Neuroimmunology
Balo concentric sclerosis
Jul. 02, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Multiple sclerosis affects every part of the neuraxis and has replaced syphilis as the great mimicker in neurology. In this article, the author describes the entire spectrum of multiple sclerosis signs and symptoms, focal and diffuse brain lesions, look-alike diseases, the overactive immune response, the complex pathology of demyelination, death and dysfunction of oligodendroglia and neurons, MRI and CSF abnormalities, the effects of vitamin D, and other comorbidities in multiple sclerosis. This update contains new comments on the 2017 revised diagnostic criteria, the evolving understanding of the demographics of and racial differences in multiple sclerosis, the recently identified prodromal state, and the increasingly well-characterized demyelinating disease myelin oligodendrocyte glycoprotein, as well as more minor updates.
|
• The incidence of multiple sclerosis is increasing around the world, but disease-modifying therapies have improved its course. | |
|
• Although historically considered primarily a disease of white young and middle-aged females, those of African ancestry in the United States carry a higher risk of developing multiple sclerosis and appear to experience a particularly aggressive disease course (256) | |
|
• Multiple sclerosis lesions cause pleiotropic focal neurologic deficits. Problems with fatigue, cognition, and bladder control are common but often overlooked. | |
|
• Diagnosis is complex and requires neurologic history, clinical and MRI examination, and sometimes spinal fluid and evoked potential analysis. | |
|
• Demyelinating diseases that mimic multiple sclerosis include neuromyelitis optica, myelin oligodendrocyte glycoprotein antibody disease, and CNS Sjögren syndrome, as well as many other toxic, inflammatory, and metabolic disorders. | |
|
• The impact of COVID-19 infection on people with multiple sclerosis is affected by multiple sclerosis immunity and multiple sclerosis therapies. |
Greek and Roman physicians did not document multiple sclerosis, but it may have been mentioned in 13th century Icelandic sagas. Saint Lidwina of Holland appears to have developed multiple sclerosis in 1396 (165). The court physician was not optimistic after examining Lidwina, stating, "Believe me, there is no cure for this illness; it comes directly from God. Even Hippocrates and Gallenus would not be of any help here." The clinical description and prognosis of multiple sclerosis have improved in the intervening 500 years, but progress in understanding its etiology is debatable.
Elizabeth Foster of Coldingham, north of the Scottish/English/North Sea border, developed intermittent paralysis, sensory symptoms, and possible optic neuritis starting in 1742 at age 18 (152). Multiple sclerosis was clearly depicted in 1822 in the diary of Sir Augustus D' Este, grandson of King George III of England (70). One of his relapses is portrayed here:
|
At Florence, I began to suffer from a confusion of sight. About the 6th of November, the malady increased to the extent of my seeing all objects double. Each eye had its separate visions. Dr. Kissock supposed bile to be the cause. I was twice blooded from the temple by leeches. Purges were administered. One Vomit and twice I lost blood from the arm. The Malady in my eyes abated, again I saw all object naturally in their single state. I was able to go out and walk” (169). |
Cruveilhier in Paris and Carswell in London illustrated CNS plaques and sclerosis in the 1840s, and Charcot characterized the demyelination in plaques and published detailed clinical descriptions of the disease. Rindfleisch described the perivascular inflammatory CNS lesions in the 1860s (40). These observers documented the intermittent and seemingly random neurologic symptoms of multiple sclerosis and the variable evolution of the disease. The history of multiple sclerosis is extensively reviewed by Murray (169).
Multiple sclerosis lesions in the brain and spinal cord damage every function of the central nervous system. Its presentation is highly variable, with clinical symptoms that vary from mild to aggressive and a course that can be either relapsing-remitting or progressive and which evolves over time. Protean symptoms include fatigue and disturbed sensory, motor, bladder, bowel, sexual, cerebellar, brainstem, optic nerve, and cognitive realms. Symptoms, especially fatigue, limit activity in three fourths of patients. The key clinical history and exam features that differentiate relapsing/remitting multiple sclerosis from other central nervous system (CNS) diseases are (1) episodic symptoms, with onset over days, lasting for weeks and then resolving to some extent, (2) onset of fatigue or fatigability that is worse with heat, (3) trouble with cognitive multitasking and short-term memory, (4) Lhermitte and thoracic cord sensory phenomena and bizarre descriptions of sensory perceptions, (5) bladder urgency or retention, (6) internuclear ophthalmoplegia, often subclinical, (7) weakness of finger extensors and iliopsoas, more than of other muscles, (8) ataxia on tandem walking, and (9) reduced vibration at toes.
In most patients, symptoms of an exacerbation arise over hours to days, typically last 2 to 6 weeks, and then remit, sometimes completely. Forty percent of these attacks cause long-lasting deficits, but 20% of patients will improve after attacks (155). Resolved symptoms can reappear transiently during infections, exercise, stress, menses, afternoon circadian temperature rise, or heat (“ghost symptoms,” Uhthoff phenomenon, pseudo-exacerbation) in up to 80% of multiple sclerosis patients (98).
The neuroanatomical location of plaques is not completely random and demonstrates a predilection for the periventricular white matter, making certain symptoms and signs characteristic of the disease. The following section describes focal and then generalized symptoms.
Optic neuritis. The optic nerves are clinically involved in approximately two thirds of patients, especially younger patients. Thirty-one percent of army recruits with multiple sclerosis have optic signs. “Asymptomatic” patients frequently have abnormal visual evoked potentials or perimetry.
Optic neuritis typically begins with subacute loss of vision in one eye, with a central scotoma described as a blurring, gray, or dark patch. Color perception and contrast sensitivity are disturbed. Subjective reduction of light intensity is often associated with an ipsilateral Marcus Gunn hypoactive pupillary response. Ninety-two percent have retro-orbital pain with eye movement.
With acute lesions and retrobulbar inflammation, the fundus is initially normal. However, the disc is usually pale after the optic neuritis resolves, typically in the temporal aspect. Using an ophthalmoscope, slit-like defects in the peripapillary nerve fiber layer appear with red-free (green) light. This axonal damage in the retina, an area free of central nervous system myelin, suggests that optic nerve pathology extends beyond central nervous system plaques. Retinal nerve fiber layer atrophy and thinning is obvious on optical coherence tomography. The fellow eye is often abnormal on OCT, though not as severely. More rarely, blurring of the disc margin or florid papillitis may develop. With papillitis, which is seen in 5%, inflammation near the nerve head causes disc swelling, cells in the vitreous, and deep retinal exudates.
Sequential optic neuritis led to multiple sclerosis in eight of 20 adults (186). Bilateral simultaneous optic neuritis led to multiple sclerosis in one of 11 adults after an interval of up to 30 years. In children, one of 17 developed multiple sclerosis after bilateral onset. Some of these may have been misclassified. Bilateral optic neuritis should raise the threshold for testing for other demyelinating pathologies, neuromyelitis optica, and myelin oligodendrocyte glycoprotein disorder. Unrecognized until recently, all of these likely altered past epidemiology and the expected prognosis of multiple sclerosis.
Visual function usually begins to improve several weeks after the onset of optic neuritis, and resolution continues over several months. Complete recovery of visual acuity is the rule, even after near blindness. However, other disturbances of vision often persist, such as visual "blurring," poor low-contrast visual acuity, and red or blue desaturation that causes colors to appear drab (“not as vivid”). There is progressive loss of color discrimination with longer duration multiple sclerosis. Depth perception is impaired and is worse with moving objects (“Pulfrich phenomenon,” where laterally moving object seems to trace an ellipse, due to light attenuation in one eye). This causes difficulties with depth perception while driving and during sports. Eye movements may cause fleeting flashes of light (“movement phosphenes”). Increased body temperature can amplify all of these symptoms and diminish visual acuity (“Uhthoff phenomenon”).
Bright lights cause a prolonged afterimage in the retina, also described as a "flight of colors," or “tracers.” This occurs because input from some retinal fibers is delayed, or due to lack of inhibition, allowing dispersion of signals and prolonged neural activation. Prolonged afterimages correlate with retinal nerve fiber thinning, lesions in optic radiations all the way to the occipital cortex, brain volume loss, and cognitive decline. Similarly, palinopsia ("again" + "seeing") is the persistence of an image after the stimulus is removed and is linked to posterior visual pathway and cortical lesions. Attribution of all visual deficits in multiple sclerosis to optic nerve lesions is an obvious impulse, but lesions of the postchiasmatic optic tract and even in the periventricular optic radiations can be responsible (214).
Additional eye pathology appearing in 1% of multiple sclerosis patients includes uveitis and pars planitis (intermediate uveitis; near where the iris contacts the vascular choroid and the sclera). Conversely, 20% of patients with pars planitis develop multiple sclerosis or optic neuritis. Some of these patients will develop macular edema, vitreous opacities, papillitis, vasculitis and vitreous hemorrhage, and cataracts. Perivenous sheathing is an inflammatory change of the retinal veins seen in one fourth of multiple sclerosis patients. Occipital cortex lesions can distort vision, eg, visual inversion. Retinal periphlebitis (see Eales disease) affects the peripheral retina in occasional multiple sclerosis patients and correlates with more severe multiple sclerosis, retinal nerve fiber atrophy, and microcystic macular edema.
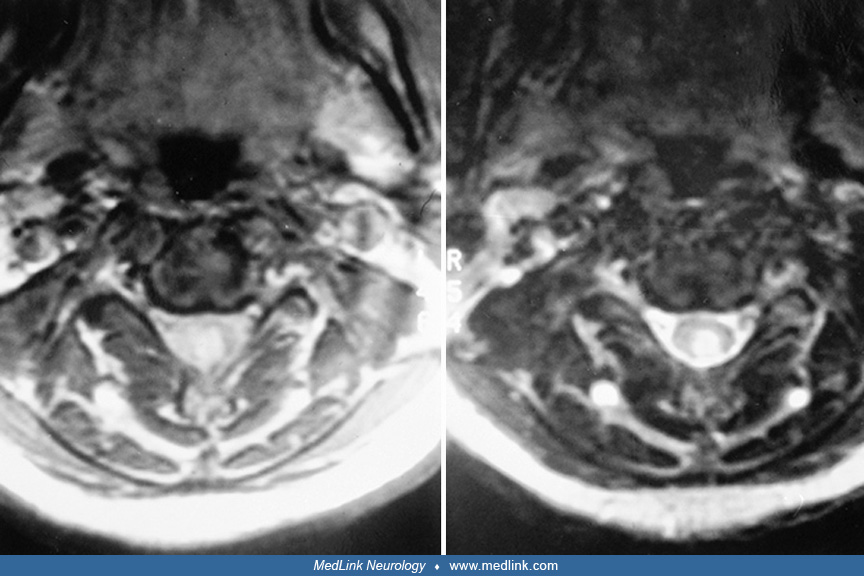
Brainstem abnormalities, including diplopia. Lesions in the brainstem disrupt intra-axial nerves, nerve nuclei, and internuclear connections as well as autonomic, motor, and sensory long tracts. Proton density MRI is best for imaging brainstem abnormalities, including plaques in the median longitudinal fasciculus.
Smell is reduced in 40% of patients with multiple sclerosis. Loss correlates with Expanded Disability Status Scale (EDSS) greater than 2, more MRI plaques, especially in temporal lobe and anterior frontal areas, and olfactory bulb or tract demyelination at autopsy. Olfactory threshold impairment is worse during exacerbations and may be correlated to CNS inflammation and could act as a biomarker for a multiple sclerosis relapse or therapy response (18). Surprisingly, olfactory receptor RNA is highly expressed in multiple sclerosis immune cells; the targets are unknown (74). Olfactory dysfunction is worse in neuromyelitis optica than in multiple sclerosis. Taste is reduced in 20%. It is lost or altered (dysgeusia) from lesions in the brainstem and sometimes in the medial ventralis posterior thalamic nucleus and recovers within 3 months.
Clinically, the third nerve is the most frequent target of brainstem lesions. Cerebellar and brainstem lesions disrupt eye movements, usually coinciding with more severe disability. Third, sixth, and rarely fourth nerve lesions cause diplopia.
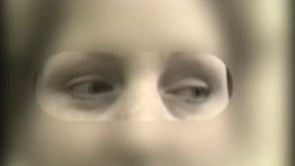
Medial rectus weakness is usually part of an “internuclear ophthalmoplegia.” In a young patient, internuclear ophthalmoplegia is nearly pathognomonic of multiple sclerosis. In older patients, infarcts, trauma, and disparate other causes are possible (125). Internuclear ophthalmoplegia is paresis or weakness of adduction ipsilateral to a medial longitudinal fasciculus lesion, along with dissociated nystagmus of the abducting eye. Lesions, usually in the pons or midbrain, cause internuclear ophthalmoplegia when they interrupt connections between the pontine paramedian reticular formation that innervates the ipsilateral abducens nucleus and the contralateral third nerve nucleus. This illustrates an important principle: plaques predominate in periventricular regions and cause characteristic signs.
Convergence may be normal despite an affected medial rectus. Medial longitudinal fasciculus lesions are seen with proton density MRI but are even more apparent with the clinical examination. Internuclear ophthalmoplegia is often worse with heat and better with cooling (79). The medial longitudinal fasciculus is near the locus ceruleus in the rostral pons, and both are near the ventricular system. An internuclear ophthalmoplegia could, thus, correlate with sympathetic abnormalities and possibly depression (Arnason personal communication 2014).
Internuclear ophthalmoplegia is subclinical or “latent” in 80% of patients (ie, “internuclear ophthalmoparesis”). Rapid eye movements can bring out this hidden, minimal oculomotor weakness, causing slowing of the early adducting saccades—an adduction lag. A related subclinical sixth nerve weakness on fast lateral movement can be seen with increased intracranial pressure (Reder personal observation). A medium rate of finger motion will detect loss of smooth pursuit and demonstrate ataxic eye movements from cerebellar lesions.
Nystagmus is common in multiple sclerosis. It is usually inconsequential, but nystagmus and oscillopsia can be severe enough to prevent reading or driving a car. Pendular nystagmus (not gaze-evoked), 4 to 6 Hz, can be unilateral and is often worse in the eye with lower acuity and in darkness. It has been treated with vibration to the face, mastoids, or skull. Memantine and gabapentin are also potential treatments.
There are reports of high T2 signal MRI lesions in peripheral fifth (in 2% of patients, with two thirds bilateral), seventh, sixth, third, and eighth nerves; overall, 8.2% of patients are affected. Fifth nerve lesions seem to have a predilection for the nucleus and tracts within the brainstem, sparing the surrounding brainstem (see trigeminal neuralgia, below). Seventh nerve lesions can mimic Bell palsy. Facial myokymia from lesions of the facial nerve in the pontine tegmentum can be reversed with carbamazepine and possibly botulinum toxin.
Hearing loss is relatively rare, but auditory processing could be slowed by brainstem and deep white matter lesions. Central hearing defects with abnormal brainstem auditory evoked potentials can help differentiate multiple sclerosis from benign positional vertigo, which has no central defect. Vertigo is common and can be so incapacitating that patients are bed-bound. It is sometimes triggered by fast lateral eye movements; these patients are very cautious on lateral gaze testing (Reder personal observation). The symptoms of mal de démyélinisation (multiple sclerosis) may resemble mal de débarquement. Isolated autoimmune disease of the auditory nerve can also cause hearing loss and vertigo. Its relation to multiple sclerosis is unclear.
Up to one fourth of patients have problems swallowing. Attacks can cause an oculo-palatal tremor or lingual dyskinesia. Horner syndrome is occasionally present.
Transverse myelitis. The cord symptoms in idiopathic transverse myelitis are generally more severe than in multiple sclerosis. Cord symptoms in neuromyelitis optica are also typically more severe than in multiple sclerosis, with more longitudinally extensive lesions and worse potential for recovery. In multiple sclerosis, a complete transverse lesion is less common than a partial cord lesion (ie, a Brown-Séquard syndrome). Progressive myelopathy can arise from solitary lesions of the cervicomedullary junction, often associated with positive CSF oligoclonal bands.
Cerebellar dysfunction and tremor. The cerebellum or its pathways are damaged in 50% of patients. "Charcot’s triad" of cerebellar signs is nystagmus, intention tremor, and “scanning” speech (in the sense of examining words carefully, “scandés” from Charcot). In 3% of patients, intention tremor of the limbs, ataxia, head or trunk titubation, and dysarthria can be totally disabling. In contrast, cognitive slowing can interrupt speech output: “My speech can’t keep up with my thoughts.”
Surprisingly, patients with severe ataxia are often strong and thin and would otherwise be fully functional. The Stewart-Holmes rebound maneuver to detect cerebellar dyssynergia does not correlate well with kinetic tremor (flex or extend at elbow) and intention tremor (finger-to-nose), suggesting damage to different anatomic pathways (259). Poor cerebellar function correlates with loss of cerebellar MRI volume. Ataxia and poor ambulation correlate with lesions of the dentate nucleus. Severe cerebellar signs correlate with poor pulmonary function. Variation in the SCN10A gene for sodium channel NaV1.8 affects coordination in multiple sclerosis.
Dystonia and parkinsonian symptoms are occasionally caused by a multiple sclerosis plaque. Extrapyramidal symptoms disappear as the plaque resolves (157).
