Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Kennedy disease is a rare, X-linked inherited, neurodegenerative disorder characterized by a progressive weakness of the proximal limbs and bulbar muscles, muscle atrophy, fasciculations (especially perioral), loss of reflexes, tremor, gynecomastia, and diabetes mellitus. It results from an excessive number of trinucleotide (CAG) repeats in the androgen receptor gene on the X chromosome. Due to its X-linked genetic association, males are predominantly affected. Particular clinical features and genetic testing can help distinguish Kennedy disease from amyotrophic lateral sclerosis. Patients with Kennedy disease generally live a normal lifespan, despite the fact that there are no treatments available to halt the slow progression of the disorder. In this updated article, the author summarizes the latest research on Kennedy disease, with particular emphasis on insights into its natural history, pathophysiological mechanisms, and potential therapeutic strategies.
|
• Kennedy disease is a rare, X-linked inherited, neurodegenerative disorder characterized by proximal limb and bulbar weakness, muscle atrophy, fasciculations (especially perioral), loss of reflexes, tremor, gynecomastia, and diabetes mellitus. | |
|
• Kennedy disease results from an excessive number of trinucleotide (CAG) repeats in the androgen receptor gene on the X chromosome. | |
|
• Due to its X-linked genetic association, males are predominantly affected. | |
|
• Particular clinical features and genetic testing can help distinguish Kennedy disease from amyotrophic lateral sclerosis. | |
|
• Patients with Kennedy disease often live a normal life span, although no treatments are available to halt the slow progression of the disorder. |
Although Kennedy disease bears the name of William R Kennedy, the first reports of this disease were likely published by LT Kurland, who described an atypical form of lower motor neuron disease in a Japanese family (47). Following the reports by Kurland, Magee provided additional descriptions of patients with X-linked spinobulbar muscular atrophy in the absence of corticospinal tract involvement (64). In 1968, Kennedy reported his experience with two large families at the Mayo Clinic in Rochester, Minnesota (41). The designation “Kennedy disease” was first introduced into the French literature in 1979 (85). The disease garnered particular interest as the first example of a polyglutamine-repeat disorder, of which there are now several other neurologic examples, including Huntington disease and several of the spinocerebellar ataxias.
The core clinical features of Kennedy disease are proximal limb and bulbar weakness, muscle atrophy, fasciculations (particularly perioral), gynecomastia, loss of reflexes, tremor, and X-linked recessive inheritance. Additional clinical features may include sensory nerve abnormalities, type 2 diabetes, infertility, and other manifestations of androgen dysfunction. These features are variably expressed in individuals of the same family and similar repeat sizes. Family history may be lacking in as many as 26% to 60% of patients (92).
Kennedy disease begins insidiously and progresses slowly. Patients typically report disease onset in their third to fifth decade of life, although subtle clinical manifestations can be overlooked until obvious symptoms, like weakness, develop. In a series of 57 patients with Kennedy disease, the most common presenting symptoms reported were cramps (32%), followed by tremor and leg weakness (23% each) (83). In a different study of 34 patients with Kennedy disease, initial onset of symptoms was demonstrated to occur in adolescence with the presence of gynecomastia (52%), premature muscle fatigue, and muscle cramps (92). Weakness was not a common initial symptom. A third study of 157 patients noted that the most common presenting symptom, hand tremor, had a median age of onset of 35 years (13). Gynecomastia, orofacial fasciculations, cramps, and fatigability were the other most common symptoms. Dysphagia and paresthesias were characteristic during the late stages of the disease. The median age at which patients started using a cane for walking was 62. Lifespan in Kennedy disease is generally normal; in a retrospective study, long-term survival for patients with Kennedy disease did not differ significantly from that of age-matched control subjects (08). However, optimal supportive care to prevent complications, such as falls or aspiration pneumonia, is required.
Motor symptoms are a prominent feature of Kennedy disease. The symptoms range from nonspecific muscle cramps and early muscle fatigue to obvious muscle atrophy and fasciculations. The majority of patients develop proximal lower extremity weakness first (44%) followed by a slow proximal-to-distal pattern of progression involving both lower and upper limbs. Motor symptoms can be asymmetric in as many as 21% to 30% of patients (87). In the legs, thigh and calf flexors are more severely affected than extensors, whereas in arm muscle groups, all muscles are affected equally (100).
