety of tissues. These mice have severe growth retardation, hypotonia, spasticity, microencephaly, failure to thrive, malnutrition attributed to abnormal bile acid formation, and severe gait abnormalities.
In the CNS, peroxisomes are more abundant in differentiating neurons and constitute important factors determining neuronal polarity and migration. Cerebellar foliation is abnormal in Pex2 mice, and there are an increased number of granular neurons that undergo apoptosis. Cerebellar layer formation is delayed, and there is increased thickness in the internal and external granular layers, compatible with delays in neuronal migration. Purkinje cell morphology is abnormal.
Pex5 null mice lacked morphologically identifiable peroxisomes and had biochemical abnormalities consistent with Zellweger syndrome. Intrauterine growth retardation, hypotonia at birth, and demise within 72 hours were prominent phenotypic features (04). The brain showed impaired neuronal migration, maturation, and extensive neuronal apoptosis. Cellular studies showed a proliferation of pleomorphic mitochondria with functional mitochondrial abnormalities (07). These might relate to increased oxidation secondary to deficient peroxisomal catalase and superoxide dismutase and were suggested as a factor in the pathogenesis of Zellweger syndrome. Of note, mitochondrial dysfunction was implicated in the first Zellweger syndrome patients described (31). Detailed analysis of the Pex5 null murine model has demonstrated that docosahexaenoic acid and isoprenoid/cholesterol deficiency are unlikely to make a major contribution to pathogenesis (36; 70).
Pex13 null mice are similar to Pex2 and Pex5 deficient mice (45). Pex11beta null mice have neuronal migration defects, enhanced neuronal apoptosis, hypotonia, and neonatal lethality. Surprisingly, there are no defects in peroxisomal matrix protein import and only mild deficiencies of beta-oxidation and ether lipid biosynthesis, challenging the idea that the clinical features are directly related to these cellular abnormalities (41).
Pex7 null mice mimic the severe rhizomelic chondrodysplasia punctata clinical and biochemical phenotype. These mice are hypotonic with growth impairment, and the majority do not survive weaning. Neuronal density is increased in the intermediate zone of the cerebral cortex; these mice also have a delay in neuronal migration (14). Endochondral ossification is also delayed.
These mouse models are useful for studying therapeutic applications. Docosahexaenoic acid supplementation in the Pex5 mouse did not result in clinical improvement (03). In contrast, bile acid supplementation of the Pex2 mouse increased survival, growth, and cerebellar development (29).
Danks has estimated a 1 in 100,000 incidence of Zellweger syndrome in Australia. When considering the full spectrum, the panethnic incidence is estimated to be 1 in 50,000 (48). PEX1 deficiency has an estimated incidence of 1 in 71,000 in North American and European populations. The other causes of Zellweger spectrum are much more rare (1 in 300,000 to 1,000,000). Population differences are known; for example, Zellweger spectrum is less common in Japan (1 in 500,000), and the most common underlying gene defect is PEX10 deficiency. Since the advent of newborn screening for X-linked adrenoleukodystrophy in New York State, 1 in 78,750 infants has been detected to have Zellweger spectrum disorders as an incidental finding (46).
Rhizomelic chondrodysplasia type1 has an estimated incidence of 1 in 100,000. A common mutation, L292X, represents a founder allele in the northern European Caucasian population (12).
Prenatal diagnosis is generally performed using cultured chorionic villus cells or amniocytes after an index case is proven to have a peroxisome assembly defect. Prenatal testing can also be performed directly on uncultured amniocytes or chorionic villi by DNA, enzyme, or immunoblot analysis. Enzyme analysis and immunoblot testing are not readily available in many places. As a result, if the gene defect is known, DNA analysis is preferred. If the familial genetic defect is known, it may be possible to perform preimplantation genetic diagnosis.
Severe hypotonia in infancy associated with eye, liver, or brain demyelination can also be seen in glutaric aciduria type II and muscle-eye-brain disease, a defect in O-mannosyl glycan synthesis. Other entities associated with severe hypotonia include Prader-Willi syndrome and congenital myopathies. The differential diagnosis for neonatal seizures also includes molybdenum cofactor deficiency, sulfite oxidase deficiency, nonketotic hyperglycinemia, Aicardi syndrome, disorders of the mitochondrial respiratory chain, and chromosomal abnormalities. Diagnoses that have been considered in Zellweger spectrum patients with an atypical presentation include Werdnig-Hoffman disease or severe spinal muscular atrophy type 1 (08), Niemann-Pick type C (63), Charcot Marie Tooth, ataxia and cholestasis (18), Usher syndrome, and late-onset leukodystrophy (48).
Historically, some patients initially diagnosed with Usher syndrome or Leber congenital amaurosis due to their physical characteristics were later shown to have assembly defects (25; 59). Others have been diagnosed with Krabbe disease, spinal muscular atrophy type 1 (08), or Prader-Willi syndrome due to their severe hypotonia and were found to have peroxisomal disorders. Still others were initially thought to have Down syndrome based on facial features and decreased tone until normal karyotype was obtained and further testing done.