Weakness. The long course of axons traveling through the cord from the motor cortex to lumbar motor neurons increases the likelihood that a random plaque will interrupt motor neuron conduction. Weakness and fatigability in two sets of muscles, finger extensor(s) and iliopsoas(es) (iliopsoai) predominate in multiple sclerosis. In the upper limbs, finger extensor weakness is the most common finding and is more sensitive than grip testing. In contrast, cord compression can cause weak grip, with normal finger extensor strength (Reder personal observation). Legs are usually affected more than arms, causing poor stair climbing tripping, and foot-drop. The hip flexors are frequently weak, likely from multiple cervical, and occasionally thoracic cord, lesions that destroy input to the iliopsoas muscle; this weakness is typically out of proportion to any weakness in other leg muscles. Patients can walk backwards more easily than they walk forward because gluteal muscles are stronger than the iliopsoas. The anatomy of the iliopsoas weakness is unclear but could parallel the cervical and thoracic sensory symptoms described above. Multiple sclerosis lesions are often subpial and could affect the corticospinal and lateral reticulospinal tracts, which are somatotopically arranged, with leg fibers lateral to arm fibers, and lesions cause increased extensor tone. Probably not involved are the rubrospinal and tectospinal tracts, which innervate arm muscles but have little input to the legs.
Hyperreflexia, spasticity, and a Babinski sign are common. Rarely, plaques interrupt intra-axial nerve roots, the deep tendon reflexes disappear, and muscles atrophy. Some muscle weakness and fatigue can be explained by a shift in myosin heavy chain isoforms and less contractile force, a result of muscle inactivity and deconditioning (82). Walking ability can be measured with a timed 25-foot walk, the 6-spot step test, and the timed 10-foot tandem walk (TTW10), which incorporate coordination and balance. Motor function can be longitudinally monitored in a patient diary with a weekly 5-minute walking test (Scott personal communication 2014).
Falls and fractures are increased in multiple sclerosis. Osteoporotic fractures double, a consequence of weakness (17). Osteoporosis arises from low vitamin D levels, less weight-bearing exercise, or as a consequence of fatigue, some medications (antidepressants, anticonvulsants), and perhaps genetic links plus environmental influences. Frequent use of steroids for exacerbations or pseudo-exacerbations can contribute to the development of osteoporosis and pathologic fractures.
Spasticity. After physical inactivity, arising, standing, and the first few steps are difficult from transient stiffness. Subclinical internuclear ophthalmoplegia often disappears after the first excursion, a similar phenomenon. Legs are usually more spastic than arms, a consequence of patchy damage throughout the spinal cord. Spasticity increases with a full bladder or bowels, pain, exposure to cold, and sometimes on the day after IFN-beta injections (an effect of cytokines or direct modification of neuronal excitability). Painful tonic spasms are common in patients with severe spasticity and can be provoked by exertion or hyperventilation.
Bladder and sexual dysfunction. Bladder dysfunction is common and markedly reduces quality of life. It is the initial symptom in 5% of patients and eventually develops in 90%. Two thirds of symptomatic patients have bladder hyperreflexia with urgency and frequency; stress incontinence can be a separate problem. Sphincter dyssynergia occurs in 50%, sometimes following an areflexic episode (20). The other third have hyporeflexic bladders and “double voiding.” Failure to empty the bladder predisposes to urinary tract infections, which, in turn, can lead to disease activity. Nocturia may be from bladder spasticity but may also be due to redistribution of fluid from swollen ankles during sleep.
A sense of fullness ascends from bladder to sacral cells ventral to the dorsal horn and then to the periaqueductal gray. This region activates the pontine micturition center in the dorsolateral pontine tegmentum, coordinating sphincters and bladder, and descends through the lateral tegmentum of the medulla to the cholinergic parasympathetic motor neurons in the sacral cord. These negative beta3-adrenergic and positive M3 muscarinic fibers innervate the bladder detrusor muscle. Parasympathetic pathways control micturition and also defecation, sexual activity, and parturition. Patients’ sensation of residual volume is often unreliable, so volume should be measured with office sonography or catheterization. Detrusor hyporeflexia is linked to pontine lesions; detrusor-sphincter dyssynergia is linked to cervical spinal cord lesions.
Glomerular filtration rate is reduced by 20% (29). This could be from chronic neurogenic bladder, urinary tract infections, antibiotics, ionic contrast agents, long-term use of non-steroidal anti-inflammatory drugs, and chronic dehydration.
Seventy percent of patients complain of sexual problems—orgasmic difficulty, poor erections or lubrication, low pleasure, low libido, poor physical movement, and genital numbness. Impotence develops in 40% to 70% of male patients, some with low testosterone levels. Fifty percent of women with multiple sclerosis have significant sexual problems and complain of loss of libido, orgasms, and genital sensation. Orgasmic dysfunction correlates with loss of clitoral vibratory sensation and cerebellar deficits (96). Difficult or no orgasm is associated with abnormal or absent (26/28) pudendal somatosensory evoked potential, although desire is normal (271). Occasional women feel diffuse orgasmic spasms, not in skeletal muscle, that last for up to 5 minutes. Others mention increased vaginal sensation and orgasmic intensity.
Sexual problems often follow or coincide with bladder dysfunction. Both are associated with loss of sweating below the waist from lesions of sympathetic pathways and disruption of genital somatosensory pathways. MRI T1 lesions in the pons correlate with sexual dysfunction, far better than other MRI measures, urodynamics, and pudendal or tibial evoked potentials. Other literature varies on the anatomical links to plaque location. Neurons in the medial preoptic areas are stimulatory; those from the ponto-medullary paragigantocellular nucleus release serotonin to inhibit erections. Antidepressants that raise serotonin levels can cause impotence. In women, right occipital lesions are linked to poor arousal; left insular lesions are linked to impaired lubrication.
Constipation. Fifty percent of clinic patients experience constipation. It is more prevalent in progressive than in relapsing forms of multiple sclerosis. Poor voluntary squeeze pressure on anal manometry, combined with little sensation of “fullness” is typical. Occasionally, insensitivity to rectal filling causes incontinence. Gastroparesis has occurred with acute medullary lesions, and half of multiple sclerosis patients have slow gastric emptying. Disruption of autonomic pathways in the medulla and cord may underlie the gastrointestinal dysfunction. Gut neurons have not been studied as direct targets of the immune system in multiple sclerosis, but the readily accessible enteric nervous system has been analyzed in Parkinson disease and diabetes. Enteric glia have more antigenic resemblance to central nervous system glia than to peripheral nervous system glia (86) and may be immune targets in multiple sclerosis. Autoantibodies against four potential target antigens derived from enteric glia and/or neurons have been identified in mouse models of multiple sclerosis. Antibodies against three of those target antigens were also present in the plasma of multiple sclerosis patients. In colon resections, there is evidence of gliosis and enteric nervous system degeneration in multiple sclerosis patients compared to non-multiple sclerosis controls (269).
Autonomic problems. Cortical, hypothalamic, brainstem, and spinal cord plaques often interrupt the sympathetic nervous system. The hypothalamus controls temperature, sleep, sexual activity, and autonomic functions. Autonomic damage causes slow colonic transit, bladder hyperreflexia, and sexual dysfunction. Less-recognized phenomena from sympathetic nervous system disruption are vasomotor dysregulation (cold, purple feet), cardiovascular changes (orthostatic changes in blood pressure and dizziness, blood pressure response to straining, and poor variation of the EKG R-R interval on Valsalva maneuver, possibly increasing risk of surgery), poor pilocarpine-induced sweating, poor sympathetic skin responses—especially in progressive multiple sclerosis (123; 02), subnormal rise in serum catecholamines on standing, pupillary abnormalities, and possibly fatigue. Rarely, plaques in brainstem autonomic pathways cause atrial fibrillation or neurogenic pulmonary edema, sometimes preceded by multiple sclerosis lesion-induced cardiomyopathy. Cold hands and feet often precede the diagnosis of multiple sclerosis, raising the possibility that undetected spinal cord lesions have already affected the autonomic nervous system.
Sixty percent of patients have pupillary reactions that are abnormal in rate and degree of constriction (50). Some pupillary defects caused by sympathetic disruption may correlate with visual-evoked potentials or history of optic neuritis. An afferent pupillary defect can follow optic neuritis.
Autonomic dysfunction correlates with axonal loss and spinal cord atrophy yet not often with cord MRI lesions. It is possible that plaques in the insular cortex, hypothalamus, periaqueductal gray of the brainstem, and cord all disrupt sympathetic pathways. The periaqueductal gray (because it abuts the ventricular system) and the insular cortex (deep in the Sylvian fissure near germinal center-like areas) are likely to be seriously damaged in multiple sclerosis. Parasympathetic and sympathetic dysfunction correlates with duration of multiple sclerosis but not with disability (97). Parasympathetic dysfunction (eg, heart rate variation with respiration, abnormal pupillary reactions) is most pronounced in primary progressive disease. Sympathetic dysfunction can worsen during exacerbations. It is tied to less response to the beta-adrenergic agonist, isoproterenol (90); dysregulated immunity (73); worse autoimmune disease in animal models and worse multiple sclerosis (122); and is common in progressive multiple sclerosis. There may be a positive feedback loop, where cord lesions damage sympathetic nuclei, remove a sympathetic brake on peripheral immune cells, and, thus, allow more aggressive attacks and disease progression (Arnason personal communication 1984).
Periodic hyperthermia is rare, but chronic hypothermia is occasionally seen and can worsen during an attack with profound decreases to 28°C/79°F (author’s observation). Cognition is surprisingly preserved with hypothermia in multiple sclerosis unless the temperature drop is extreme. Hypothalamic or thalamic plaques presumably cause the abnormal temperature regulation. Hypothermic patients are at high risk for infection because immunity is compromised at low temperature. Worsening hypothermia also can forecast an infection. Interferon-beta elicits fever by direct effects on thermosensitive neurons in the preoptic or anterior hypothalamus, without elevation of other pyrogenic cytokines.
Sensory symptoms. Sensory symptoms are common in multiple sclerosis. Patients often have difficulty describing these sensations because they are spontaneous or distorted perceptions of everyday stimuli modified by areas of demyelination and ephaptic connections unique to each patient.
Positive sensory symptoms are common and range from subtle to marked loss of sensation, which can be patchy or span the entire body. They are described as tingling, numbness, a tight band (usually at T6-T10, the “multiple sclerosis hug”), pins and needles, a dead feeling, “ice” inside the leg, standing on broken glass, and something “not right.” Paresthesias typically begin in a band (a “multiple sclerosis hug”) around the trunk at T6-T9 (often from a cervical plaque). They sometimes start in a hand or foot and progress over several days to involve the entire limb. The sensations then resolve over several weeks.
Poor perception of vibration in the feet, but spared position sense, is present in more than 90% of multiple sclerosis patients. Vibratory loss can be quantified with a tuning fork and sometimes improves with drug therapy. Impulses from the rapidly oscillating tuning fork are not transmitted by central sensory paths from a combination of demyelination and cytokines that interfere with axonal conduction (234). Poor vibration perception is a sensitive measure for differentiating high versus low risk of developing multiple sclerosis; greater than genetic and environmental risk factors such as HLA alleles and smoking, vitamin D levels, obesity, EBV infection, and migraine (270).
Lhermitte and other spinal cord signs. In 1924, Lhermitte described an electric discharge following flexion of the neck in multiple sclerosis. Forty percent of multiple sclerosis patients have Lhermitte sign (symptom, phenomenon), and 95% of these have cervical cord MRI lesions. This rapid and brief "electric shock," “zapping,” or "vibration" runs from the neck down the spine. Intensity of the pain is directly related to the amplitude and rapidity of neck flexion. In an instinctive protective reflex, the patient may straighten her neck. Patients often have a complete or hemi-circumferential sensory band at around T7, a “multiple sclerosis hug.” A thoracic sensory sign can be elicited, especially when the lesion is active. With the neck immobile, the patient flexes (crunches) at the thorax, and pain or paresthesias radiate around the chest (203). The longitudinal/vertical Lhermitte sign and the circumferential thoracic crunch sign are from mechanical stimulation and ephaptic conduction of irritable demyelinated axons. They are similar to ulnar nerve trauma triggering the “funny bone” at the elbow. Cord compression can also generate these signs.
Trigeminal neuralgia. Trigeminal neuralgia is relatively rare in multiple sclerosis (occurring in 0.5% to 1% of patients), but incidence is higher than in the normal population (220). Bilateral trigeminal neuralgia may be pathognomonic of multiple sclerosis (118).
In the elderly, trigeminal neuralgia can be caused by arteries compressing the trigeminal nerve at the junction of the central and peripheral nervous system (root entry zone) (164). Vascular compression causes demyelination and remyelination, sometimes aberrant, allowing ephaptic conduction between active and silent nerve fibers and between light touch and pain fibers (154).
Trigeminal neuralgia of multiple sclerosis, however, is from a plaque in the fifth nerve nucleus (180) or the brainstem entry zone of nerve fibers, suggesting that therapies will differ between the elderly and that multiple sclerosis forms of trigeminal neuralgia. Coexisting demyelination and vascular compression could synergize. The brainstem or cisternal (peripheral) fifth nerve enhances on MRI in 3% to 7% of multiple sclerosis patients, but this is usually clinically silent (171). Herpes infections, which are latent in the majority of trigeminal ganglia, potentially enhance inflammation. Neural ganglia in the majority of autopsies contain T cells and macrophages, interferon-gamma, and interferon-induced markers (248); the immune response is likely to suppress virus replication but could enhance inflammation elsewhere in multiple sclerosis. Activation of multiple sclerosis is not certain, however. After facial nerve injury, IFN-gamma increases, but pituitary adenylyl cyclase-activating polypeptide (PACAP) recruits anti-inflammatory and neuroprotective Th2 cells.
Brainstem plaques can cause glossopharyngeal neuralgia. Radicular pains in multiple sclerosis, especially if lancinating, may have a similar mechanism.
Pain. Up to two thirds of patients with multiple sclerosis have pain at some time during the course of their disease (243), although pain was regarded as rare in much of the older literature. The pain is chronic most of the time. Pain is more common in older women with spasticity or myelopathy and in multiple sclerosis of long duration. It is often worse at night and when the ambient temperature changes suddenly.
Pain can be chronic with fluctuation, acute, or intermittent. It is most often bilateral but can be generalized, unilateral, or focal. Legs are affected in 90% and arms in 30% (183). Radicular symptoms arising from a posterior cord lesion are often painful, but anterior plaques are not. A classic finding is a thoracic band of several dermatomes, usually near T6, and is often described as a “multiple sclerosis hug.” A common MRI finding in multiple sclerosis pain is a central cord lesion between T1 and T6 (177). In contrast, pain in neuromyelitis optica is often bilateral or thoracic, and it is less likely to affect the limbs. Fibromyalgia, with severe tenderness between the shoulder blades, can confuse the pain exam and is common.
The spectrum of pain includes central neuropathic pain from focal demyelination (eg, trigeminal neuralgia (above), dysesthesias (sometimes associated with paresthesias and nonspecific pain), dysesthesias from ephaptic transmission (Lhermitte symptom, radicular pain, tonic seizures), inflammation or swelling (optic neuritis, headaches; pain occurs in neuromyelitis optica and the experimental autoimmune encephalomyelitis model of multiple sclerosis), visceral pain from chronic constipation or painful bladder spasms, abnormal motor activity (tonic seizures, spasms, clonus), or simple orthopedic musculoskeletal pain.
Lesions causing disinhibition of central pain pathways, abnormal sodium channel redistribution, perhaps with fewer NaV1.7 and NaV1.8 channels, or maladaptive neural plasticity during plaque repair may cause the central pain. The periaqueductal gray surrounds the aqueduct of Sylvius. Because it abuts the ventricular system, it is likely to be seriously damaged in multiple sclerosis. The periaqueductal gray controls pain, and it is possible that lesions in periaqueductal gray cause pain and headaches. Chronic back pain can arise as a secondary consequence of multiple sclerosis, causing unilateral weakness or spasticity and, in turn, poor posture and accelerated degenerative disc disease.
In optic neuritis, a swollen, inflamed optic nerve puts pressure on the dural sheath. Pain in or behind the eye sometimes precedes the visual loss. The frequently seen pain in optic neuritis and can be present at rest, on voluntary eye movement, and with pressure on the globe. Vasoactive amines, prostaglandins, and kinins released by inflammatory cells may magnify the pain in optic neuritis and in trigeminal neuralgia. Half of non-multiple sclerosis patients with autoantibodies to voltage-gated potassium channels have pain, and most respond to immunotherapy (128). Because antibodies to voltage-gated potassium channels appear in 5% of children with demyelinating disease, a search for these antibodies in serum could be considered in multiple sclerosis and neuromyelitis optica pain syndromes.
Headaches. Headaches are more common in multiple sclerosis (27%) than in matched controls (12%) (258). They can herald exacerbations. Headaches may reflect cortical inflammation near the meninges.
Seizures and paroxysmal symptoms. Epileptic seizures double in incidence in multiple sclerosis and are more common in later stages. They may arise from new or enhancing lesions in the cortex or subcortical areas. They also can be triggered by 4-amino pyridine therapy or rapid reductions in baclofen.