In most cases, limb weakness is followed by bulbar involvement, leading to varying degrees of facial and tongue weakness, dysphagia, and dysarthria. Fasciculations are ubiquitous in Kennedy disease and, distinctively, are almost always present in perioral regions of the face (92). Recurrent laryngospasm may be an under-recognized symptom in up to half of patients with Kennedy disease and may distinguish Kennedy disease from other forms of motor neuron disease (91). Initial reports examining the association between polyglutamine repeat length and onset of weakness were conflicting (34; 04). Subsequent studies with larger cohorts have demonstrated an inverse correlation between repeat length and both age at onset and degree of weakness (17; 86; 87). A correlation between CAG repeat size and compound muscle action potential (CMAP) amplitude, but not sensory nerve action potential (SNAP) amplitude, has been described (42). A patient with a very large (72 CAG) repeat expansion has been described as having a severe phenotype (63). This inverse link is similar to other triplet-repeat diseases, though it is still not known how CAG-repeat length influences the progression and prognosis of CAG-repeat diseases (05). However, repeat length should not be considered prognostic for any individual patient; expression of Kennedy disease is too variable to rely on this relationship in the clinical setting.
The lack of sensory symptoms in early case reports suggested pure motor involvement in Kennedy disease. However, on examination, loss of reflexes is a consistent finding, and sensory nerve conduction studies show decreased or absent sensory responses in most patients (20). The possibility of subclinical involvement of the autonomic nervous system in Kennedy disease is also under investigation (84). A study of monozygotic twins, both with Kennedy disease but with phenotypical differences, found evidence of small-fiber neuropathy, confirmed by Sudoscan, in the twin with more sensory features (76).
Tremor was part of the initial description of Kennedy disease and remains a common finding (65% to 80%) among large published cohorts (34; 66). It is best characterized as a postural tremor that worsens with activity, similar to essential tremor, and it responds similarly to alcohol or beta-blocker treatment (16). Patients can have tremor as their initial symptom in a minority of cases (6%) (92). Peripheral factors, such as subclinical sensory disturbances or reductions in motor unit numbers, may contribute to tremor genesis in Kennedy disease (29).
Although CAG polymorphism length within the androgen receptor (AR) gene has been associated with an increased risk of heart disease, Kennedy disease is generally regarded to have cardiac manifestations only rarely at later stages of the disease. These may include ST-segment abnormalities, Brugada syndrome, dilated cardiomyopathy, hypertrophic cardiomyopathy, or sudden cardiac death. It is possible that these findings are related to the progression of age alone and are unrelated to Kennedy disease itself (23).
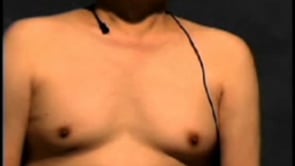
Affected individuals may also manifest endocrinopathies. Three patients in Kennedy’s initial report had gynecomastia, and two had diabetes (41). Since then, the prevalence of these two features has been investigated. In a study of the early symptoms of Kennedy disease, gynecomastia was found to be the most common initial symptom or sign (52%) (92), followed by testicular atrophy, decreased libido, and subfertility (33). Adult-onset diabetes is found in approximately 10% of patients (66). In regard to hormonal profile, most affected men demonstrate partial androgen resistance with unusually high testosterone levels (33).
Sleep disturbances, including obstructive sleep apnea, nocturnal hypoventilation, and REM sleep without atonia, may be more frequent in patients with Kennedy disease (49).
Autophony (the perception of one’s own voice being too loud), presumably from bulbofacial muscle weakness, was the sole presenting symptom in a patient with Kennedy disease (54).
Although X-linked recessive disorders are often asymptomatic in women, female carriers of the Kennedy disease mutation are occasional exceptions. Lyonization, a general mechanism by which selective deactivation of one of the two X chromosomes in heterozygous women occurs, explains how an X-linked recessive disorder may become expressed in a female carrier. The most common clinical features in women are mild muscle cramps (58%) and fatigue (66). Electrophysiologic studies may demonstrate decreased motor unit number estimation and EMG abnormalities, including high-amplitude or polyphasic potentials in female carriers, along with clinical manifestations of mild muscle weakness in neck flexion and a slow walking speed (98).