Other causes of chondrodysplasia punctata include defects in arylsulfatase E (CDPX1) (16), sterol isomerase (CDPX2) (11), maternal vitamin K deficiency or warfarin use, and maternal autoimmune disease (75).
|
• Any phenotype, in theory, can have any mutation, so plasma very-long chain fatty acid analysis, phytanic and pristanic acid biochemical testing to start evaluation. | |
|
• Genetic testing (by single gene, panel, or via whole exome/genome sequencing) may also be done to confirm diagnosis. |
Biochemical investigation of peroxisomal function is warranted if the clinical picture includes abnormalities in the following systems: craniofacial, neurologic (hypotonia, seizures, peripheral neuropathy), neurosensory (visual or hearing abnormalities), hepatic, skeletal, hypocholesterolemia, and failure to thrive (54). A patient suspected to have a Zellweger spectrum disorder should have a fasting or preprandial blood collected for the measurement of plasma very-long chain fatty acids and the branched-chain fatty acids, phytanic and pristanic acid (49). False-positive results can be associated with non-fasting specimens, sample hemolysis, or patients on a ketogenic diet. The branched-chain fatty acids accumulate with dietary exposure to dairy, meat, and other food sources containing phytol. Thus, branched-chain fatty acids are normal in the newborn period. Additional studies on the blood should be performed if the plasma fatty acid analysis is consistent with a defect in peroxisomal beta-oxidation (increased C26:0, C26:0/C22:0, and C24:0/C22:0) or alpha-oxidation (increased phytanic acid), or if the biochemical results are equivocal and the clinical features are suggestive of Zellweger spectrum. These studies include measurement of erythrocyte plasmalogen levels, plasma pipecolic acid, and plasma bile acids. An expanded fatty acid profile usually shows a deficiency in docosahexaenoic acid. In addition, urine can be submitted for organic acid analysis (2-hydroxysebacic acid and epoxydicarboxylic acids), pipecolic acid measurement, and bile acid studies. Zellweger newborns generally have very high urine pipecolic acid levels due to an inability to reabsorb this metabolite, but this reverses after the first 6 months of life. DNA analysis can be useful for confirmation of the diagnosis, carrier testing of at-risk relatives, and prenatal testing. If DNA testing is not informative, further characterization of peroxisome abnormalities can be performed in cultured skin fibroblasts. Useful studies include measurement of very-long-chain fatty acid content, beta-oxidation (substrates: C24:0, C26:0, or pristanic acid), phytanic acid alpha-oxidation, total plasmalogen synthesis, or the single enzymes needed for plasmalogen biosynthesis, or catalase solubility. In addition, immunohistochemistry can be used to visualize peroxisomes and matrix proteins to determine the extent of the peroxisome assembly defect. If the only demonstrable defects involve fatty acid metabolism, then it is more likely the patient has a single enzyme defect.
Patients suspected of rhizomelic chondrodysplasia punctata should have erythrocyte plasmalogen levels measured (34). If plasmalogens are deficient, it is advisable to measure plasma very-long-chain fatty acid and phytanic acid (these are typically part of one test). Although plasma phytanic acid is elevated only after dietary exposure to phytol, it is important to demonstrate plasma very-long-chain fatty acids are normal due to some clinical overlap between this disorder and Zellweger spectrum. DNA analysis is available and can be used to determine the patient’s mutation(s). A variety of common alleles are associated with PEX7 exons 7 and 9 (10). If genetic testing result is unclear, a skin fibroblast cell line can be established to document the defect in plasmalogen synthesis and to measure phytanic acid oxidation. If phytanic acid oxidation is deficient, then the patient has a PEX7 defect.
Studies have identified that very-long chain fatty acid levels may help predict severity, especially the C26:0 and C26.0/C22.0 ratio (67).
|
• For Zellweger spectrum disorders, the majority of therapies focus on identifying and treating underlying complications including seizures, liver dysfunction, and adrenal dysfunction. | |
|
• Other therapeutic approaches (eg, dietary support, medications) are determined by the individual patient’s phenotype. |
At diagnosis (and if considered) individuals with peroxisomal disorders should undergo the following systematic evaluation that includes visual and auditory evoked response testing, comprehensive eye examination, brain MRI, renal and cardiac ultrasound, skeletal survey, and evaluation of hepatic enzymes and clotting factors. ACTH and cortisol levels should be measured to determine the extent of adrenal dysfunction.
There is no known specific therapy for the assembly/biogenesis peroxisomal disorders (Zellweger disorders) that correct the underlying abnormality. Any intervention or therapy should take into account that features of Zellweger spectrum disorders begin prenatally and may not be responsive to post-delivery interventions. Thus, some families may choose to focus on palliative care for their affected infant or child.