Paroxysmal symptoms last seconds to minutes. Paroxysms include visual complaints, diplopia, convergence spasm, vertigo, dysarthria, facial and limb myokymia, tonic motor seizures, tonic spasms, dystonia, restless legs, akinesia, spontaneous or kinesigenic choreoathetosis, hyperekplexia, rapid eye movement sleep disorders, ataxia, itching, altered taste, and pain and paresthesias (eg, trigeminal neuralgia, Lhermitte sign). They are triggered by hyperventilation (eg, 20 deep breaths), stress, cold, touch, metabolic abnormalities, exercise, or acute exacerbations. Transverse spread between demyelinated axons (ephaptic transmission) is a likely cause. It is probably amplified by cytokines, extracellular potassium, dysfunction of ion channels, and heterogeneity of new sodium channels. Lesions are in internal capsule, cerebral peduncles, and spinal cord.
Fatigue from central lesions and the role of sleep. Generalized physical and mental fatigue is the number one problem in two thirds of patients (204). Patients describe fatigue as “profound”; it “disrupts life” and it is “different from any other experience.” They say that because of the fatigue, “each day of the week at work is cumulatively harder.” It is usually worse with heat or humidity.
The normal motor fatigue that follows muscular exertion is magnified after sustained or repetitive muscle contractions and after walking (“walking in quicksand”). “Fatigability” develops rapidly after only minimal activity (in 75% of patients). It is most commonly focused in the intrinsic hand muscles, easily demonstrated with repetitive squeeze of spread finger extensors, often despite normal grip strength (Reder personal observation). Fatigability is distinct from weakness and may not correlate with strength of individual muscles (228). Cold temperature, which can increase spasticity and effort to move, will increase fatigability.
Another type of fatigue is sometimes unprovoked (“lassitude,” “asthenia,” or “overwhelming tiredness,” in 20%), and this can interfere with motivation. Fatigue also interferes with cognition, sometimes out of proportion to motor fatigability, and it limits prolonged neuropsychological testing. Rating scales of multiple sclerosis fatigue are difficult to design and correlate poorly with function because these symptoms are so multidimensional. Self-reports often do not correlate with clinical measurements of muscle and cognitive fatigue.
Fatigue is an essential dimension of the neurologic history. Fatigue can be the only symptom of an exacerbation or one of many. It is least common in primary progressive multiple sclerosis. Thirty percent of multiple sclerosis patients report fatigue before the diagnosis of multiple sclerosis (Berger personal communication 2011). MRI may show atrophy of the primary sensory cortex, frontal lobes, basal ganglia, and particularly the thalamus, caudate, putamen, and pons in patients with fatigue. Diffusion tensor imaging may show disruption of white matter connections between these brain regions. Fatigue seen on the Sickness Impact Profile Sleep and Rest Scale (SIPSR) predicts later brain atrophy (159). It is associated with reduced event-related potentials, with low prefrontal activity on PET, with posterior parietal cortical atrophy on MRI, and with low N-acetylaspartate in frontal lobes and basal ganglia on magnetic resonance spectroscopy.
“Primary fatigue” in multiple sclerosis can’t be explained by other factors such as depression and apathy, but it is intertwined with lack of sleep. Fatigue usually is worse in heat, in high humidity, and in the afternoon; body temperature is slightly higher in all these situations. This extreme sensitivity to heat is termed “Uhthoff phenomenon,” wherein a minimal elevation of body temperature, less than 0.5 degrees C, interferes with impulse conduction by demyelinated axons because of their lower “safety factor.” This is a measure of the amount of current available over the amount needed to activate at the node of Ranvier, about five in healthy neurons, but about one in demyelinated axons. Cytokines in the CNS are likely to play a role, but correlative studies have been difficult to perform. In 1824, Charles Prosper Ollivier d’Angers reported that a hot bath induced right leg numbness and loss of sensation and dexterity in a multiple sclerosis patient. Heat is associated with much lower central conduction times and commonly induces fatigue, weakness, and cognitive problems in multiple sclerosis. Motor function, best at 1 AM, and serum cortisol, highest at 1 AM, have a circadian relationship. In the afternoon, fatigue increases, strength diminishes, cortisol is low, and body temperature is highest.
Secondary fatigue from comorbid conditions that are frequent in the general population can add to multiple sclerosis fatigue. Obstructive sleep apnea is common in multiple sclerosis, and an elevated apnea/hypopnea index (AHI) is associated with brainstem multiple sclerosis plaques.
CNS fatigue has been attributed to decreased Na+/K+ ATPase in multiple sclerosis plaques, high energy demands of the large number of sodium channels redistributed on axons, disruption of the Kv1.3 potassium channel in mitochondria, inflammatory cytokines (IL-6, prostaglandins, tumor necrosis factor-alpha [TNF-alpha], CRP, and interferon-gamma [IFN-gamma]; this cytokine storm is worse after sleep deprivation), excess ammonia, serotonin, serum and spinal fluid neuroelectric blocking factors. A report of a specific brain sodium channel blocker could not be confirmed (25; 45). Other potential causes of primary fatigue in multiple sclerosis are demyelination and shadow plaques, with fewer wraps around the axon and different lipid composition (Szuchet, Molina personal communication 2024), axonal injury and poor axonal conduction, impaired glial function, poor perfusion of deep gray matter area, and neuronal dysfunction and metabolic exhaustion from the need to use wide areas of the cortex. Functional MRI for physical and cognitive tasks shows widespread compensatory (inefficient) reorganization of the damaged CNS, with increased demand on remaining neurons. In “non-primary fatigue,” contributors to fatigue and central conduction block are acidosis, lactate, and heat after exercise; the circadian rise in body temperature in the afternoon paralleling a circadian drop in serum cortisol; a half-degree centigrade rise in body temperature during the luteal phase post-ovulation; pain; poor sleep (daytime fatigue with waking at night, “middle insomnia,” often caused by need to urinate, and also spasms, itching, and high incidence of sleep-related movement disorders); depression—perhaps more likely with early morning fatigue; low levels of dehydroepiandrosterone (DHEA) and its sulphated conjugate (DHEAS); and low levels of vitamin D. Insula lesions in stroke cause underactivity and tiredness; the insular cortex atrophies in secondary progressive multiple sclerosis. Spasticity amplifies fatigue by creating resistance to movement, complicating routine actions. Fatigue is associated with restless leg syndrome, circadian rhythm disruption, periodic limb movements, and hypersomnolence on sleep studies. Medications, hypothyroidism, anemia, and muscle deconditioning can contribute to fatigue. Other diseases can cause fatigue, including Parkinson disease, amyotrophic lateral sclerosis, post-polio syndrome, myasthenia gravis, stroke, and traumatic brain injury.
Sleep disorders in multiple sclerosis are heterogeneous, often profound, and often unexplained. One third of multiple sclerosis patients complain of insomnia and severe daytime fatigue. Pain, depression, and nocturia impair sleep. Hypothalamic plaques in corticotrophin-releasing factor pathways are common and likely to damage orexin-containing neurons. This would reduce input to the suprachiasmatic nucleus and disrupt circadian clock genes, leading to insomnia and disrupted sleep. In small studies, however, CSF hypocretin (orexin) is normal in multiple sclerosis, except for scattered cases of hypothalamic plaques with hypersomnia, unlike the frequent low levels in narcolepsy. Brainstem lesions correlate with more apnea-hypopnea (AHI), and progressive multiple sclerosis correlates with abnormal central sleep indices (23).
Restless legs syndrome is four times more common in multiple sclerosis than in the general population. The urge to move the legs, which is accompanied by uncomfortable sensations in the legs, predominates in the evening or night, begins or worsens with rest or inactivity, and is relieved by movement. Iron and ferritin levels are often low; correction of low iron is sometimes therapeutic.
Shift work at a young age increases the risk of developing multiple sclerosis by 2-fold (102). Shift work may disrupt circadian rhythm, restrict sleep, elevate cortisol, activate viruses, and lead to proinflammatory immunity.
Cognitive function. Charcot described "conceptions that formed slowly" and “enfeeblement of memory” in multiple sclerosis. Higher cortical functions, language skills, and intellectual function usually appear normal to casual observers. However, careful clinical observation and sensitive neuropsychological tests find slight to moderate cognitive slowing (common), slow information processing (common), word-finding difficulties but otherwise intact language function, poor recent “explicit” memory, heightened distractibility, poor clock-drawing, poor executive function (17%), poor recognition of negative facial emotional expression, and decline in effortful attention in 50% of patients (201; 09).
Warm outdoor temperatures impair cognition. Cognitive impairment appears at the earliest stages of multiple sclerosis. At onset, one third of children are impaired and by year 5, two thirds have cognitive loss. Up to half of adult patients with clinically isolated syndromes are significantly abnormal on some cognitive tests.
Complaints range from “I always forget where I put my keys” and “the lights are off in the factory” to “I am no longer able to perform cube roots in my head.” Subcortical signs of slow processing speed often appear during complex tasks (especially with concurrent use of affected limbs), speeded responses, working memory, and when multiple visual and sensory stimuli confront the patient: “I feel like I live in an IMAX theater.” The simple question, “Do you have trouble walking through a shopping mall?" is often met with an anguished, "Yes, it’s too overwhelming.” Patients should be screened at the first examination, for cognitive problems and cognitive slowing (information processing speed, multitasking, sustained and complex attention, working memory). Patients with normal cognition tend to maintain their cognitive function, whereas mild cognitive deterioration predicts progressive decline in cognition over 3 years.
Healthy United States army recruits who later develop multiple sclerosis score six points lower on IQ tests than non-multiple sclerosis army peers. This indicates that there are early subclinical changes, likely paralleling clinically undetected inflammation and MRI lesions. A quarter of patients with clinically isolated symptoms have cognitive impairment. If untreated, more than half will be impaired 5 to 10 years later. This ominous natural history underscores the importance of using those multiple sclerosis therapies with proven effects on cognition and reflects the major improvement in multiple sclerosis care from therapies for multiple sclerosis (190).
The symbol digit modalities test (SDMT) is the best single screening measure, recommended by the Brief International Cognitive Assessment for MS (BICAMS) group (141). It tests processing efficiency and speed. Because it does not use language, it is excellent for cross-cultural studies. It can be supplemented with the California Verbal Learning Test (CVLT, memory) and the Brief Visuospatial Memory Test (BVMT). All three can be performed in a small center by staff members who may not have neuropsychiatric test training. Neuropsychological evaluation can review residual strengths and weaknesses that impact employment, social function, and driving ability; evaluation detects depression and leads to therapy. The widely used PASAT also measures mental processing speed but requires math skills and is very susceptible to practice effects. The Minimal Assessment of Cognitive Function in Multiple Sclerosis (MACFIMS) has seven component tests and takes 90 minutes but provides a relatively complete cognitive assessment. It shows cognitive decline in 60% of multiple sclerosis patients. A computer-based version, the processing speed test, is available.
Slow cognition commonly causes mood swings, irritability, and frustration. Cognitive decline impairs job performance, employment, and daily life. Slowed processing speed can cause slurred speech: “My mouth can’t keep up to my mind.” Patients have more difficulty walking while performing cognitive tasks. The family may notice impairment before the patient does, and patient complaints of cognitive decline may suggest depression. Cognitive deficits are most pronounced in secondary progressive disease but often do not correlate with physical disability. Cognition is least affected in primary progressive multiple sclerosis. Patients with more cognitive reserve are protected against decline, especially early in the disease. Greater maximum lifetime brain growth and enriched environments are protective. Risk factors for cognitive decline include cognitive problems at onset of multiple sclerosis, increased duration of multiple sclerosis, higher Expanded Disability Status Scale (EDSS), secondary progressive multiple sclerosis, lesion in the mesial temporal cortex, thalamic atrophy, black African ancestry (perhaps confounded by socioeconomic status), smoking, fatty liver, and perhaps the AA genotype of Apo E.
Decision making is compromised from slower learning plus impaired emotional reactivity. Occasionally, patients go through a phase of wildly illogical thinking that later resolves as the disease progresses. “Low anxiety” leads to inconsistent, risky decisions in a Gambling Task and predominates in early multiple sclerosis (127). Impulsivity correlates with loss of anterior corpus callosum integrity in cocaine-dependent subjects and possibly also in multiple sclerosis. Multiple sclerosis patients may have more health-adverse behaviors before diagnosis.
Some patients have nearly normal neurologic examinations yet are unable to walk from poor and disrupted speed of sensory feedback, motor impulses, and patterning of leg movement and gait. Electrophysiological tests confirm this apraxia and show impaired input to the motor cortex and to pathways involved in motor planning. Spinal learning may also be impaired (09). The EDSS score correlates better with cortical lesions (r = 0.6) than with white matter lesions (0.36). The whole brain NAA/Cr ratio, a measure of neuronal function and survival, correlates with cognition at r = 0.62.
Patients with mild cognitive impairment have cortical thinning on MRI. Chronic cases have extensive hippocampal demyelination (87). The small cornu ammonis 2 to 3 and the dentate gyrus CA1 and subiculum correlate with more depression and higher cortisol levels. Third ventricular width, or thalamic volume, perhaps the best measure at this time, has a direct correlation with the degree of cognitive loss. Corpus callosum atrophy, basal ganglia hypointensity and atrophy (brain parenchymal fraction), T1 and also T2 brain lesion load, and decreased fractional anisotropy on diffusion tensor imaging all correlate modestly with poor cognition. Lesions in the thalamus and frontal lobes impair executive function, and damage to the corpus callosum slows cognitive speed and math performance. A high cortical lesion volume triples the risk of cognitive impairment. Retinal nerve fiber layer thickness correlates quite well with symbol digit modality tests (r = 0.754) (252). Global N-acetyl aspartate has a moderate correlation with cognitive loss. Decreased attention correlates with lower N-acetylaspartate in the locus ceruleus in relapsing-remitting patients. Vitamin B12 and methylmalonate should be checked as abnormal serum levels can hurt cognitive function and cause brain atrophy.
On functional MRI (fMRI), decreased activation of the cerebellum correlates with poor motor learning. Conventional MRI and fMRI abnormalities correlate with slow psychomotor speed and more accidents while driving. Excessive activation (poorly focused) in the supramarginal gyrus, insula, and anterior cingulum correlates with poor episodic memory (Rao personal communication 2005). Excess activation also links to poor hand dexterity, suggesting greater and inefficient allocation of cognitive resources. Cognitively normal multiple sclerosis patients have increased activation in the parahippocampus and anterior cingulate, suggesting functional reorganization and adaptation to brain lesions, but some cognitively impaired patients have less activation in these areas (113). These regions all have less resting state functional connectivity. Paradoxically, with severe cognitive impairment, functional connectivity within the thalamus is increased, perhaps because normal complex thalamic integration is now pruned to a simple dysfunctional state. Resting state functional connectivity on fMRI for the default mode network is disrupted in multiple sclerosis. The shift out of the default mode network during tasks becomes difficult in multiple sclerosis patients with severe cognitive impairment. Positron emission tomography shows cortical hypometabolism above subcortical plaques. Cognitive impairment in rats with experimental allergic encephalomyelitis lasts long after the inflammatory lesions resolve.
Exacerbations can reduce cognition and processing speed, sometimes as the sole symptom. This decline can be detected with the symbol digit modalities test (SDMT). Arnason argues that memory problems appear during exacerbations in early multiple sclerosis, coincident with T cell inflammation in the CNS. Later in the disease, cognition is increasingly impaired, coincident with greater monocyte and microglial activation (09). Cognitive decline can be independent of other disease variables (57), suggesting that therapies could impact cognition independently of effects on relapses.
Low bone density is associated with cognitive impairment (Weinstock-Guttman personal communication 2011). This may be a consequence of an underlying cytokine or vitamin D-linked abnormality, or possibly from loss of CNS trophic input to bone.
Visual memory declines in multiple sclerosis. Visual pathways course from optic nerves, around the ventricles to the occipital cortex, and back around the ventricles to temporal memory areas. Visual pathways are interrupted by periventricular plaques and by inflammatory cytokines (191). IFN-beta therapy benefits visual memory (below).
Aphasia is rare in multiple sclerosis but can arise in acute disseminated encephalomyelitis.
Depression. The incidence of depression is increased 2- to 3-fold in multiple sclerosis patients (greater than 50%) and their families. Severe, short-duration multiple sclerosis is associated with more depression, but primary progression is associated with less depression (09). Plaques and hypometabolism in the left arcuate fasciculus (supra-insular white matter) (199), right temporal (19), and left temporal and inferior prefrontal areas (65) are associated with depression. However, depression does not correlate with MRI burden of disease or atrophy, disability, or cognitive deficits.
The dexamethasone suppression test reflects neuroendocrine function in depression. It is abnormal during active multiple sclerosis (207; 64), possibly from chronic inflammation, cytokine stress, and induction of CRH/AVP in hypothalamic neurons.
During attacks, depression is strongly correlated with cytokine levels, TNF-alpha, IFN-gamma, and interleukin-10 (119), possibly because IFN-gamma increases serotonin transporter and indoleamine dioxygenase levels, lowering serotonin.