Life expectancy does not appear to be affected by Kennedy disease (08). The disease is progressive in a nearly linear fashion over decades in most patients, although the disease course can occasionally be variable. From data collected on 223 patients with Kennedy disease, investigators found that patients with longer CAG-repeat size developed onset of disability in activities of daily living (ie, requirement of a handrail, use of a cane, use of a wheelchair) at an earlier age, but the rate of subsequent declines was similar regardless of triplet repeat size (05).
Efforts have been made to establish a reliable biomarker to monitor disease progression in patients with Kennedy disease. Investigators found that urinary levels of the oxidative stress marker 8-hydroxydeoxyguanosine (8-OHdG) correlated with the severity of motor dysfunction in patients with genetically confirmed Kennedy disease (65). Patients with Kennedy disease have insulin resistance, and the degree of this resistance has been shown to correlate with disease severity. The possible mechanism may relate to the expression (or reduction thereof) of insulin receptors in skeletal muscle (70). In a clinical study, the 6-minute walking test was found to be a reliable, objectively measurable parameter for tracking motor impairment in patients with Kennedy disease (94).
Patients with Kennedy disease are at risk for aspiration pneumonia, although the incidence of this complication is unknown. Bulbar involvement can contribute to mortality in rare advanced cases. Falls can place the patient at risk for fractures, head injury, or other musculoskeletal injuries. However, most patients with Kennedy disease develop only mild neurologic impairment, maintain good ambulatory function years after diagnosis, and do not become subject to debilitating dysphagia or respiratory dysfunction (08).
A 73-year-old man presented with a 2-year history of “tiredness” in the calves and tingling paresthesia in the fingertips. Over the last few years, he had noticed that his shoulder muscles fatigued quickly when he was clipping the hedges in his garden. On specific questioning, he reported that he had felt since his youth that he was weaker than others his age.
The patient was otherwise healthy and took no medications. As a young man, he had been investigated for infertility and was found to have “low sperm count.” His father died of prostate cancer at the age of 89, and his mother died of “malnutrition” at the age of 69. One older brother, who died at the age of 78, had been followed for years for undiagnosed “motor neuron disease.” A younger brother, aged 71, was recently evaluated by a neurologist after he began having difficulty manipulating the gearshift of his car. The patient also had two healthy sisters.
Neurologic examination revealed facial fasciculations, particularly around the mouth. He was unable to whistle. His tongue was mildly atrophied. Neck strength was slightly diminished. He had atrophy of the shoulder girdle and interosseous muscles, and 4/5 weakness of the proximal muscles in the upper and lower extremities. Reflexes were attenuated. Sensory examination was normal. The remainder of his physical examination was notable for gynecomastia but otherwise was normal.
Blood tests showed a serum creatine kinase level of 880 IU/L, but otherwise were normal. Nerve conduction studies revealed decreased CMAP and SNAP amplitudes with normal conduction velocities. On needle examination of several proximal and distal muscles, there were motor unit potentials of increased amplitude and duration, without abnormal spontaneous activity. A blood sample sent for genetic testing demonstrated the presence of 43 CAG repeats within the androgen receptor gene (normal: 11 to 36), confirming the diagnosis of Kennedy disease.
Kennedy disease is caused by a trinucleotide CAG repeat expansion in the gene encoding the androgen receptor. The androgen receptor gene is located on the X chromosome (Xq11-12) (51). This excessive number of trinucleotide CAG repeats occurs at the 5’ end of the first exon, near the transcriptional activation region. This portion of the gene encodes a series of glutamine residues. The normal range for the number of trinucleotide repeats is 11 to 36; patients with Kennedy disease typically have 38 to 62 repeats (OMIM entry 313200). Paternal transmission of the mutation is typically associated with larger expansions in CAG repeat number (86).
The androgen receptor protein is a member of the steroid and thyroid hormone receptor family. It acts as a ligand-dependent transcriptional factor. The androgen receptor protein has three major domains: (1) an N-terminal transactivating domain; (2) a DNA-binding domain; and (3) a ligand-binding domain. Androgen receptor is expressed not only in sexual organs but also in nonreproductive organs, including the kidney, skeletal muscle, adrenal gland, skin, and nervous system.
The polyglutamine repeat expansion in the androgen receptor is responsible for the clinical manifestations of Kennedy disease. Precisely how this mutation produces motor dysfunction and androgen insensitivity remains uncertain. Both loss and gain of function of the mutated androgen receptor have been implicated as underlying mechanisms of Kennedy disease (59; 24; 07). To account for this purported dual effect of the Kennedy disease mutation, some authors attribute the endocrine symptoms of the disorder to loss of function and the neurologic symptoms predominantly to gain of function of the androgen receptor (02; 73).