Cholbam (cholic acid) has been approved to treat liver disease in the Zellweger spectrum disorders. This approach was first reported by Noetzel (51). It is used to increase cholic acid amounts. Increased cholic acid works through a feedback loop, which inhibits production of the toxic C27 intermediate bile acids. The long-term impact on overall survival is unknown and is currently under study. This medication must be used with caution because it can lead to worsening of liver function if fibrosis and cirrhosis already exist, but studies show improvement in patients treated with it (02).
Dietary approaches to resolve specific biochemical abnormalities include dietary restriction of metabolites that accumulate and replacement of those that are deficient. This approach has been used in adult Refsum disease by decreasing phytanic acid. The degree of benefit is unclear but appears to improve skin, sensory neuropathy, and ataxia (05; 72). Due to the benefit of dietary phytanic acid restriction in adult Refsum disease, this is often prescribed for surviving peroxisome biogenesis disorder patients; however, there is no evidence of benefit. For the most part, dietary restriction and intermediate replacement approaches are mostly anecdotal and have not been studied systematically in patients. For example, repletion of erythrocyte plasmalogen levels has been achieved by dietary administration of plasmalogen precursors such as batyl alcohol, but this has not improved the clinical status. Patients have received docosahexaenoic acid supplementation, but there is not yet a proven clinical benefit to this therapy (44; 53).
An infantile Refsum disease patient has received an orthotopic liver transplant, and their follow-up (17 years later) showed stabilization of biochemical parameters, hearing, vision, and neurodevelopmental status (69; 19). Of the two other liver transplanted in this cohort with mild infantile Refsum disease, one died following transplant, and one had only a short-term follow-up of 9 months (19).
Supportive management of affected children must be tailored to disease severity. Infants may require gastrostomy tube placement to ensure appropriate nourishment. Vitamin K supplementation and water-miscible preparations of fat-soluble vitamins should be provided. It is recommended that cataracts be extracted in early infancy to preserve some visual function. Orthopedic referral may be useful to follow skeletal defects.
Pediatric care in early childhood should include monitoring of growth and nutrition, yearly screening of thyroid, hearing, and visual function, and periodic visits to genetics and neurology clinics.
Regular liver function analysis. Following synthetic proteins (albumin), coagulation factors (ie, prothrombin and partial thromboplastin time), total/free bilirubin, and liver transaminases should be done at least annually. Ultrasounds or fibroscan should also be done at least annually.
Developmental assessments are recommended not only in infancy but also as patients approach school age and through school age. Semiannual dental exams are recommended. Bone disease can be problematic and so often is followed using dual-energy x-ray absorptiometry and evaluation of calcium and vitamin D status with adequate supplementation. One study demonstrated benefit of bisphosphonates (61). Early intervention, family support services, and hospice care may be required.
Seizures can be treated using standard antiepileptic drugs.
Individuals tolerate and, on the standard, schedule should receive all immunizations, including annual influenza and respiratory syncytial virus vaccines. Older individuals with Zellweger spectrum disorders are at increased risk for hyperoxaluria and should be screened for stones and renal dysfunction.
Evaluation of patients with rhizomelic chondrodysplasia punctata should include eye examination, auditory evoked responses, renal and cardiac ultrasound, and skeletal survey. These patients do not have abnormal liver or adrenal functions. Supportive management should be instituted as discussed above for the Zellweger spectrum.
Outcomes depend on severity of disease, which frequently correlates with age of onset. In most cases, therapy focuses on mitigating the complications of the disease. There are currently no cures and few peroxisomal-specific therapies.
Caregivers and parents often utilize palliative care support and frequently require respite care.
Peroxisomal disorders (with some exceptions, such as X-linked adrenoleukodystrophy) are inherited in an autosomal recessive manner such that couples who are both carriers have a 25% recurrence risk of having an affected child.
X-linked adrenoleukodystrophy is X-linked, and 50% of offspring of females will inherit their family variant (change), but their phenotype often is impacted by their sex. Males who inherit the variant, having only a single X chromosome, are often more affected than females. Males with a genetic change can only pass their affected X chromosome to their female children.
Prenatal diagnosis is possible by biochemical or molecular testing. Biochemical testing has a higher sensitivity if cultured fibroblasts from the index case are characterized. It is possible to perform biochemical testing on pregnancies considered at-risk based on fetal abnormalities detected during routine prenatal care. Molecular testing should be performed only if the familial causative mutations have been identified in the index case, or the parents are shown to be carriers of pathogenic mutations, or both.
Provided coincident infections or coagulopathies are treated, no specific risks for anesthesia or surgery have been documented for patients with disorders of peroxisome assembly. Attention to respiratory status is critical due to the extreme hypotonia in Zellweger spectrum and restrictive lung disease in rhizomelic chondrodysplasia punctata.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Michael F Wangler MD
Dr. Wangler of Baylor College of Medicine has no relevant financial relationships to disclose.
See Profile
Andrea Gropman MD
Dr. Gropman of St. Jude Children's Research Hospital has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026