Therapy with IFN-beta can occasionally trigger depression. Interferon elevates indolamine-2,3-dioxygenase, which lowers levels of tryptophan and serotonin. IFN-beta therapy as well as antidepressants could elevate brain serotonin by decreasing IFN-gamma levels. Both agents induce brain-derived neurotrophic factor. Surprisingly, patients taking anti-depressants have lower BDNF levels in circulating immune cells (100), possibly because depressed multiple sclerosis patients have low BDNF levels before antidepressant therapy.
Suicide is elevated 7-fold in multiple sclerosis. Suicidal patients are more likely to have a family history of mental illness, to abuse alcohol, to be under social stress or be depressed, and to live alone. Confused thoughts and occasionally psychosis can be seen with multiple sclerosis exacerbations. Epidemiological studies show a 1.5- to 2-fold increase in depression, anxiety, bipolar disorder, and schizophrenia in multiple sclerosis compared to non-multiple sclerosis Canadians (160). Pseudobulbar affect (PBA, pathological laughing and crying, involuntary emotional expression disorder) can be disabling. Disinhibition is from multiple supratentorial plaques; based on PBA in primary lateral sclerosis, lesions may involve white matter connections of frontotemporal cortex, transverse pons, and middle cerebellar peduncle. PBA is occasionally associated with hiccups and paroxysmal dystonia. Euphoria, despite concurrent neurologic problems, was described by Charcot. It is possible the euphoria is cytokine-mediated, akin to “spes phthisica”—a feeling of hopefulness for recovery seen in patients with tuberculosis.
Of note, depressed patients are more likely to start, but less likely to continue, therapy.
Associated diseases. In multiple sclerosis, there are links between inflammatory bowel disease and thyroiditis. Bone mass is low. Other autoimmune diseases are not associated with multiple sclerosis—and may be less prevalent than in the general population. Many reported associations are likely from the strong autoimmune proclivity in Devic disease or CNS Sjögren disease, variants that comprise 5% of “multiple sclerosis” patients in some series. Cancer and allergy incidence is likely reduced, perhaps from an overactive, Th1-biased immune system. A review of all papers from 1953 to 2010 showed a lower incidence of most types of cancer in multiple sclerosis patients (161). Parkinson and Alzheimer diseases occur at half the expected rate.
Comorbid diseases double the risk of hospitalizations in multiple sclerosis. The incidence of diabetes, hypertension, and ischemic heart disease is rising in the general North American population and faster in multiple sclerosis patients. The risk of death in multiple sclerosis patients increases with diabetes, cardiovascular disease, chronic lung disease, depression, and anxiety. Clinical trials of multiple sclerosis drugs seldom systematically document comorbidities.
Conversely, the disability associated with multiple sclerosis itself may contribute to the development of additional comorbidities. Inactivity and resultant deconditioning are clearly linked to the development of osteoporosis and vascular disease. These are environmental effects, which are independent of the immunology of multiple sclerosis or potential deleterious effects of treatments.
Natural history. The course of multiple sclerosis varies. Heterogeneity over time complicates the use of stage-specific therapies. Classification is important because no therapies are effective in the progressive forms. These categories are not immutable; patients frequently drift from one type of multiple sclerosis to another, become stable, or suddenly develop active disease.
At onset, at an average of 28 years old, multiple sclerosis is relapsing-remitting in 85% of patients. This form predominates in young women. Attacks typically occur every 2 years. The mortality rate in multiple sclerosis is 3-fold higher than in age-matched controls. Survival is decreased by 8 to 10 years but can be prolonged by IFN-beta-1b therapy (95).
The prodromal phase of the disease has been of increasing interest, with attempts to characterize the period prior to diagnosis. In 2018, a matched cohort study was published using data from linked health administrative and clinical databases from four Canadian provinces (264). Hospital, physician, and prescription use data from people with multiple sclerosis and matched general population controls were assessed in the 5 years before the first demyelinating disease claim or clinically reported symptom onset; a few cases were in the pre-MRI era. The primary outcome was all-cause use of health care. The study demonstrated more frequent use in patients with multiple sclerosis than in controls in the 5 years before a first demyelinating event, specifically for visits comprising nervous, sensory/pain, musculoskeletal, psychiatric, hematologic/anemic, fewer pregnancies, and genito-urinary complaints. This suggests the existence of a measurable multiple sclerosis prodrome (also seen in rheumatoid arthritis and inflammatory bowel disease), which could ultimately allow an earlier window of opportunity to identify and potentially treat multiple sclerosis, potentially in combination with MRI findings. Progressive multiple sclerosis could have a distinct prodrome as well that could assist in prognostication and disease course. Earlier identification of disease could also lead to a better understanding of the instigating immune mechanisms of the disease.
Fifty percent of relapsing-remitting patients become “secondary progressive” after 10 years, and 89% by 26 years. Relapses in the first 2 years predict earlier onset of progression. However, relapses after the first 2 years predict a lower chance of becoming progressive (227), suggesting that immune dysregulation evolves and modifies the course of multiple sclerosis. Also, arguing for sequential changes in immune response, there is a 10-year delay in the onset of progression in patients treated with IFN-beta (61). Progression has features of an age-dependent degenerative process (134). Progression begins, on average, at age 39 in both primary and secondary multiple sclerosis. Short telomere length, a biomarker for aging, has been associated with disability progression in multiple sclerosis (136).
The number of neurologic systems affected in the initial attack, and not recovery from the attacks, predicts the chance of developing progressive disease. The onset of progression is a watershed event that strongly determines the outcome of multiple sclerosis. Older age with the first attack leads to an earlier onset of secondary progression by the age of 40 or 50 years (226). Once progression appears, the rate of decline is constant and is unaffected by the prior duration of relapsing multiple sclerosis.
Ten percent to 15% are progressive from onset, at an average of 38 years old, with continuing deterioration and without obvious exacerbations or remissions, although the rate of decline fluctuates. Compared to a 10- to 19-year-old patient, the relative risk of primary progression is 2.3 at age 25, 8.1 at 35, 19 at 45, and 47-fold higher at age 50 to 59 years (241). Primary progression is considered a unique form of multiple sclerosis, but 28% of these patients will eventually have exacerbations (133), sometimes after 20 years of pure progression. Faster progression is linked to frequent relapses before or after progression starts, female sex, onset of progression over 50 years old, and lack of multiple sclerosis therapy for progressive/relapsing bout onset multiple sclerosis.
The progressive form affects the spinal cord predominantly (in 90%), begins at a later age than the relapsing form, and is approximately as common in men as in women. There is progressive paraparesis and loss of vibration and pinprick sensation in the legs, and typically a small, spastic neurogenic bladder. Brain MRI lesions are six times less frequent in primary progressive multiple sclerosis, compared to relapsing-remitting patients who become progressive later on (250). White matter that appears normal on conventional MRI, however, has low magnetization transfer ratio and N-acetyl aspartate levels, reflecting widespread neuronal loss or dysfunction (69).
Exacerbations contribute to disability by an average of 0.2 to 0.6 EDSS points at more than 30 days after the exacerbation. Forty-two percent to 49% have residual gain of 0.5 EDSS points at 2 to 4 months, and 28% to 33% have a loss of one or more EDSS point (155; 109). However, some improve: 19% have a 0.5-point decrease or improvement in EDSS and 10% have a one point decrease (155). In 700 placebo-treated patients from 11 clinical trials, worsening after exacerbations was nearly equivalent to improvement (60). The authors conclude that disability could not be used as an outcome measure in most (short-term) clinical trials. Attacks in older patients have less complete recovery, likely due to higher burden of lesions and effete repair.
The clinical course was clarified in 2013 (156). Residual symptoms after an attack should be called “worsening.” Progression,” independent of relapses, is not uniform in its rate and is a retrospective clinical term. Progressive disease can have four characteristics: (1) associated with active relapses, (2) without exacerbations, (3) largely stable for a period with superimposed exacerbation, and (4) largely stable, without exacerbations. These definitions are important in interpreting data on “progression” in clinical drug trials where cumulative average residual problems arise from incomplete recovery after attacks and not from true progression.
Occasionally, patients have acute fulminant multiple sclerosis (Marburg variant). This malignant form of multiple sclerosis is possibly associated with developmentally immature myelin basic protein (266). Patients with rapid accumulation of disability at early stages and later, at EDSS of 6, tend do poorly.
Tumefactive multiple sclerosis lesions, larger than 2 cm, can masquerade as tumor or abscess. They are rarely recurrent and had a surprisingly good prognosis in the first large series (126). They often contain T1 black holes within the leukoencephalopathy. After an initial tumefactive lesion, two thirds of patients will develop multiple sclerosis, whereas one third do not (06); occasionally patients develop Balo-like lesions.
Twenty percent of patients have “benign multiple sclerosis,” defined as a Kurtzke disability score of 3/10 or lower. After 20 years, 6% of the overall population is still benign—largely comprised of those with Kurtzke EDSS score of 2 or lower at 10 years (101). Autopsy studies indicate a large reservoir of undetected and, therefore, benign multiple sclerosis (205). Some patients with benign multiple sclerosis have surprisingly large lesion loads on MRI (244). Clinical or MRI dissociation is typical when correlating MRI with clinical activity (r is only 0.25). Auspicious predictors include young onset, past pregnancy, Caucasian, monosymptomatic, few attacks, no cord symptoms, good recovery after attacks, and few MRI lesions. Cognitive function, fatigue, and pain should be included in assessment of a propitious course as 50% of patients with “benign” multiple sclerosis based on motor disability have cognitive decline.
Unsuspected and asymptomatic cases. Multiple sclerosis is sometimes unsuspected during life yet found at autopsy. Twelve unsuspected cases of multiple sclerosis were found in 15,644 autopsies in Switzerland. Only two had no reported neurologic signs during life (85). There were five diagnosed cases of multiple sclerosis in 2450 autopsies in London and Ontario (88). In autopsy studies, the calculated prevalence of unsuspected multiple sclerosis would be about 31 in 100,000 in Paris (3 in 9300) (32); 90 to 128 in 100,000 in Switzerland (85); and 204 in 100,000 in Ontario (88). This suggests that the number of undiagnosed "normal" people with multiple sclerosis approximates the number of patients diagnosed with multiple sclerosis. Of asymptomatic “normal” first degree relatives, 4% to 10% have MRI lesions indistinguishable from multiple sclerosis (53). This suggests that “benign” multiple sclerosis is itself a spectrum and sometimes should not be treated with immunomodulators.
Clinically isolated syndromes (CIS). Clinically isolated syndromes include optic neuritis, transverse myelitis, and solitary demyelinating brainstem lesions. Clinical symptoms appear when the demyelinating lesions are in optic nerve, spinal cord, or clustered in critical areas of the brain (clinically eloquent or expressive areas). About one third of cases never evolve to multiple sclerosis (see idiopathic optic neuritis and transverse myelitis). Clinically isolated syndrome evolves into multiple sclerosis most often when the MRI T2 lesion load is high, when there is brain atrophy, when the CSF reflects inflammation, when evoked potentials are delayed, and when serum vitamin D is low (162). When clinically isolated symptoms appear in parallel with non-enhancing MRI lesions plus at least one enhancing lesion, 70% to 80% of patients will have another gadolinium-positive lesion within 6 months. Partial cervical myelopathy, without brain MRI lesions, often evolves into clinically definite multiple sclerosis if evoked potentials and CSF are abnormal (16).
Childhood and pediatric multiple sclerosis. An attack before the age of 16 happens in 4% of all multiple sclerosis patients and in 1% with onset before age 10. The incidence is higher in boys under the age of 10 but switches to girls at puberty. The incidence of demyelinating diseases, including multiple sclerosis, optic neuritis, transverse myelitis, and ADEM, is 1.63/100,000 and is higher in blacks than in Asians, Hispanics, and whites in Southern California (145). In girls, obesity increases the risk of multiple sclerosis, clinically isolated syndrome, or transverse myelitis by 1.6; massive obesity by 3.8 (143). The combination of obesity, HLA-DRB1*15, and absence of HLA-A*02 increase multiple sclerosis incidence by over 16-fold (104). Other risk factors are optic nerve lesions, EDSS worsening, two new MRI lesions over 2 years, low serum vitamin D, smoking, and lack of exercise. Early onset before the age of 11 is more common in males (the ratio is switched at puberty) and is linked to brainstem lesions. Older onset leads to faster accumulation of disability. Diagnosis is difficult because of the rarity of multiple sclerosis in children and the clinical overlap with childhood infections and other diseases. A family history is more common than in adult forms. Ninety-eight percent of children have a relapsing/remitting course, but slowly expanding MRI lesions are common. Red flags for a false diagnosis of multiple sclerosis in children include consanguinity or family history of developmental neurologic problems, early developmental delay, onset before the age of 1, gradual progression, and disease of other organ systems.
Sensory symptoms and optic neuritis are common (approximately 50%, even though these symptoms may sometimes not be reported by children). Brainstem and cerebellar symptoms, polysymptomatic disease, and seizures are more frequent than in adult-onset multiple sclerosis, but recovery from exacerbations is better (229; 218). One third of patients have cognitive problems. As in adult forms of multiple sclerosis, sphincter involvement and a (rare) progressive course have a poor prognosis. Boys predominate over girls between 8 and 10 years of age, but the girl-to-boy ratio is 2:1 after 10 years. Relapses are a bit more frequent in childhood (every 1.6 years versus every 2 years in adults) but are only 4 weeks long versus 7 weeks in adults (173). The course is slower than in adult-onset multiple sclerosis (232), and the median time from onset to secondary progression is 28 years. Nonetheless, with continuous exacerbations they become disabled at a younger age than adult-onset patients. Primary progression is exceptionally rare (2% of an already uncommon event).
MRI, EEG, and visual-evoked potentials are each abnormal in 80% of children with multiple sclerosis (14). Obvious cortical lesions are less common than in adults. However, intracranial volume and head size is reduced. One episode of demyelination will diminish brain growth in children. CSF is abnormal in 66% of patients, and CSF IgG levels are lower in children. Bands are positive in only 29% of acute disseminated encephalomyelitis but in 64% of acute multiple sclerosis and in 82% of multiple sclerosis at later times in a medium-sized series (47). In first demyelination in children, CSF contains molecules that localize to the node of Ranvier but not myelin membrane proteins (55). The prolonged relapsing-remitting course suggests multiple sclerosis therapies may be more effective in children than in adults. With more effective treatments, persistent disability has been reduced by 60% in recent years.
Quality of life and clinical scales. Multiple sclerosis has direct and indirect costs. For multiple sclerosis patients compared to non-multiple sclerosis patients, direct costs are $25,000.00 more per year, employment is 3.3 times less likely, and patients spend 4 days more in bed per year (30).
Responses by 433 patients were used to generate the 59-question Functional Assessment of Multiple Sclerosis (FAMS) quality of life scale (34; 33). A factor analysis demonstrated that multiple sclerosis had independent effects on several important factors that impact patients’ lives. Separate axes with little overlap included the following:
(1) Mobility. This correlated highly with the neurologic examination (Kurtzke Expanded Disability Status Score, Scripps Numerical Rating Scale, and Ambulation Index) but not with the other subscales.
(2) “Emotional well-being” and “general contentment,” which negatively correlated with psychiatric measures of anxiety and depression.
(3) “Symptoms.”
(4) Family and social well-being.
(5) “Fatigue” plus “thinking,” an indicator of cognitive function. Fatigue is highly prevalent; cognitive loss has the most important impact on quality of life.
Neurologic and social function, fatigue, mood, and cognition are important components of clinical multiple sclerosis that are often more disabling than inability to walk. Because these factors do not correlate, different pathogenic mechanisms are likely. For example, difficulty walking could arise from damage to long tracts or oligodendroglia, and fatigue may be caused by inflammatory cytokines in the CNS. Different pathological causes may also vary in responses to drugs; they should all be evaluated in therapeutic trials. There are trends or significant improvement in quality of life for all approved therapies.
The Kurtzke Extended Disability Status Score is a central clinical measure in most trials. It is based on the neurologic examination and ranges from 0 to 10, where 0 = normal, 4 = walks unaided for greater than 500 meters, 5 = walks unaided for greater than 100 meters, 6 = needs a cane to walk 100 meters, 7 = walks less than 5 meters with aid, 8 = perambulated in wheelchair, and 10 = death. Cognitive problems, fatigue, sexual function, job capabilities, and social factors do not weigh heavily in this scale. This scale is not linear; transition between stages 4 and 6 is fastest. The EDSS-Plus incorporates a timed 25-foot walk and 9-hole peg test and may double detection of progression. The MS Prediction Score, MS Progression Discussion Tool, and SPMS nomogram are tools for the difficult to detect transition to secondary progression. Progression independent of relapse activity is discussed in article on multiple sclerosis pathology. Frailty impacts, and is caused by, multiple sclerosis, especially in older patients.
The Multiple Sclerosis Functional Composite Scale (MSFC) evaluates motor function of legs and arms and cognition. It adds information to the Kurtzke Expanded Disability Status Score and was used in a phase 3 clinical trial of intramuscular IFN-beta-1a (37). Correlation between the Kurtzke scale and the multiple sclerosis Functional Composite scale is only r = -0.15. One component to the MSFC is the Timed 25-Foot Walk (T25FW). An improvement of 15% to 20% reflects a clinically meaningful change based on patients’ self-reports. Slowed walking speed of 6 to 8 seconds over 25 feet is associated with occupational disability and use of a cane. Speed of greater than 8 seconds correlates with collecting disability payments and use of a walker (92).