Further insights into the pathophysiology of the mutated androgen receptor protein have been made possible with transgenic animal experiments. Several experiments indicate that the neurotoxic mechanisms underlying Kennedy disease depend on the presence of circulating androgen. In one study, male transgenic mice containing a mutated human androgen receptor with an expanded polyglutamine tract developed a profound motor neuron disease phenotype that reversed with castration. Female mice were only mildly affected by the mutation but deteriorated significantly with administration of testosterone (38). In human subjects, one case report describes the motor deterioration of a patient with known Kennedy disease after initiation of testosterone treatment for preventing osteoporosis. The patient returned rapidly to his baseline motor status after discontinuing hormonal therapy (45).
Nuclear localization of the mutant protein seems to be necessary for the development of neuronal cell dysfunction and degeneration in many of the polyglutamine diseases. Takeyama and colleagues demonstrated that in Kennedy disease, translocation of the mutated androgen receptor into the nucleus yielded a neurotoxic effect, whereas trapping of the mutated androgen receptor in the cytoplasm attenuated neuronal cell death (95). Katsuno and colleagues showed that testosterone facilitated the nuclear translocation of the mutated androgen receptor by accelerating its dissociation from heat shock proteins (37; 40). Leuprorelin, an androgen receptor antagonist, inhibited nuclear accumulation of mutant androgen receptors and prevented motor dysfunction in male transgenic mice. Thus, it would appear that testosterone is necessary for the development of neurotoxic effects in Kennedy disease through translocation of the mutated androgen receptor into the nucleus, where it may form toxic aggregates or alter transcriptional activity.
Pathological study of motor fibers shows neuron loss at multiple segments throughout the cord and lower brainstem. All sizes of motor neurons (large, medium, and small) are affected, unlike amyotrophic lateral sclerosis, in which small motor neurons are relatively preserved (96). Larger dorsal root ganglion cells are shrunken and contain mutant androgen receptor mRNA, confirming the involvement of sensory neurons in Kennedy disease (58). The ocular motor nuclei are relatively spared pathologically (as opposed to severe neuronal depletion in other brainstem motor nuclei), in keeping with their typically normal clinical function, consistent with rodent brainstem histology, which shows relatively few androgen-concentrating cells in the ocular motor nuclei (vs. facial and hypoglossal nuclei). However, asymptomatic slowing of horizontal and vertical saccades was documented in a patient with Kennedy disease, consistent with involvement of ocular motor neurons, and possibly internuclear neurons (97).
The presence of nuclear inclusion bodies is a pathological hallmark of the polyglutamine diseases. The nuclear inclusions in Kennedy disease contain the mutated, truncated androgen receptor and are most frequently found within motor neurons in the brainstem and spinal cord (56). These nuclear inclusions can also be found in skin, testes, and many other organs (57). Although most inclusions are localized in the cell nuclei, widespread inclusions are also seen in the cytoplasm of certain cells, such as islet cells of the pancreas, and likely contribute to the pathophysiology of Kennedy disease (01).
Poort and colleagues investigated the morphology of neuromuscular junctions in two mouse models of Kennedy disease (79). Contrary to their expectations, no evidence of denervation was found in either model, but junctions in both models showed pathological fragmentation and an abnormal synaptophysin distribution consistent with functionally weak synapses. The ultrastructure of these neuromuscular junctions revealed additional pathology, including deficits in docked vesicles presynaptically, wider synaptic clefts, and simpler secondary folds postsynaptically. They concluded that these observed pathologies of neuromuscular junctions in Kennedy disease model mice are likely the morphological correlates of defects in synaptic function, which may underlie motor impairments associated with Kennedy disease.
Surprisingly, in a study of transgenic mice, overexpression of wild-type androgen receptor solely in skeletal muscles, and not in motor neurons, resulted in a Kennedy disease phenotype (68). Cortes and colleagues developed a mouse model of Kennedy disease and demonstrated that the muscle-specific expression of mutant androgen receptor alone accounted for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy (14). In a clinical trial, six of eight patients with Kennedy disease showed myogenic changes in their muscle biopsy, and all patients were found to have higher plasma CK levels than expected in degenerative diseases. Both neurogenic and myopathic changes were also observed in female carriers by biopsy (88), though only neurogenic changes were observed by needle EMG, and myopathic changes were absent in another study (98). These findings challenge the notion that the symptoms of Kennedy disease are caused exclusively by expression of polyglutamine-expanded androgen receptor in motor and sensory neurons and indicate a key role for muscle fiber pathology in clinical expression of disease (82).
Expansion of the polyglutamine (polyQ) tract of the androgen receptor ultimately leads to Kennedy disease. One study demonstrated that a Leu-rich region preceding this polyQ tract influences its conformation (18). This finding suggests that the residues flanking these repetitive sequences may represent viable therapeutic targets.
There is evidence for mitochondrial dysfunction in Kennedy disease (81). Mitochondrial dysfunction in Kennedy disease is a result of complex interactions of elongated poly-Q AR with mitochondria, mitochondrial proteins, nuclear or mitochondrial DNA, causing oxidative stress, decreased mitochondrial membrane potential, or activation of the mitochondrial caspase pathway. As mitochondrial disease may mimic Kennedy disease, therapeutic approaches may depend on modifications of mitochondrial pathways (22).
Kennedy disease is a rare disorder. Studies of disease prevalence have been performed in distinct populations with varying results. The Vaasa region of Western Finland was found to have a surprisingly high prevalence of 13 out of 85,000 male inhabitants (99). In contrast, the prevalence was found to be 3.3 per 100,000 males in a Northern Italian province (26) and 0.83 out of 100,000 inhabitants in Ireland (55). In the United States, the prevalence of Kennedy disease is estimated to be approximately one case in 40,000 men. One publication described the first case of Kennedy disease in the Brazilian Amazon (03). In a Russian study, the incidence of Kennedy disease in infertile men was found to be one in 3000 (11).
Whereas early reports suggested that all patients with Kennedy disease originated from a single common ancestor, subsequent studies have confirmed that CAG repeat expansions derive from multiple founder mutations worldwide (62). There is a general impression that Kennedy disease may be underdiagnosed, owing in part to misdiagnosis and to the mild symptomatology exhibited by some patients (74; 92).
The differential diagnosis for Kennedy disease is broad, encompassing a number of other motor neuron diseases, as well as forms of neuropathy and myopathy that are phenotypically similar to Kennedy disease. There is an average delay of 5 years from the onset of weakness to reaching a diagnosis of Kennedy disease (83). However, as long as the possibility of Kennedy disease is considered, the availability of genetic testing now makes a confident diagnosis relatively straightforward.
Amyotrophic lateral sclerosis is perhaps the most common misdiagnosis. In one study, 2% of patients initially diagnosed with amyotrophic lateral sclerosis and being followed in amyotrophic lateral sclerosis clinics were found to have a molecular diagnosis of Kennedy disease (74). Another study found that 13% (six of 47) of patients with Kennedy disease received an initial misdiagnosis of amyotrophic lateral sclerosis, and 32% (15 of 47) overall received an initial misdiagnosis (83). In amyotrophic lateral sclerosis, both upper and lower motor neuron involvement is typically found, whereas in Kennedy disease, only lower motor neuron involvement is clinically seen (101). Unlike in amyotrophic lateral sclerosis, sensory nerve conduction studies are usually abnormal in Kennedy disease, even in the absence of sensory symptoms (28). Progression of disease is faster, and life span is shortened in amyotrophic lateral sclerosis compared with Kennedy disease.
Kennedy disease can also be mistaken for other disorders causing lower motor-neuron weakness, particularly those presenting with predominantly proximal weakness. The clinical phenotype of limb-girdle muscular dystrophies or inflammatory, endocrine, or toxic myopathies may resemble Kennedy disease. Patients with fascioscapulohumeral muscular dystrophy have slowly progressive facial and limb weakness, but inheritance is autosomal dominant, and androgen insensitivity does not occur. Patients with oculopharyngeal muscular dystrophy present with bulbar weakness but can be largely distinguished by the presence of symptomatic ocular findings not seen in Kennedy disease. The common occurrence of elevated serum CK values in patients with Kennedy disease may further prompt an initial misdiagnosis of myopathy.