Patient-rated scales provide important information about independent factors that are missed when examinations are limited to assessment of mobility. Telephone and self-administered scales correlate well (r = 0.9) with physician examinations.
The global multiple sclerosis Severity Scale (MSSS) combines disease duration with the Kurtzke score to combine rate and severity (215). Many of the patients who defined the MSSS were on therapy, so untreated progression rates are probably even higher than the table indicates.
The overall life expectancy for untreated multiple sclerosis patients is at least 7 years less than normal, or 80% of expected survival (260; 221). Mortality increases with more disability. Case fatality ratios are 1:5 for patients with Kurtzke disability scores of 7 or lower, but 1:1 for those with scores greater than seven. One multiple sclerosis therapy, IFN-beta-1b, had a pivotal study design that allowed assessment of long-term survival. Five years of IFN therapy decreased mortality by 47% and extended life by about 7 years (95).
The cause of death in 50% of a clinic population and in approximately 75% of all multiple sclerosis patients is from complications of multiple sclerosis, usually pneumonia (222; 206). Most patients die as disability scores approach 8.0. Brainstem lesions occasionally cause loss of inspiratory drive, causing the patient to stop breathing; this is most common at night. Deaths from malignancy are less common than in age-matched controls (222).
The cumulative lifetime dose of disease-modifying therapy improves prognosis. Interferon-beta-1b extends survival by at least 6 years (see multiple sclerosis therapy below).
The suicide rate is increased 7.5-fold in multiple sclerosis (222). The lifetime risk of suicide is 2%. Less-disabled young patients within 5 years of diagnosis are the most likely actors (221).
The future course of multiple sclerosis is poor if there are cerebellar or pyramidal symptoms, slow timed-walk test at baseline, early sphincter symptoms, multi-site onset, frequent early attacks, development of progression or a primary progressive course, and age over 40 years at onset. In addition to not smoking, lack of obesity, adequate vitamin D levels, healthy diet, and marriage, good prognostic signs include optic neuritis, sensory symptoms, and continuing exacerbations and remissions (260; 188). Attacks after the first 2 years correlate with better prognosis, perhaps indicating that the patient has relapsing and not progressive multiple sclerosis. The course is a more important predictor than age of onset. Development of a progressive course is the strongest predictor of poor outcome. The second strongest predictor is the number of relapses in the first 2 years. After a first demyelinating episode, a second attack is more likely in younger patients with abnormal CSF, with more than eight T2 MRI lesions (dissemination) or one Gd-enhancing lesion (activity). Complete recovery is more likely with mild severity and mono-lesional exacerbations as well as in young patients. Clinic-based cohorts have more severe multiple sclerosis than population-based groups, where many patients remain stable or progress minimally over 10 years (189). Multiple sclerosis reduces quality of life throughout its course. Cognitive loss amplifies the negative effect along with disability, fatigue, depression and anxiety, and medical comorbidities.
Patients with relapsing-remitting multiple sclerosis take 15 years from onset to reach an EDSS score of 6.0 (using a cane to walk 100 meters), based on longitudinal studies in Ontario, Canada (59). Those with primary progressive multiple sclerosis take 8 years to use a cane; early progression and multi-system symptoms hasten the rate of progression. Patients with one attack in the first 2 years do not need a cane for 20 years; those with five or more attacks need a cane within 7 years. Once a patient becomes unable to walk 500 m (EDSS =4; typically after 11 years), progression is no longer affected by relapses. Later-onset multiple sclerosis is more common in men and is often primary progressive where incidence in men and women is equal. The transition from relapsing-remitting disease to secondary progressive multiple sclerosis is earlier in men than in women.
The average rate of decline is similar in all groups once multiple sclerosis becomes progressive (including primary progressive at onset, “bout onset progression,” or at the transition from relapsing-remitting to secondary progression) (59; 134). This happens on average at the age of 40, preferentially targets the corticospinal tract, and is not obviously influenced by prior relapses. Within the progressive group, however, rates vary (“sooner to cane, sooner to wheelchair”), even though the average rate for progression is the same for secondary and primary progressive multiple sclerosis after progression starts. Long-duration, low-disability multiple sclerosis is likely to remain stable.
On MRI, bad prognostic signs include a large number of T2 lesions or high T2 volume, T1 hypointensities, many enhancing lesions, low magnetization transfer, lesions in juxtacortical, infratentorial, and periventricular sites, and brain atrophy--especially early atrophy. Gray matter damage, loss of volume in the gray matter faction, weakly predicts worse disability (odds ratio = 0.79) (68). Good signs are little tissue damage and sparing of important regions. An MRI with a few large lesions gives a better prognosis than one with the same volume of many, smaller lesions (126; Zivadinov personal communication 2005). In a 20-year follow-up of 107 relapse-onset clinically isolated syndrome patients, lesion growth was 0.80 cc/year in those who were relapsing-remitting but was 2.89 cc/year in secondary progression (72). These baseline characteristics will influence prognosis. Baseline lesions and clinical activity must be matched in randomized cohorts to evaluate apparent responses to drugs (94).
In clinically isolated syndromes (lesions in optic nerve, brainstem, or spinal cord), total T2 lesion volume on MRI at onset correlates with disability at 10 years (r = 0.45). All patients with clinically isolated syndrome who have a total lesion volume greater than 3 cc progress to definite multiple sclerosis by 4 years (224). Thalamic atrophy and ventricular enlargement or new T2 MRI lesions 3 months after the first symptoms also strongly predict clinically definite multiple sclerosis – 88% after a positive MRI versus 19% after a negative MRI (24). In this patient cohort at 20 years, 79% of patients with clinically isolated syndrome and no MRI lesions had not had a second attack (72). A second attack had occurred in 21 of those with no MRI lesions at baseline, 80% with one to three lesions, 85% with four to nine lesions, and 81% with more than 10 lesions. The latter were much more likely to need a cane.
Efforts to formalize the diagnosis of multiple sclerosis have evolved over time since its initial characterization by Charcot, followed by Schumacher (1965), Poser (1983), and, most recently, several iterations of the McDonald criteria (249). At their core, criteria require dissemination of lesions in space and time (DIS, DIT), as well as the elimination of a more likely diagnosis.
The current clinical criteria to make a multiple sclerosis diagnosis in a person who has experienced a typical attack/clinically isolated syndrome are as follows:
|
• Two or more attacks and clinical evidence of two or more lesions; OR two or more attacks and clinical evidence of one lesion, with clear historical evidence of prior attack involving a lesion in a different location |
None. DIS and DIT have been met. | |
|
• Two or more attacks and clinical evidence of one lesion |
DIS shown by one of the following criteria: | |
|
• additional clinical attack implicating a different CNS site | ||
|
• One attack and clinical evidence of two or more lesions |
DIT shown by one of the following criteria: | |
|
• additional clinical attack | ||
|
• One attack and clinical evidence of one lesion |
DIS shown by one of the following criteria: | |
|
• additional attack implicating different a CNS site | ||
|
DIT shown by one of the following criteria: | ||
|
• additional clinical attack | ||
The current clinical presentation additional criteria in a person who has had steady progression of disease since onset are as follows:
|
• 1 year of disease progression (retrospective or prospective) |
DIS shown by at least two of the following criteria: | |
|
• one or more typical multiple sclerosis T2 lesions (periventricular, cortical, juxtacortical, or infratentorial) | ||
Current and historical criteria have been helpful in elucidating the potential for progress from clinically isolated syndrome to multiple sclerosis. In 532 patients with clinically isolated syndromes followed for up to 9 years, definite multiple sclerosis developed in only 35% of those with no asymptomatic baseline lesions but in 74% of those with three of four of the following lesions: (a) one enhancing or nine T2, (b) three periventricular, (c) one juxtacortical, or (d) one infratentorial (McDonald criteria) (131). In the BENEFIT clinically isolated syndrome study, nine baseline T2 lesions (hazard ratio = 1.6) or three periventricular lesions (hazard ratio = 1.7), or when one lesion changed to more than one, predicted conversion to multiple sclerosis. Multiple sclerosis is most likely to develop when the lesions are in the corpus callosum (hazard ratio of 2.7) or in fibers that control motor function. In patients with clinically isolated syndrome and monofocal non-cord symptoms who do not fulfil the McDonald criteria, a subclinical spinal cord MRI lesion increases the risk of developing definite multiple sclerosis by over 14-fold (236). When clinically isolated symptoms appear in parallel with non-enhancing MRI lesions plus at least one enhancing lesion, 70% to 80% of patients will have another gadolinium-positive lesion within 6 months. Partial cervical myelopathy, without brain MRI lesions, often evolves into clinically definite multiple sclerosis if evoked potentials and CSF are abnormal (16). There remains an important but arguably overlooked nuance to the criteria, which is that multiple sclerosis is best diagnosed by a clinician with experience in multiple sclerosis. Although the criteria form an important framework for decision making, it is ultimately most appropriately employed as an aid to clinical judgement.
Children who are female and obese and with low vitamin D levels, isolated symptoms at onset, multiple (multifocal) MRI lesions, elevated IgG index, and oligoclonal bands in CSF are more likely to develop multiple sclerosis than those with polyclonal onset and negative CSF tests. Organic solvents (103) and poor air quality have also been proposed as risk factors for pediatric multiple sclerosis (146). Adults develop multiple sclerosis when the MRI is abnormal, the CSF reflects inflammation, evoked potentials are delayed, and serum vitamin D is low (162).
After starting thrice-weekly interferon beta-1a therapy, first-year MRI lesions increase the chance of progression at 2 years by 4%. Clinical relapses after the first year increase the chance of progression by 49% (237).
Brain atrophy is often present in mild-to-moderate multiple sclerosis. Atrophy is most likely to progress when there are Gd-enhancing lesions at baseline (231). Ventricles in relapsing-remitting multiple sclerosis increase in size by 5% per year, compared to 1% to 2% in normal controls. In most cases of multiple sclerosis that come to medical attention, disease activity never sleeps, and atrophy progresses relentlessly in all multiple sclerosis subtypes. However, there are a large number of subclinical cases with benign courses and presumably much milder inflammation. Lesions lower the threshold for dysfunction when neurons are lost with age, well-illustrated with Steve Keiger’s topographical model of multiple sclerosis.
Magnetic (motor) and electric evoked potentials do not correlate with disability at first presentation. However, abnormal evoked potentials do predict disability at 2 years (r = 0.6 with motor and visual potentials) (80) and at 5 years (r = 0.5 with motor and sensory) (120).
CSF with a high white cell count predicts future Gd+ MRI lesions (216). CSF with high numbers of natural killer cells and monocytes augurs slower progression (35), although subsets of both of these cell types can damage oligodendroglia. High levels of IgG predict faster progression. B cells and plasma cells and a high B cell to monocyte ratio in CSF predict more disease activity and faster progression, as do increased myelin basic protein, increased IgM oligoclonal bands, and IgM synthesis (spinal cord involvement is 8-fold more likely), homozygous HLA-DRB1*1501, and polymorphisms in multiple genes (78). Antibodies to myelin basic protein in clinically isolated syndromes were highly predictive for development of multiple sclerosis in one study, but other labs have not been able to replicate these findings. Conversely, the 10% of multiple sclerosis patients with negative oligoclonal bands in CSF are more likely to have progressive forms of multiple sclerosis, non-specific “supratentorial” symptoms, lower cells and IgG in CSF, and less well-defined MRI lesions (233).
Other markers may be helpful in the future. Some of these include molecular indicators of interferon efficacy, B7 costimulatory molecules (CD8, CD86, PD-1, PD-L1) and adhesion molecule expression on mononuclear cells, serum cytokines or cytokine receptors, kallikrein proteases and matrix metalloproteases, IgM antibodies to glycans (anti-GAGA4), antibodies to CNS proteins (galactocerebroside, myelin basic protein, myelin-oligodendrocyte glycoprotein, neurofilaments, proteolipid protein), heat shock proteins, increased CSF 14-3-3 protein (neuronal loss) and myelin basic protein (oligodendrocyte damage), and CSF or serum soluble intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM) (correlate with MRI activity).
The lifetime cost of multiple sclerosis in the United States is high compared to other neurologic diseases. Several years after the first disease-modifying drugs for multiple sclerosis were introduced, multiple sclerosis had a total lifetime cost of $2,200,000 and an annual cost of $34,000 to $47,000. Multiple sclerosis therapy was more expensive than ischemic stroke ($100,000 lifetime) and Alzheimer disease ($49,000 to $490,000 lifetime) (262). Expense in diseases of long duration accrues from earnings lost, rehabilitation, drug therapy, “shadow costs” in monitoring the therapy, medical equipment, and formal and informal care. Fifty-three percent is direct (40% drugs, 3% inpatient hospital care), 10% is informal care, and 37% is productivity loss (reduced work time and retirement) (129). Costs increase dramatically with more severe disease. A British cost-effectiveness analysis suggests that each relapse avoided during interferon therapy costs 28,700 British pounds (185). Relapses are easy to count but have weak correlation with progression--a better indicator of disability. Longer survival with interferon beta-1b has not yet been factored into these analyses.
A 28-year-old woman began to stumble when walking. Her right leg was slightly stiff and was weak, especially after exercise and hot showers. These symptoms developed over 3 days and gradually disappeared over 4 weeks.
She was on the college swim team before these symptoms arose. When she was 21 years old, she developed a unique and extreme type of fatigue that differed from the usual fatigue after intense swimming workouts. The extreme fatigue disappeared after several weeks but reappeared again when she was 28 years of age. A maternal aunt had multiple sclerosis.
An MRI scan showed multiple periventricular lesions. Her spinal fluid had elevated IgG levels and three oligoclonal bands (normal, less than two).
One year later, 10 days after a “cold,” she developed blurred vision in her right eye and her visual acuity dropped to 20/200. She had moderate pain behind her eye when she looked to either side. The pain and visual loss gradually disappeared over 6 weeks. Two years later, both legs gradually became weaker and spastic, and she was sprinting to the washroom nearly every hour to urinate. These symptoms slowly progressed over the next 10 years, with occasional exacerbations affecting other areas of the brain. IFN-beta was begun in the middle of the relapsing and progressive phase, and the frequency of attacks and rate of progression slowed. She is now walking with the help of bilateral ankle and foot orthoses. She has been aided by minor modifications of her workplace and by treatment of multiple sclerosis symptoms, and she continues to work as a business executive.
The prevalence of multiple sclerosis in the United States was 400,000 in 1975. A 2019 revision by the National Multiple Sclerosis Society reported that the prevalence was 914,000 (256). In 2010, the U.S. prevalence was 309 per 100,000 people (727,000 cases); 450 per 100,000 women; 160 per 100,000 men; female:male = 2.8; 375 in 100,000 Whites, 77% of cases; 298 in 100,000 Blacks, 10%; 161 in 100,000 Hispanics, 7%; and 198 in 100,000 Others, 4%. Incidence increased by 16 cases per 100,000 with each degree of latitude (110). Worldwide, the incidence rose over decades but may be plateauing, and the course became milder with use of disease-modifying therapies and perhaps less smoking and more vitamin D use. Goodin argues that only a small proportion of the population is able to develop the disease and that a truly random mechanism plays a role, perhaps T- and B-cell receptor hypermutation (93).
Geographic variation. The incidence and symptoms of multiple sclerosis are different around the globe. Multiple sclerosis is uncommon at the equator (prevalence 2 to 10 per 100,000) and increases with distance from the equator (up to 300 per 100,000 at latitudinal extremes). This suggests environmental factors influence the incidence. However, emigrating northern Europeans did tend to stay in temperate climates, suggesting genetic influence. Multiple sclerosis is rare in Asia (4 per 100,000) (139). Multiple sclerosis in Japan, China, Malaysia, in black Africans, and in some groups of Canadian Aboriginals often resembles neuromyelitis optica disease because it typically affects the optic nerves and spinal cord and occurs at an earlier age than the Western form of multiple sclerosis (42; 188). The incidence is changing, with higher rates worldwide, especially in black women in the United States. Multiple sclerosis odds in England are slightly higher (1.15x odds ratio) for young Blacks than Whites, but not in those over 40 (0.65x) (56). The clinical profile also changed in Japan over 30 years, with an 8-fold shift from an Oriental/Devic-like presentation, to Western, multiple sclerosis-like symptoms and signs. Environmental changes in smoking, diet, and vitamin D levels are likely to play a role. Details on the incidence and prevalence of multiple sclerosis are in the immunopathology of multiple sclerosis chapter. Epidemiologic updates are on https://www.atlasofMS.org.
There is no known cause for multiple sclerosis. Viruses are often implicated as the primary cause, but in no case has this been substantiated. There is no apparent risk of transmitting multiple sclerosis to spouses or medical personnel.
Symptoms and exacerbations frequently follow virus infections (230; 184; 62), bacterial and bladder infections (202), prostatitis, and decayed, abscessed, or missing teeth (43). Panitch found that exacerbations sometimes occurred 1 to 2 weeks before overt virus infections (184). Some virus proteins block interferon signaling and could reduce the already low level of serum type I IFNs in multiple sclerosis. Exacerbations during systemic infections are twice as likely to lead to sustained clinical deficits, even though there is no difference on MRI (28). It is wise to prevent immune activation with good bladder and dental hygiene and to minimize exposure to people with upper respiratory infections. Hospital-treated infections increase disability by 0.2 EDSS points and are worse in progressive disease and while treated with “interferon/glatiramer” (grouped together despite their different mechanisms) versus rituximab (111).