Patients with chronic inflammatory demyelinating polyradiculoneuropathy can exhibit symmetrical proximal muscle weakness, hyporeflexia, facial weakness, and bulbar dysfunction in advanced cases. Nerve conduction studies with features of demyelination and cytoalbuminologic dissociation on lumbar puncture are the hallmarks of this disorder, distinguishing it from Kennedy disease. Forms of spinal muscular atrophy, such as type I and II, will have a much younger onset of disease, without features of androgen insensitivity.
Disorders of the neuromuscular junction can sometimes present similarly to Kennedy disease. In Lambert-Eaton myasthenic syndrome, patients develop slowly progressive proximal muscle weakness with diminished deep tendon reflexes. The presence of associated autonomic disturbances, as well as characteristic electrophysiological abnormalities, can help distinguish this entity from Kennedy disease. Occasionally, patients with myasthenia gravis can resemble those with Kennedy disease and present with proximal muscle weakness and bulbar-predominant features in the absence of symptomatic ocular impairment. Furthermore, decremental responses of compound muscle action potentials to slow (3 Hz) repetitive nerve stimulation have been found in some patients with Kennedy disease (35).
Other disorders that could be mistaken for Kennedy disease include scapuloperoneal neuropathies or neuronopathies, cervical spondylosis, and hereditary motor neuropathies. The latter group of disorders includes a distal form of neuropathy, characterized by spinal and bulbar muscular atrophy and vocal cord paralysis that results from mutations in the dynactin gene (78). Very late-onset Sandhoff disease has been mistaken for Kennedy disease (10), and Kennedy disease has been misdiagnosed as POEMS syndrome (103) and polymyositis (69).
In electrophysiological studies by Ferrante and Wilbourn of patients with Kennedy disease, compound muscle action potential amplitudes were reduced in 37% to 46% of cases (20). Needle examination showed long-duration, large-amplitude motor unit potentials indicative of chronic denervation and reinnervation in all symptomatic patients. Sensory nerve action potentials were diminished or absent in 82% to 95% of patients. In some cases, decremental responses to repetitive nerve stimulation, as are commonly found in disorders of the neuromuscular junction, have been reported (44). Motor nerve conduction block has also been detected in patients with Kennedy syndrome and has been linked to patient reports of chronic fatigue (71).
Elevations in serum creatine kinase (often 500 to 1500 IU) can be found in patients with symptomatic Kennedy disease and may even precede symptom onset by 10 years or more (89; 09). Serum testosterone levels are usually normal or elevated (02). When the diagnosis is in question, an elevated serum myoglobin may help differentiate Kennedy disease from amyotrophic lateral sclerosis (27).
Although CAG polymorphism length within the androgen receptor (AR) gene has been associated with an increased risk of heart disease, it is unclear whether patients with Kennedy disease should undergo regular cardiac screening investigations (23). A more recent publication recommends cardiac screening with ECG, testosterone levels, and potentially cardiovascular magnetic resonance imaging to assess cardiac risk factors. Patients with spinal and bulbar muscular atrophy may show abnormal ECGs (Brugada pattern, early repolarization, or fragmented QRS) and structural abnormalities (left ventricular hypertrophy, low end-diastolic ventricular volumes, diffuse myocardial fibrosis), which may explain an increased risk of sudden death (93).
A 2016 study compared magnetic resonance imaging alterations in the cortex and white matter in patients with Kennedy disease and those with amyotrophic lateral sclerosis (21). Patients with Kennedy disease showed degeneration of the pontine crossing fibers, right frontotemporal and fronto-occipital tracts, and right cingulum. Also noted was subtle cortical thinning of the inferior frontal gyrus bilaterally, left premotor regions, middle temporal gyrus, and right precuneus. Compared to patients with Kennedy disease, patients with ALS showed greater involvement of the corticospinal tracts, corpus callosum, external capsule bilaterally, and left superior longitudinal fasciculus.
Sensory impairment secondary to dorsal root ganglion neuronopathy is a common, though typically subclinical, finding in Kennedy disease. Cross-sectional area, as measured by ultrasound, of nerves of patients with spinal and bulbar muscular atrophy was significantly smaller than that of patients with axonal neuropathy or controls (75).
Proton magnetic resonance spectroscopy demonstrated a significant reduction in the N-acetyl-aspartate (NAA) to phosphocreatine (Cr) ratio in the motor cortex of patients with Kennedy disease compared to normal controls, indicating involvement of the motor cortices in patients with Kennedy disease (53).