There is a 1.6x relative risk of development of multiple sclerosis in cigarette smokers (107). Smokers are more likely to develop a progressive course (58), possibly from an effect on the blood-brain barrier or an influence of chronic bronchitis. Smoking increases the exacerbation rate by 60% and doubles the rate of brain atrophy. This raises an ethical question—should a person who refuses to stop smoking be placed on a $50,000.00 per year disease-modifying therapy, which will be canceled by the smoking?
Vaccinations and immunizations usually do not cause exacerbations of multiple sclerosis (230; 11) and may actually reduce exacerbations, with relative risk of 0.71 following vaccination for tetanus and hepatitis B (38). Measles, mumps, rubella, and human papilloma virus vaccines are considered safe. Tetanus and diphtheria vaccinations are associated with a reduced chance of developing multiple sclerosis (0.67) (105), as is Bacille Calmette-Guèrin vaccine (212). Influenza vaccination should be encouraged in order to obviate the risk of exacerbation during influenza infections.
There is debate about exacerbations from recombinant hepatitis B vaccine (106). Live virus vaccines, however, are likely to induce a cytokine storm and should be administered with caution. Yellow fever vaccinations, for instance, increase the risk of exacerbations 9-fold in relapsing-remitting multiple sclerosis. Some multiple sclerosis therapies reduce responses to H1N1 swine flu vaccination. Protective levels are reached in 44% of controls, and multiple sclerosis patients on IFN-beta therapy, compared to 24% on natalizumab, 22% on glatiramer, and 0% on mitoxantrone (181). Fingolimod induces a normal percentage of protection above threshold, but overall titers are reduced by 60%.
Diet and environment appear to affect the development and course of multiple sclerosis. In Norway, cod liver oil and fish intake (omega-3 fatty acids, vitamins A and D) reduces risk of developing multiple sclerosis (121). High-risk groups such as unaffected relatives of multiple sclerosis patients could benefit from a dirty environment, a diet rich in polyunsaturated fats (evening primrose and flaxseed oil), weight loss, not smoking, sunlight, and vitamin D (Bielby personal communication 2005; 193). The MIND diet may offer some protective effect against brain atrophy (124). Other dietary approaches (ketogenic, Wahls) have not been validated. Lifelong exposures, the “exposome,” and environmental risks include Epstein-Barr virus and possibly HHV-6A infection, obesity, smoking (synergy with obesity), low vitamin D, night shift work, and ozone exposure and microplastics in water. In contrast, protective factors include cytomegalovirus, HIV (50% lower rate of multiple sclerosis), polyfluorinated substances, a fish diet, social drinking, and nicotine.
Theoretical reasons exist to avoid several drugs, but therapeutic need may outweigh theory. Cimetidine, a histamine H2 blocker (07), and melatonin (39) enhance immune function, and may oppose the ability of H1 agonists to reduce blood-brain barrier permeability. Beta-adrenergic blockers inhibit suppressor cell function (122). Occasional patients have worse multiple sclerosis symptoms with fluoroquinolone antibiotics (eg, ciprofloxacin) (Reder personal communication 1986), which in addition to their antibacterial effects induce inflammatory cytokines (210). These antibiotics double the risk of peripheral neuropathy. Some patients are extremely sensitive to low doses of carbamazepine; weakness is probably caused by blockade of Na+ channels in demyelinated axons. Immune checkpoint inhibitors for cancer can trigger autoimmune encephalitis. Anti-TNF agents can precipitate new multiple sclerosis, transverse myelitis, and optic neuritis.
Stress does not provoke attacks of multiple sclerosis in controlled trials, despite widespread anecdotes of stress worsening multiple sclerosis (91). Some patients have brief episodes of worsening and then complete recovery over several hours. They often deny temperature elevation, but brief viremia or pulmonary/gut barrier breakdown, including a surge of clostridium perfringens epsilon toxin, are possible causes.
Exercise, physical therapy, dancing, social contacts, better diet, and drug treatment of symptoms can improve motor function and coordination, and quality of life. All of these interventions allow patients to realize that they can control some of the symptoms of multiple sclerosis.
Monosymptomatic illnesses from focal or multifocal CNS insults can mimic the first attack of multiple sclerosis. These illnesses and cover a wide spectrum of neurologic diseases.
Postinfectious and postvaccinal encephalomyelitis follow inflammation-induced sensitization to myelin antigens. These reactions cause inflammatory demyelination that is localized (eg, transverse myelitis, optic neuritis) or diffuse (eg, encephalomyelitis). Symptoms often develop after upper respiratory tract infections (usually viral or mycoplasma) or vaccinations, leading to acute disseminated encephalomyelitis. Spontaneous autoimmune encephalitis is associated with multiple autoantibodies. CSF oligoclonal bands are less common than in multiple sclerosis and, if present, often disappear. MRI lesions should all be of the same age at onset, but several weeks afterward, partially resolved lesions can appear to be different ages. The perivascular inflammation and demyelination is similar to the histopathology in multiple sclerosis, but these fever-associated disorders are monophasic.
Experimental allergic encephalomyelitis is the animal model for postinfectious encephalomyelitis. Multiple sclerosis patients vaccinated with porcine myelin basic protein can develop encephalomyelitis. This non-recurring demyelinating disease suggests (1) a primary response to the antigen and (2) that myelin basic protein does not trigger multiple sclerosis.
Recurrent symptoms that mimic relapsing-remitting multiple sclerosis may be caused by focal repetitive insults such as transient ischemic attacks or exacerbations of connective tissue disease. Vascular insults usually have a rapid onset (99).
In hypertensive vascular disease, MRI lesions are in the centrum semiovale instead of periventricular sites, are not in cerebellar outflow pathways, and do not spare subcortical U fibers. Some authors have implicated anticardiolipin antibodies and granulomatous angiitis in multiple sclerosis-like syndromes. CADASIL, Binswanger disease, cryopyrins, hemiplegic migraine, Sjögren syndrome, and Behçet disease can cause episodic, multifocal central nervous system lesions that can be confused with multiple sclerosis clinically and on MRI.
Progressive cord symptoms can be caused by subacute combined degeneration, adrenoleukodystrophy, chronic fatigue syndrome, and tropical spastic paraparesis from human T cell lymphotropic virus infection. Damage from a spinal dural arteriovenous fistula, cavernous hemangioma, or tumor can cause indolent cord symptoms. Progressive symptoms also suggest metabolic problems (copper, vitamin B12, vitamin E, or folate deficiency—often from complications of gastric bypass surgery), genetic disorders (adrenoleukodystrophy, sometimes with a late inflammatory phase; hereditary spastic paraplegia; PLP1 gene mutation causing hereditary spastic paraplegia; Wilson disease), and mitochondrial disorders. Magnetic resonance spectroscopy can help differentiate plaques from central nervous system tumors.
“Phenocopies” of multiple sclerosis appear on MRI scans. CADASIL, hypertensive vascular disease, Susac syndrome, leukodystrophies, vanishing white matter disease, Alexander disease, sarcoid, and migraine all overlap with the MRI appearance of multiple sclerosis.
Transverse myelitis can be an isolated event or the first sign of multiple sclerosis or neuromyelitis optica. The cord lesion in multiple sclerosis is more often partial, patchy, and asymmetric, but in idiopathic transverse myelitis it is symmetrical and bilateral and longitudinal demyelination may be extensive, and CSF abnormalities are less common than in multiple sclerosis.
Optic neuritis is often the first sign of multiple sclerosis. Isolated optic neuritis involves only the optic nerves, without dissemination of lesions in time and space. CSF oligoclonal bands are less common than in multiple sclerosis. Nonetheless, one third of patients with optic neuritis will eventually develop multiple sclerosis. Optic neuritis must be differentiated from ischemic optic neuropathy, increased intracranial pressure causing papilledema, vitamin B12 deficiency, vasculitis (temporal arteritis), viral infections, and Devic disease.
Conditions most commonly confused with multiple sclerosis are migraine, fibromyalgia, psychogenic disorders, neuromyelitis optica spectrum disorder (Devic), and myelin oligodendrocyte glycoprotein disease. Errors stem from missing atypical symptoms (see red flags, below) and atypical MRI findings, or conversely, over-reliance on MRI without integrating the clinical picture. Differential diagnosis in early onset includes acute disseminated encephalomyelitis, myelin oligodendrocyte glycoprotein antibody disease, and neuromyelitis optica and in late onset includes neurosarcoid, tumors, vascular disease, and vitamin deficiencies, plus more comorbidities (112).
Mimics include the following:
• Acoustic neuroma. Nerves VIII, V, and VII are compressed by slowly expanding neuroma or meningioma.
• Acute disseminated encephalomyelitis (ADEM) often follows an infection or vaccination (postinfectious or postvaccinal encephalomyelitis). ADEM is more likely to occur in children and is one fourth of pediatric demyelinating disease. It has a polysymptomatic or multifocal presentation, plus systemic symptoms that are uncommon in multiple sclerosis. Patients develop sudden pyramidal symptoms, bilateral optic neuritis (unilateral optic neuritis is seldom or never seen and suggests multiple sclerosis). Importantly, there is also fever, headache, vomiting, shooting pains, meningismus, encephalopathy, altered consciousness, EEG changes, and blood and CSF pleocytosis. Oligoclonal bands in CSF are uncommon (0% to 30%), but protein is often greater than 100 mg/dl. Many of these patients have serum antibodies to myelin oligodendrocyte glycoprotein.
MRI lesions in ADEM are large, “fluffy,” enhancing, often in ring patterns, often in a diffuse bilateral pattern, often in the corpus callosum and thalamus (thalamic gray matter T2 lesions are rare in multiple sclerosis), but seldom in a Dawson finger shape, less often periventricular, and seldom cause T1 black holes (47). Macrophages and sleeves of (later) demyelination surround venules in many small and large inflammatory lesions. Axons are relatively preserved. Myelin loss is more pronounced in multiple sclerosis than in ADEM and experimental allergic encephalomyelitis. All lesions are approximately the same age in the initial attack. A storm of many cytokines appears, with high granulocyte colony stimulating factor, but no IL-17.
ADEM is usually monophasic, and MRI lesions are of similar age. This basic form lasts up to 3 months. Symptoms can fluctuate, and MRI may transiently show Gd-enhancing and nonenhancing lesions as ADEM evolves. If similar lesions reappear 3 months later, it is termed “recurrent ADEM.” In “multiphasic ADEM,” new brain areas are involved during a second episode. Many of these NMO-IgG negative patients have serum antibodies to myelin oligodendrocyte glycoprotein. It is possible that abrupt discontinuation of steroid therapy allows recrudescent ADEM lesions; in this case, the disease is not truly recurrent. Therapy with high-dose glucocorticoids and then a prolonged taper is advised in ADEM.
Multiple sclerosis eventually develops in 20% to 33% of ADEM patients. Thus, most patients with ADEM (66% to 80%) do not develop multiple sclerosis and should not be treated for multiple sclerosis when oligoclonal bands are negative. A second attack is less likely after initial encephalopathic symptoms, in males, and with later age or onset.
• Acute hemorrhagic leukoencephalitis of Weston Hurst appears to be a more severe form of ADEM but may have a distinct etiology. Polymorphonuclear infiltrates are common, and the histopathology differs.
• Acute anterior ischemic optic neuropathy has a sudden onset, is usually painless, may progress over several days with an unremitting course, and is typically seen in patients 60 to 100 years old. Visual loss involves the central fixation area but is altitudinal, with sharp borders. Ischemic optic neuropathy is likely to cause disc swelling, pallor, arterial attenuation, hemorrhage, and permanent loss of vision.
• Acute necrotizing encephalopathy (ANE) of childhood, seen in East Asian countries, follows several days of fever with a respiratory or gastrointestinal virus infection, which can then be followed by seizures and neurologic deficits. Lesions are hypointense on T1 and hyperintense on T2 MRI in the bilateral thalami (classic for this condition) but are not present in the basal ganglia and cerebral cortex.
• Adrenoleukodystrophy (X-linked adrenomyeloneuropathy and cerebral adrenoleukodystrophy, both with brain MRI lesions often in corticospinal tract, temporal pole, and splenium) with high very-long-chain unbranched fatty acids from mutation in the ABCD1 gene. In adults, it is important to also consider Alexander disease, chloride channel ClCN2 coding for ClC2 protein and connexin-32 mutations, fatty acid 2-hydroxylase (FAH2), globoid cell leukodystrophy (Krabbe), GM1/GM2 gangliosidoses, Kearns-Sayer, LMNB1-related leukodystrophy (lamin B1), metachromatic leukodystrophy, Pelizaeus-Merzbacher and Hypomyelination of early myelinating structures (HEMS) (both from PLP1 mutation), and vanishing white matter disease.
• Adult-onset autosomal dominant leukodystrophy. From a lamin B1 mutation, the onset is at approximately 40 years, with autonomic symptoms and then cerebellar and pyramidal signs. T2 MRI shows symmetric frontoparietal and cerebellar white matter lesions but sparing of the periventricular areas.
• Alexander disease can appear in adults (type II) with a myelopathy and medullary signs. MRI shows patchy medullary and brainstem lesions with atrophy of medulla and cord.
• Alzheimer occurs at half the expected rate in multiple sclerosis, and there is less amyloid beta than expected in multiple sclerosis plasma.
• Aminoaciduria: 3-methylglutaconic aciduria type I causes adult-onset leukoencephalopathy.
• Amyloid angiopathy causes cerebral microhemorrhages but also a leukoencephalopathy that involves the periventricular trigone area and U-fibers. Iron in lesions can be seen on T2-weighted MRI.
• Aneurysm of intracranial blood vessels.
• Astrocytopathy--autoimmune, associated with CSF and serum antibodies to glial fibrillary acidic protein (GFAP), causes necrotizing meningo-encephalitis. It is associated with ovarian teratomas in one fourth of patients. Enhanced MRI shows thin radial perivascular vessels and speckled cord lesions.
• Atopic myelitis, idiopathic eosinophilic myelitis, hyperIgEaemic myelitis.
• Autoimmune thyroid disease--often familial, causes tremor, and seizures. Spinal cord involvement may suggest overlap with neuromyelitis optica.
• Balo concentric sclerosis has large concentric lesions with successive, centrifugal waves of demyelination and remyelination. MRI shows ring enhancement with Gd; increased white matter signal on FLAIR, importantly, sparing the U-fibers; ring lesions on diffusion-weighted imaging; and related changes in apparent diffusion coefficient imaging. With time, lesions may evolve to look like tumefactive multiple sclerosis. There is loss of myelin-associated glycoprotein and oligodendrocyte apoptosis. Demyelinated areas are high in nitric oxide synthetase (iNOS). Hypoxic conditioning may explain the rings, where sublethal hypoxia provides resistance to later injury. Hypoxia-inducible factor1alpha (HIF1a) is increased in the outer edge of preserved tissue (240); it is also increased in normal-appearing white matter in multiple sclerosis.
• Behçet disease – CSF pleocytosis, large MRI lesions in upper brainstem and basal ganglia, and occasional punctuate parenchymal enhancement. Behçet disease causes intermittent cranial nerve deficits. Biopsies show polymorphonuclear cells and eosinophils surrounding arterioles. It is associated with rash, arthralgia, genital and oral ulcers, uveitis, cerebral vein thrombosis, and meningoencephalitis. Optic neuropathy is relatively rare in Behçet disease. Twenty percent of patients with Reiter syndrome (reactive arthritis) have uveitis.
• CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) is caused by a mutation of the NOTCH3 gene. NOTCH controls lymphocyte development, oligodendrocyte maturation, and pericyte function. Here, NOTCH3 protein aggregates around vascular pericytes, which retract from capillaries or die. Astrocyte end feet detach from cerebral microvessels, also allowing leak of plasma proteins into the CNS. The intermittent strokes in midlife on a background of patchy MRI T1 holes plus diffuse increased white matter T2 signal can be confused with multiple sclerosis. T2 lesions are large and confluent and often significant in the anterior temporal lobes. It is diagnosed with a forearm skin biopsy for granular osmiophilic material in arterioles or with DNA analysis for the NOTCH mutation. Related syndromes include autosomal dominant retinal vasculopathy with cerebral leukodystrophy, CARASIL (below), hereditary cerebral amyloid angiopathy (CAA), COL4A1 angiopathy, inflammatory CNS leukoencephalopathy with calcifications and high IP-10, and perhaps interferon (NOTCH1 mutation), neuronal intranuclear inclusion disease (NIID; NOTCH2NLC mutation), and vascular leukoencephalopathy mapping to chromosome 20q13.
• Cancer--primary and secondary brain tumors. Hemophagocytic lymphohistiocytosis, Langerhans cell histiocytosis, and neoplastic angioendotheliosis can be confused with multiple sclerosis. Compared to glioma, patients with tumefactive multiple sclerosis are younger women, with smaller lesions that always enhance, sometimes with an open ring.
• CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) is caused by a mutation of the HTRA1 gene, which codes for a serine protease that controls TGF-beta signaling. MRI and presentation is similar to CADASIL, but with the “arc sign” on T2 MRI from pons to middle cerebellar peduncle, CARASAL has more confluent and symmetrical white matter lesions, from mutation of the CTSA gene coding for cathepsin A, which inactivates endothelin-1.
• Carcinomatous polyradiculopathy is associated with adenocarcinoma of breast and lung, lymphoma, or melanoma.
• Cavernous sinus thrombosis affects cranial nerves III, IV, V, and VI.
• Celiac disease can cause myelopathy and encephalopathy. Celiac disease was increased in relapsing multiple sclerosis patients (11%, which is 5x above normal) and families (“32%”) (213). Many autoantibodies, as well as anti-tissue transglutaminase-2, were elevated in eight of 72 multiple sclerosis patients, but only one had diarrhea and five were constipated. This study needs replication.
• Cerebellar degeneration and Friedreich ataxia can mimic progressive cord symptoms.
• Cerebrotendinous xanthomatosis (progressive myelopathy in a young patient, but with cataracts, diarrhea, ankle tendon xanthomas, and cerebellar dentate lesions on MRI).
• Cervical compression (disc, spondylosis, or tumor) can cause a progressive paraparesis, gait disorder, and bladder dysfunction. MRI shows a spindle shape or “pancake sign” on T2 with Gd enhancement of the central hub, located slightly caudal to the compression.
• Charcot-Marie-Tooth disease (brain MRI lesions and progressive course).
• Chemotherapy can cause a leukoencephalopathy and cognitive decline (Ara-C, cisplatin, 5-fluorouracil, 5-florauracil+levamisol); neurotoxicity is worse with radiotherapy and progresses over time.
• Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) with optic neuritis.
• Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS) has enhancing lesions centered in the pons, sometimes with spread to cord with extensive lesions, also cerebellum, basal ganglia, and small juxtacortical lesions. Severe, approximately biannual attacks cause brainstem and cerebellar, and sometimes cord, symptoms. CD4 T cells infiltrate the lesions in this non-demyelinating disorder of unknown etiology. One patient has developed CLIPPERS after natalizumab withdrawal. A rare variant is supratentorial lymphocytic inflammation with parenchymal perivascular enhancement responsive to steroids (SLIPPERS).
• Cogan syndrome (vestibulo-auditory problems). Inflammation is predominantly CD4 T cells.
• Combined central and peripheral demyelination and encephalomyeloneuropathy appears in multiple hereditary and degenerative diseases (147). Many patients with combined central and peripheral demyelination have antibodies to neurofascin, a CNS and PNS adhesion molecule in the nodes of Ranvier and paranodes. Others have antibodies to neutral glycosphingolipids.
• Congenital adrenal hyperplasia (diffuse brain white matter and corpus callosum abnormalities).
• Connective tissue diseases can mimic multiple sclerosis. They can cause vasculitis with neuropathy, cranial nerve damage, and CNS destruction. Systemic lupus erythematosus causes a severe, progressive thoracic myelopathy also called “lupoid sclerosis” or “acute lupus myelopathy.” It is acute or subacute with scattered white matter lesions in the cortical or subcortical junction but can affect gray and white matter. Twenty percent of patients have optic neuropathy in one or both eyes. When the cord is involved, the damage extends over many segments or the entire cord. This historic entity is likely to have been from neuromyelitis optica associated with connective tissue disease. These cases are likely to fall within the Devic/CNS Sjögren overlap syndrome described below.
Most patients with systemic lupus erythematosus, however, have no cord lesions. Treatment of connective tissue disorders diametrically differs from multiple sclerosis therapy, as interferons could cause worsening. (Sjögren syndrome is described below.) Craniofacial scleroderma is associated with bilateral-enhancing MRI lesions and oligoclonal bands.
• Copper deficiency causes a progressive myelopathy and neuropathy, often related to gastrointestinal disorders, post-gastric bypass, and zinc excess (similar to the cuprizone model in rodents).
• Cortical blindness must be discriminated from optic neuritis.
• Cranial arteritis (temporal arteritis) can affect the posterior optic nerve, without papilledema, in 70- to 80-year-old patients. It causes devastating visual loss; temporal pain, fever, weight loss, headache, fever, and high erythrocyte sedimentation rate and is linked to polymyalgia rheumatica.
• Devic disease, or neuromyelitis optica, is a demyelinating, sometimes necrotic, inflammatory disease of the spinal cord and the optic nerves. Attacks are more severe and more frequent than in multiple sclerosis. In Asia and South America and in Native American Indians, Devic disease is more common than multiple sclerosis. In Europe and the United States, multiple sclerosis is far more common. Devic disease traditionally accounted for less than 1% of occidental demyelinating disease, but a test (NMO-IgG) suggests that approximately 5% of “multiple sclerosis” cases with optic neuritis and longitudinal cord lesions are the Devic variant.
This IgG1 neuromyelitis optica antibody recognizes aquaporin-4, a water transport channel that is localized in astrocyte foot processes over paranodal axons and near blood vessels. Aquaporin-4 is present in high levels in renal tubules, possibly explaining the low glomerular filtration rate in progressive multiple sclerosis (29). Antibodies to aquaporin predict development of neuromyelitis optica and a more severe course. Although this antibody is present at all times, the attacks are intermittent. Some patients do not have serum NMO-IgG antibodies but do have antibodies to myelin oligodendrocyte glycoprotein. The latter form of neuromyelitis optica is milder but exhibits more ataxia.
Neuromyelitis optica is associated with connective tissue disease, rheumatoid arthritis, pernicious anemia, hypothyroidism and antibodies to thyroid antigen, and myasthenia gravis. The odds ratio of having another autoimmune disease is 10. Linkage of multiple sclerosis to these autoimmune disorders becomes unlikely when neuromyelitis optica disease cases are eliminated from the “multiple sclerosis” pool. CNS Sjögren disease is similar to neuromyelitis optica (detailed below), but only 40% are NMO-IgG positive.
Neuromyelitis optica is monophasic in one third of patients (265). In the remaining two thirds, neuromyelitis optica has severe and frequent relapses and does not become progressive. The damage in the optic nerves and cord is diffuse, often total, and includes significant axonal loss and cervical cord atrophy. Because axons are lost out of proportion to the demyelination, lesions cause pronounced clinical symptoms, but heat sensitivity is less prominent than in multiple sclerosis. Thoracic pain is common (above). Lesions are sometimes cavitary and necrotic and are often hypointense on T1-weighted MRI scans. The MRI lesions only infrequently surround veins, unlike multiple sclerosis, where more than 95% have a central vein that is clearly seen. The spinal lesions in neuromyelitis optica extend over more than two vertebral segments and are usually cervical. Cervical MRI images in neuromyelitis optica are similar to CNS Sjögren disease (below). Optic nerve lesions are also longer, averaging 2 cm, and if greater than 4 cm, they are diagnostic. These lesions tend to be chiasmal and bilateral (200) (20%). In the early, strict definition, MRI scans of the brain are normal, and there is no demyelination in the brain, brainstem, or cerebellum (158). However, 20% later evince large brainstem and hypothalamic lesions that are poorly demarcated and which sometimes enhance on MRI. CSF protein and neurofilament heavy chain levels are elevated. Seventy-five percent of patients have CSF pleocytosis, and 35% have greater than 50 cells/mm3; polymorphonuclear cells are often present. CSF IgG synthesis is usually normal, and CSF IgG1 is not elevated (it is elevated in multiple sclerosis). Oligoclonal bands are less common (40% or less) than in multiple sclerosis (97%) and can disappear over time.
Neuromyelitis optica lesions in parenchyma and meninges have more IgM than IgG. There are few T cells but prominent eosinophils and granulocytes, likely related to excess IL-17 (115). CD8 cells that produce Th1-like cytokines are elevated in optico-spinal multiple sclerosis (175). Aquaporin-4 is modulated off the astrocyte foot processes in neuromyelitis optica but not in multiple sclerosis.
Rituximab is therapeutic. Plasmapheresis may also reduce symptoms. Newer drugs that affect IL-6 and complement have been approved and are quite effective. Interferon therapy, however, causes worsening (117; 257), but because this disease is so active, some reported adverse interferon effects could be spurious. Serum interferon levels are high in neuromyelitis optica and the neuromyelitis optica /CNS Sjögren overlap syndrome, and they are low in multiple sclerosis, with an odds ratio of 35 (67). Interferon levels are also high in systemic lupus, where type I interferon also triggers exacerbations. It is dangerous and not logical to administer interferons in neuromyelitis optica and systemic lupus erythematosus.
• Eales disease, a syndrome of retinal perivasculitis and recurrent intraocular hemorrhages, is infrequently associated with neurologic abnormalities (7 of 17 patients).
• Encephalitis (anti-NMDA receptor encephalitis), in 3% of cases, has prominent MRI or clinical features of multiple sclerosis or neuromyelitis optica.
• Erdheim-Chester disease causes sclerosis and bone pain, diabetes insipidus, and histiocytic infiltration of the meninges, and T2 positive, enhancing lesions of cerebellum and brainstem. It is very sensitive to interferon-alpha therapy.
• Folate deficiency can cause encephalopathy and spastic paraparesis.
• Fragile X-associated tremor ataxia syndrome, from an FMR1 gene CGG repeat expansion, causes tremor, ataxia, and a sensory peripheral neuropathy. MRI has hyperintensity of the corpus callosum, splenium, and the middle cerebellar peduncle in two thirds.
• Gadolinium encephalopathy (MRI lesions).
• Genetic--see storage disease plus other genetic disorders.
• Gerstmann-Sträussler-Scheinker syndrome is a prion disease that can cause ataxia, MRI, and CSF findings similar to multiple sclerosis.
• Guillain-Barré syndrome, Miller Fisher variant (below).
• Hemophagocytic lymphohistiocytosis (associated with Epstein-Barr virus, a perforin mutation, infections, malignancies, and rheumatologic disorders). Loss of perforin function prevents termination of immune response and inflammation of brain and peripheral nerves. Symptoms include fever, seizures, meningismus, and motor symptoms.
• Hepatic encephalopathy, with symmetric high MRI T1 signal in the globi pallidi, likely from manganese deposition.
• Hereditary diffuse leukoencephalopathy with axonal spheroids is from mutations in the CSF1R gene, causing depression, progressive loss of memory and executive function, seizures, tremor, bradykinesia, apraxia, rigidity, and gait dysfunction. MRI shows confluent frontoparietal periventricular T2 lesions that spare U fibers and cortex, plus corpus callosum lesions. Possibly related to POLD.
• Hereditary spastic paraplegia (versus primary progressive multiple sclerosis).
• Hypothyroidism--can cause constipation and fatigue even though the motivation to act remains. In multiple sclerosis, abulia is sometimes associated with fatigue.
• IgG4-related disease cause progressive spasticity and dementia, plus cortical and subcortical MRI lesions.
• Increased intracranial pressure and normal pressure hydrocephalus can cause visual and long tract findings.
• Infection. Tuberculosis, tuberculomas, fungus such as Cryptococcus, syphilis, Lyme disease (Borrelia burgdorferi), chlamydia, mycoplasma, toxoplasmosis (in 80% of French, 30% world-wide), brucellosis (abscesses, occasionally intramedullary, can involve spinal cord), familial Mediterranean fever, Listeria monocytogenes (cervical cord lesions), CNS Whipple disease, with brain and cervical MRI lesions, toxocariasis, purulent leptomeningitis, or intraocular inflammation. Inflammation of the paranasal sinuses seldom causes optic nerve inflammation.
• Inflammatory bowel disease with brain lesions.
• Leber hereditary optic neuropathy is a hereditary, maternally transmitted mitochondrial disease (base pair 11778 of mitochondrial DNA, causing loss of function of the reduced form of nicotinamide adenine dinucleotide dehydrogenase 4). It causes bilateral, often sequential, painless visual loss with dilated, tortuous eye vasculature. In females, but not in males, it is sometimes indistinguishable from multiple sclerosis because of associated multiple sclerosis-like symptoms, destructive white mater lesions containing macrophages and CD8 T cells, widespread and periventricular white matter lesions on MRI, abnormal visual evoked potentials, and sometimes abnormal CSF (132). In men, diffuse white matter lesions are not present, but affected optic nerves show abnormalities on short-term inversion recovery MRI. The symptoms are often unilateral but eventually are bilateral. Visual loss is permanent and untreatable. Although neurons (third-order retinal neurons) are the main targets in Leber disease, a parallel mitochondrial defect theoretically also could make oligodendroglia more susceptible to damage in multiple sclerosis. Other ocular disorders include Eales disease and retrobulbar vasculopathy of Susac (261; 246).
• Leukoencephalopathy with vanishing white matter. In adults, this causes slow neurologic deterioration and white matter lesions. Autosomal recessive mutation in eukaryotic initiation factor 2B (eIF2b) increases unfolded proteins and cell death. It occurs rarely in adults, with progressive symptoms. White matter signal is diffusely increased on T2 and FLAIR and low on apparent diffusion coefficient MRI.
• Lyme disease is occasionally associated with unilateral or bilateral optic neuritis or ischemic optic neuropathy (in addition to retinal vasculitis), internuclear ophthalmoplegia, deafness, and multiple other neurologic signs.
• Lymphoma can form white matter MRI lesions. Tumor-filled centrum ovale venous streaks can occur on MRI. Intravascular large B cell lymphoma, angioendotheliomatosis proliferans sytematica is a non-Hodgkin variant that infiltrates CNS and skin.
• Maculopathy or macular degeneration.
• Macrophage activation syndromes can be confused with multiple sclerosis in very young patients and include familial hemolymphophagocytic lymphohistiocytosis, Chediak-Higashi disease, Griscelli disease, and Purtillo syndrome. Crystal-storing histiocytosis can mimic multiple sclerosis in adults.
• The Marburg variant of multiple sclerosis has axonal loss and severe widespread diffuse or multifocal demyelination. Lesions can be of different ages and contain massive macrophage and CD8 cell infiltrates; in one case, inflammation preceded the demyelination. Several cases showed active vasculitis including polymorphonuclear cells, suggesting overlap with acute hemorrhagic leukoencephalitis. There may also have been cases of neuromyelitis optica spectrum disorder in the older literature. Death is within a year. Death in 1 month is frequent, often from brainstem or upper cord lesions. Steroids and chemotherapy prolong life to 3 months; plasmapheresis may be helpful. Large tumefactive lesions, with much better prognosis, may be confused with the Marburg variant.
• Marchiafava-Bignami disease causes demyelination of the corpus callosum, usually the medial sector, sometimes with spread to periventricular white matter and internal capsules. There is a clinical pattern of hemispheric disconnection. Severe malnutrition and cheap red wine are classic precipitants.
• Metastasis from cancer.
• Metronidazole causes transient cerebellar dentate lesions.
• Migraine can cause visual disturbances, vertebrobasilar symptoms, and disseminated T2 lesions on MRI. In women, when migraines are frequent and associated with aura, MRIs show small cerebellar infarcts—the location is more lateral than the cerebellar peduncle lesions of multiple sclerosis. Occasionally deep white matter lesions are seen in the centrum semiovale, likely from associated hypertensive disease (135). One third of headache patients with MRI lesions actually fit the McDonald criteria for multiple sclerosis. Importantly, they do not have multiple sclerosis symptoms.
• Miller Fisher syndrome (subacute ataxia and ophthalmoplegia from Guillain-Barré syndrome) affects cranial nerves and eye movements. There are rare cases of this disorder and chronic inflammatory demyelinating polyneuropathy along with multiple sclerosis.
• Mitochondrial disorders. Genetic mitochondrial syndromes can cause strokes, diffuse periventricular T1 holes and T2 white matter lesions, including the spinal cord. Non-syndromic mitochondrial disorders can cause scattered white matter lesions on FLAIR MRI. Intrinsic mitochondrial defects could affect onset or course of multiple sclerosis.
• Myasthenia gravis can cause diplopia and muscle weakness.
• Myelin oligodendrocyte glycoprotein antibody disease (MOG, MOGAD). Previously falling under the umbrella of neuromyelitis optica spectrum disorder, myelin oligodendrocyte glycoprotein antibody disease is an independent clinical entity that is often is confused with multiple sclerosis (253). It is more common than multiple sclerosis prepubertally. Of the patients with neuromyelitis optica spectrum disorder who test negative for the aquaporin-4 antibody, approximately 40% have anti-MOG antibodies directed against myelin oligodendrocyte glycoprotein on the outermost myelin membranes throughout the CNS. The disease typically targets the optic nerves, with bilateral involvement in over 50%, often with papilledema, eye pain, and headache, and the spinal cord, in short or long segments. MRI lesions are often similar to multiple sclerosis lesions. CSF can have lymphocytic pleocytosis and normal or mildly elevated protein but rarely has oligoclonal bands (10%; those with oligoclonal bands are closer to multiple sclerosis). Serum antibodies to Epstein-Barr virus are less common than in multiple sclerosis. Antibodies are best detected with a cell-based assay in the serum. Myelin oligodendrocyte glycoprotein disorder may be monophasic, particularly in children, and is typically highly steroid responsive, with better recovery and less disability than either multiple sclerosis or neuromyelitis optica. Unrecognized past cases could have affected the epidemiology of optic neuritis.