Ten patients with genetically confirmed Kennedy disease demonstrated glucose hypometabolism in the frontal areas of the cerebrum by (18)F-fluorodeoxyglucose-positron emission tomography (FDG-PET). This disturbance of cerebral glucose metabolism in patients with Kennedy disease is evidence against Kennedy disease being a pure lower motor and sensory neuron syndrome. Mutations in the androgen receptor gene likely have a more widespread effect in the cerebrum than previously recognized (48).
Magnetic resonance imaging of the cervical and thoracic spinal cord in 19 patients with Kennedy disease revealed spinal cord atrophy when compared to the spinal cords of control subjects and patients with other disorders involving lower motor neurons (90). A whole-body muscle MRI study of 81 patients with Kennedy disease demonstrated that the posterior calf was the first muscle to show fat infiltration in this population, and that thigh muscle involvement most significantly correlated with a patient’s 6-minute walk test distance (43).
The bright tongue sign (tongue hyperintensity seen on brain sagittal T1-weighted MRI), most commonly seen in bulbar amyotrophic lateral sclerosis, may also be present in Kennedy disease (60). In a study to determine the prevalence and features of fatty liver disease in patients with Kennedy disease, nonalcoholic liver disease was diagnosed by laboratory analysis and liver imaging in nearly all patients (25).
It has been suggested that a finding of gynecomastia on chest CT may facilitate the diagnosis of Kennedy disease (46).
For definitive diagnosis, DNA analysis of a blood sample to detect a CAG triplet repeat expansion in the androgen receptor gene is available. This test also reliably distinguishes Kennedy disease from other motor neuron disorders, including amyotrophic lateral sclerosis (74). The normal repeat number ranges from 11 to 36 CAG trinucleotide repeats; in patients with Kennedy disease, the repeat varies from 38 to 62 CAGs. Presymptomatic testing for at-risk male relatives or carrier testing for female relatives of an affected family member is possible.
Management of Kennedy disease is mainly supportive. No effective disease-modifying therapy is available to slow or cure the disease (80). The natural history of Kennedy disease, including its slow progression and tendency to plateau, renders its clinical status difficult to evaluate in therapeutic trials. Furthermore, the low incidence of the disease renders recruitment for clinical trials demanding (102). Because of the slow progression of Kennedy disease, it has been proposed that future clinical trials assess patients using a unique scale developed specifically for patients with Kennedy disease (the KD 1234 scale), rather than the previously used ALSFRS scale (61).
The major focus of experimental studies of treatment for Kennedy disease has been on the interaction of the androgen receptor with its ligand, testosterone. Elimination of testosterone in animal models through castration appears to be protective and may restore some lost motor function (38; 12). This finding led to interest in the use of antiandrogenic therapies for treating humans with Kennedy disease. However, two randomized, placebo-controlled trials did not demonstrate a significant effect of dutasteride (a 5alpha-reductase inhibitor) or leuprorelin (a gonadotropin-releasing hormone agonist) on the progression of muscle weakness or swallowing dysfunction in patients with Kennedy disease treated for 24 to 48 months (39; 19). Interestingly, a 2016 case study described a 40-year-old male-to-female transgender Kennedy disease patient who developed full disease manifestations despite undetectable levels of androgens. It was postulated that spironolactone, the anti-androgen the patient used for 15 years, promoted nuclear localization and toxicity of the mutant protein, potentially explaining the disease manifestations in the patient (50).
Nevertheless, interest in androgen-modulating therapy as a treatment for Kennedy disease continues (33), and a study using long-term treatment (8 years) with leuprorelin appeared to delay functional decline and reduce the incidence of pneumonia and death (30). A 1-year observational study similarly showed improved pharyngeal function in patients treated with leuprorelin (36). The thiazole class of antibiotics was identified as neuron-selective androgen receptor (AR) signaling inhibitors, sparing androgen receptor signaling in muscles; thus, they hold potential in the prevention or treatment of Kennedy disease symptoms (72).
The androgen receptor protein has three major domains: (1) an N-terminal trans-activating domain, (2) a DNA-binding domain, and (3) a ligand-binding domain. The N-terminal domain contains the polyglutamine tract. Expansion of the polyglutamine (polyQ) tract of the androgen receptor ultimately leads to Kennedy disease. A study demonstrated that a Leu-rich region preceding this polyQ tract influences its conformation. This finding suggests that the residues flanking these repetitive sequences may represent viable therapeutic targets (18).
Treatment of SBMA knock-in mice with clenbuterol, a beta-agonist, decreased the accumulation of polyglutamine-expanded androgen receptor, improved muscle pathology, ameliorated motor function, and extended survival, suggesting a novel therapeutic strategy for Kennedy disease (67).
The activation function-2 (AF2) domain of the androgen receptor may be a potential drug target for selective modulation of toxic androgen receptor activity. 1-[2-(4-methylphenoxy)ethyl]-2-[(2-phenoxyethyl)sulfanyl]-1H-benzimidazole (MEPB) has shown promise in a mouse model of spinal bulbar muscular atrophy in this regard, demonstrating a dose-dependent rescue from loss of body weight, rotarod activity, and grip strength as well as MEPB ameliorated neuronal loss, neurogenic atrophy, and testicular atrophy (06).
Heat-shock protein 70 (Hsp70) serves in cellular protein quality control, facilitating the degradation of misfolded proteins. In Kennedy disease, expanded CAG repeat length in the androgen receptor leads to misfolding and aggregation of mutant proteins. Thus, there is interest in the development of small molecules designed to enhance Hsp70 function in order to treat polyglutamine diseases, including Kennedy disease (15).
There have been significant advances in nucleic acid therapeutics for hereditary neurologic disorders. Spinal-bulbar muscular atrophy is a trinucleotide repeat expansion disorder and affects both motor neurons and skeletal muscle. Thus, there are many potential targets for nucleic acid-based therapies in spinal-bulbar muscular atrophy (32).
AJ201 is a novel small molecule designed to reduce mutant AR protein levels and stimulate protective cellular pathways (eg, Nrfl/Nrf2 proteostasis/antioxidant pathways) and has been granted fast-track designation by the U.S. FDA based on the results of a Phase 1/2a randomized, double-blind, placebo-controlled study evaluating its safety, tolerability, pharmacokinetics, pharmacodynamics, and early clinical signals in adult patients with Kennedy disease. Early results reported meaningful functional signals and reductions in mutant AR biomarkers.
Although antisense oligonucleotide (ASO) therapy targeting the androgen receptor remains a promising treatment approach for Kennedy disease (52), no clinical trials of anti-AR ASO therapy in patients with Kennedy disease have been initiated to date.
Current treatments focus on alleviating symptoms rather than modifying disease progression. Many symptomatic interventions for Kennedy disease mirror symptomatic treatments for ALS, and, like for ALS, referral to a multidisciplinary clinic may improve quality of life and prolong survival. Tremors may be treated with beta-blockers. There are no FDA-approved medications for the alleviation of cramps. Off-label medications that are sometimes used include benzodiazepines, anticonvulsants, muscle relaxants, or quinine sulfate. There is a lack of controlled studies regarding the efficacy of these treatments, and all have potential side effects, including prolonged QT syndrome for quinine sulfate. Early foot drop can be aided by an orthotic device. Occupational and physical therapy are helpful for addressing mobility concerns and maintaining function for as long as possible, and they help minimize the risk of falls later in the disease course. Bulbar dysfunction can also be problematic. Sialorrhea may be treated with tricyclic antidepressants, scopolamine patch, and sympathomimetics such as pseudoephedrine, botulinum toxin B injection of the salivary glands, or alternatively, low-dose radiation therapy to the salivary glands. Later in the disease course, dysphagia may require placement of a percutaneous endoscopic gastrostomy tube for feeding. Aspiration precautions and involvement of speech pathology are also important to reduce the risk of life-threatening pulmonary complications. Frequent, moderate-intensity aerobic conditioning has shown little beneficial effect in patients with Kennedy disease (77).
A regular exercise routine, as long as the patient’s degree of weakness allows, is recommended. The goal should be to maintain the range of motion of joints and to promote cardiovascular health. However, recommendations generally also advise avoiding severe exercise to the point of muscle exhaustion (to avoid muscle breakdown). Hirunagi and colleagues proposed a potential mechanism underlying the benefits of exercise (31). This study showed that exercise reduced polyQ-expanded androgen receptor proteins in rat muscle cells, leading to improvement in muscle function.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Robert W Pratt MD
Dr. Pratt of the University of Colorado has no relevant financial relationships to disclose.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University received a consultant honorarium from Roche and Ovid Therapeutics.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Peripheral Neuropathies
May. 12, 2026
Developmental Malformations
May. 08, 2026