• Myotonic dystrophy types 1 and 2 (brain white matter lesions on MRI).
• Neoplastic (lymphoma, intravascular lymphoma, primary or metastatic central nervous system tumor; can involve eyes also).
• Neuroretinitis. This is a form of papillitis with associated deposits of lipids and protein. These deposits radiate from the macula and form a stellate pattern at the macula or a half star between the macula and the disc. The "macular star" is formed as fluid from leaking disc capillaries accumulates within the Henle layer around the fovea. The macular star may take up to 2 weeks to form after the onset of papillitis. Symptoms are similar to those in typical optic neuritis, but neuroretinitis seldom progresses to multiple sclerosis, suggesting that another etiology has been confused with multiple sclerosis.
• Nutritional neuropathy includes Jamaican neuropathy and Cuban epidemic neuropathy.
• Optic nerve glioma ("benign" glioma of childhood–a pilocytic astrocytoma; malignant glioblastoma is more common in adults).
• Orbital pseudotumor with proptosis, pain, and ophthalmoplegia, but infrequent visual loss.
• Paraneoplastic: anti-CV2 antibodies for optic neuritis. Also possible are limbic encephalitis (often with slow cognitive processing speed), brainstem encephalitis, cerebellar degeneration, and lobar encephalopathy with large MRI lesions.
• Parasites can migrate into the CNS and cause focal symptoms and must be excluded in patients from endemic areas, eg, cysticercosis.
• Parkinson disease occurs at half the expected rate in multiple sclerosis. The 0.6 rate of Parkinson disease in systemic lupus erythematosus suggests it will also be less common in neuromyelitis optica because of overlaps between neuromyelitis optica and connective tissue disease.
• Pelizaeus-Mertbacher disease (brain MRI lesions and progressive course), including proteolipid protein-1 (PLP-1) mutation.
• Pigmented orthochromatic leukodystrophy mutations (POLG-1 = mitochondrial DNA polymerase gamma; the CSF1 receptor is also implicated) can cause progressive CNS signs in childhood or late teens, psychiatric symptoms, and bilateral periventricular lesions on MRI similar to leukoariosis. The CSF-1R mutation also causes hereditary diffuse leukoencephalopathy with axonal spheroids.
• Polyglucosan body disease in adults begins at approximately 50 years and causes neurogenic bladder, spastic paraplegia with vibration loss, and a peripheral axonal neuropathy (167). A glycogen-branching enzyme mutation causes accumulation of polyglucosan bodies throughout the nervous system and cerebral myelin loss. T2 and FLAIR MRI shows symmetrical white matter lesions in periventricular areas, posterior internal capsule, external capsule and medial lemniscus of pons with atrophy of medulla and spinal cord. There is a mutation in glycogen branching enzyme.
• Porphyria. Hereditary coproporphyria can cause progressive CNS and PNS symptoms.
• Posterior reversible leukoencephalopathy. Increased T2 MRI signal in white more than gray matter. Triggers include eclampsia, acute renal failure, hypertensive encephalopathy, and immunosuppressive drugs such as cyclosporin (high dose) and methotrexate.
• Progressive multifocal leukoencephalopathy. Progressive symptoms such as cognitive loss, occipital visual loss, and hemiparesis. MRI lesions are usually large and rarely enhance. Optic neuritis or cord lesions are very likely.
• Progressive necrotizing myelopathy provoked by mycoplasma pneumoniae, m Tb. In rats with experimental allergic encephalomyelitis, it is provoked by tilorone, an IFN-alpha/beta inducer.
• Pseudotumor cerebri (visual loss).
• Pseudoxanthoma elasticum (brain MRI lesions and vascular disease).
• Psoriasis frequency may be double the expected rate in multiple sclerosis based on several clinic series.
• Psychogenic. Multiple sclerosis symptoms are often confusing to patient and doctor because they are generated by aberrant CNS conduction and can be elaborated or misreported. Actual psychogenic symptoms can easily be confused as multiple sclerosis but can also be difficult to challenge when the patient is invested in the diagnosis of multiple sclerosis. “Pseudo-multiple sclerosis” or “therapeutic mislabeling” should not be supported by the neurologist (21).
• Radiation necrosis or radiation-induced optic neuropathy (not necessarily neuritis). The threshold for radiation-induced damage may be much lower when there is prior optic neuritis or tumefactive multiple sclerosis, possibly ameliorated with corticosteroids and hyperbaric oxygen.
• Raeder paratrigeminal syndrome is unilateral facial pain in the V1 and V2 branches of the trigeminal nerve; it is associated with ptosis and miosis from a parasellar mass and must be differentiated from trigeminal neuralgia due to multiple sclerosis.
• Sarcoidosis can involve single or multiple cranial and peripheral nerves, the brainstem (base of the brain is the most common site), hypothalamus, spinal cord—sometimes with longitudinal lesions, and meninges. MRI lesions are nodular “string of pearls” in the spinal cord and can persist for over 3 months; 20% have CSF oligoclonal bands.
• Schilder disease (diffuse sclerosis) causes large hemispheral demyelinating lesions.
• Sjögren syndrome. When associated with central and peripheral nervous system lesions, classic Sjögren symptoms (sicca and rheumatic) are less common than in primary Sjögren disease. Serum antibodies to Sjögren syndrome A and B proteins (SSA and SSB) are positive in only one third of patients and are more often negative than in Sjögren disease without neurologic symptoms. A lip or parotid biopsy is needed to clinch the diagnosis (05; 225; 117).
In CNS Sjögren syndrome, one third have MRI, CSF, or evoked potential evidence of cerebral abnormalities; one third have longitudinal spinal cord lesions; the rest have optic neuritis and diffuse symptoms such as seizures, cognitive loss, and encephalopathy (54). There are small white matter MRI lesions in two thirds (occasionally in basal ganglia, infrequently in corpus callosum), oligoclonal bands in one third, and abnormal visual evoked potentials in about two thirds (51). PNS Sjögren disease can cause sensory ganglionitis, painful sensory neuropathy, and distal sensory motor axonopathy.
Adil Javed has described a new Sjögren-related entity, seen predominantly in young black women (116). Patients with severe destruction from optic neuritis and longitudinal cervical cord lesions resemble patients with Devic disease, but NMO-IgG levels are positive in only 40%. However, minor salivary gland biopsy is positive for Sjögren disease: inflammation grade 4+/4 in 85%, and 2+ in 100%, often when SSA and SSB serology is negative. Other autoimmune diseases (myasthenia gravis, primary biliary cirrhosis) are often associated. Treatment differs from multiple sclerosis therapy, as interferons may cause worsening. Mycophenolate mofetil provides some benefit, but the best responses are with rituximab (Javed personal communication 2007).
• Storage disorders and other genetic diseases versus childhood multiple sclerosis. Leukodystrophies are usually confluent and bilateral on MRI. Juvenile metachromatic leukodystrophy and late onset Tay-Sachs disease have MRI signatures that could be confused with multiple sclerosis. Also to be considered are Alexander disease (frontal, cerebellar, brainstem, and spinal cord T2 MRI lesions), childhood ataxia with cerebral hypomyelination (eIF2b mutation), late-onset Canavan disease (mutations of aspartoacylase gene, restricted to oligodendrocytes, with accumulation of the substrate molecule N-acetyl-aspartate), Fabry disease (periventricular lesions, but non-multiple sclerosis clinical symptoms), globoid cell leukodystrophy/Krabbe disease, hereditary diffuse leukoencephalopathy with axonal spheroids (see above), Pelizaeus-Merzbacher disease (PLP mutation and dysmyelination; “jimpy” mouse is model). In adults, consider polyglucosan body disease (autosomal recessive leukodystrophy; glycogen branching enzyme deficiency), Refsum disease (ataxia and MRI lesions), tuberous sclerosis, and Wilson disease.
• Subacute myelo-optic neuropathy from halogenated hydroxyquinolines, including Entero-Vioform, diodoquin, and clioquinol.
• Subacute sclerosing panencephalitis--T2 MRI lesions in periventricular and subcortical white matter, but this progressive disorder follows measles infection, with high titers of anti-measles antibodies.
• Susac syndrome. Retrobulbar vasculopathy of Susac causes encephalopathy, branch (distal) retinal artery occlusions, and hearing loss (261). It affects 20- to 40-year-old women and is associated with headaches, hearing loss, tinnitus, pseudobulbar speech, and encephalopathy. There are microangiopathic infarcts in gray and white matter, and bilateral branch artery occlusions in the retina. MRI shows many multifocal white matter lesions of the central corpus callosum, plus lesions in deep gray, posterior fossa, brain parenchyma, and occasionally the leptomeninges. Acute large “snowballs” and multiple older small “punched-out” areas riddle the central corpus callosum (246). Lesions are less likely to have a central vein than in multiple sclerosis. Intravenous immunoglobulin and corticosteroids sometimes improve hearing and MRI. Natalizumab blocks binding of pathogenic cytolytic T cells to endothelial cells and prevents recurrences.
• Syphilis also has huge variety in its presentation.
• Thyroid ophthalmopathy can cause diplopia.
• Tobacco-alcohol amblyopia.
• Tolosa-Hunt syndrome. Painful ophthalmoplegia with subacute boring eye pain, palsy of extraocular muscles, V1 sensory loss, sympathetic denervation of pupils, and rapid response to 100 mg prednisone.
• Trauma; direct or after anterofrontal deceleration.
• Tuberculomas in the brain parenchyma.
• Tuberous sclerosis can cause subcortical tubers that appear in white matter on MRI.
• Tumor necrosis factor receptor-1-associated periodic syndrome (familial Hibernian fever) is from a mutation in the p55 receptor for TNF. It occasionally has onset and MRI features similar to multiple sclerosis (138). It does not respond to multiple sclerosis therapies but improves with anti-TNF therapy.
• Vaccination (polio and possibly influenza). The associations reported in a few papers are likely spurious, as the vast majority of studies find no link. Some find a 3-fold increase in the incidence of multiple sclerosis after vaccination with recombinant hepatitis B vaccine but not with vaccines against other viruses (106), yet others report no increase. Confusion clouds this issue from statistical and reporting problems--combining data on recombinant and nonrecombinant vaccines, written versus computer records, and date of onset versus date of diagnosis. Most experts urge caution with live virus vaccines (measles, mumps, rubella, varicella/zoster, and yellow fever vaccines).
• Vascular disease lesions (small vessel disease) are usually small, spherical and located in the centrum semiovale instead of “fingers” radiating outward from the corpus callosum. Cord, corpus callosum, and juxtacortical white matter are usually spared. Vascular lacunes are common in the basal ganglia and central pons. Some lesions in hypertensive elderly patients are periventricular and are quite similar to multiple sclerosis lesions. Vascular lesions with aging tend to be smaller and random but sometimes symmetrically involve the periventricular white matter in confluent posterior ischemic damage (10). Vascular malformations and cavernous hemangiomas show persistent Gd enhancement. Moyamoya syndrome has paroxysmal symptoms and can be confused with multiple sclerosis on MRI.
• Vasculitis (temporal arteritis, angiitis, cranial arteritis, Churg-Strauss syndrome, eosinophilic vasculitis). Isolated angiitis can occur in children.
• Virus infections or viral encephalitis can mimic multiple sclerosis clinically and on MRI. Reports include EBV, chickenpox, COVID-19, cytomegalovirus, hepatitis A and B, HHV-6 encephalomyelitis, HHV-7, VZV, herpes zoster vasculopathy, acute HIV infection, HTLV-I (also associated with Sjögren syndrome), infectious mononucleosis, Japanese encephalitis (a flavivirus with bilateral thalamic lesions and polio-like flaccid paralysis), measles, mumps, rubella, post-measles autoimmunity and subacute sclerosing panencephalitis, mumps, rubella, poliomyelitis (central cord lesions on MRI), West Nile virus (flavivirus) with a polio-like presentation, and Zika virus with uveitis and CNS lesions. Note, HIV patients, usually on highly active retrovirus therapy, have a reduced risk of multiple sclerosis.
Japanese macaque encephalitis causes multifocal inflammatory demyelinating plaques of varying ages. It is caused by a gamma-2 herpesvirus with 50% homology to human Kaposi sarcoma-associated virus.
• Vitamin B8 (biotin) deficiency from biotinidase defect causes a neuromyelitis optica–like syndrome with optic neuritis and longitudinal cord lesions.
• Vitamin B12 deficiency causes subacute combined degeneration with centrocecal scotomata, optic atrophy, MRI lesions around the corpus callosum (not Dawson fingers), partially reversible leukoencephalopathy, and long tract signs from cord degeneration. Methionine synthase deficiency is a rare cause of similar symptoms with symmetric periventricular leukoencephalopathy.
• Vitamin E deficiency causes ataxia, myelopathy, and neuropathy.
Multiple sclerosis is traditionally described as clinical symptoms or signs of two CNS lesions separated in time and space that are not caused by another CNS disease. These lesions are detected with a history and neurologic examination--the sine qua non of a diagnosis of multiple sclerosis. MRI and CSF analysis strengthen the diagnosis (195; 163) and are each abnormal in more than 95% of definite multiple sclerosis cases. MRI is essential to rule out mimics of multiple sclerosis. A standardized brain and cord imaging and reporting protocol from the (Consortium of Multiple Sclerosis Centers) (CSMC) enhances diagnosis, clinical trial assessment, and monitoring of disease activity and damage. CSF shows ongoing inflammation and oligoclonal bands, and it can exclude alternate causes.
Criteria that define “multiple sclerosis” have been revised several times and, despite evolving diagnostic criteria, allow for earlier diagnosis at the time of the first attack. It is still important to note that there is temporal variation in the duration of enhancement, plus some regional differences that interfere with comparison of Gd+ and Gd- lesions. For instance, Gd+ MRI lesions in white matter are easier to see than lesions in the cortical gray matter. Similarly, Gd enhancement lasts 2 weeks (median) to 3 weeks (mean), but there is temporal variation in the duration of enhancement. Gd+ ring-enhancing lesions with central pallor correlate with disease severity and are larger and last longer than homogenously enhancing lesions. With this temporal variation, there could be a tail of enhancing lesions several weeks after a clinically isolated syndrome or ADEM, so “old” and “new” lesions could still be from the initial insult.
Widened inclusion criteria for a diagnosis of multiple sclerosis after a clinically isolated demyelinating syndrome lead to spurious improvement in prognosis, the “Will Rogers phenomenon” (239). The new criteria also affect clinical trials. Patients with milder disease have been allowed into recent studies, necessitating larger patient cohorts to see any drug benefit. Drugs that benefit MRI more than clinical activity will seem to be more effective. Furthermore, drug efficacy can be measured by induction of a “disease activity free” state that can be achieved in a subset of patients, but not all. This state is much more dependent on MRI than clinical activity and is more stringent when scans are frequent, with triple-dose gadolinium, and on high-Tesla MRI magnets. Importantly, drug effects on MRI do not necessarily correlate with clinical relapses or with long-term prognosis (08).
MRI in diagnosis. On monthly scans, 90% of untreated relapsing-remitting patients will have MRI activity over 9 months (150). Early, active MRI lesions are typically hyperintense on T2 with a hypointense ring, possibly containing activated macrophages. Large, fluffy T2 lesions often have preserved axons and repletion of oligodendroglia. Some acute plaques enhance with gadolinium, but early on, they can be almost isointense on T2 MRI (27). Late active lesions are less hyperintense on T2 and sometimes hypointense on T1-weighted images (“black holes”). Inactive lesions (demyelinated or myelinating) are hypointense on T2 scans and normal or hypointense on T1 scans. Black holes are not uniform. They range from slightly hypointense (“gray holes” would be more apt) to CSF-like hypointense (black), indicating a spectrum of axonal loss and demyelination. Variation in blackness is not usually analyzed.
Certain MRI features are typical of multiple sclerosis, such as multiple ovoid-shaped bright lesions on T2-weighted MRI, abrupt loss of T2 signal as a lesion approaches the gray matter (“open ring” sparing U-fibers), and periventricular lesions, often radiating up from the corpus callosum or out from the ventricle (called “Dawson fingers”), especially near the body and posterior horn of the lateral ventricle.
Other typical features are a lesion greater than 5 mm; lesions in the corpus callosum, brainstem, temporal horn, or cortical gray matter; and lesions below the tentorium, especially in the cerebellar peduncle or hemisphere (176). Corpus callosum lesions can also be caused by vascular disease, CADASIL, Susac syndrome, tumor, Marchiafava-Bignami disease, echovirus 9, and adrenoleukodystrophy.
Twenty percent of Gd+ lesions are reoccurrences at prior sites; these are larger and have lower magnetization transfer values than new lesions. T2 volume, MRI “burden of disease,” increases by 5% to 10% per year. A leak through the tight junctions of the blood-brain barrier is usually invoked as the cause of Gd enhancement. It is likely that T cells constantly activate endothelial cells. Activated, enlarged endothelial cells can pinocytose gadolinium and gadolinium-protein complexes and could cause a capillary blush on MRI (26; 36). Treatment with glucocorticoids blocks MRI enhancement, probably through a direct effect on endothelial cells. IFN-beta and antibodies to VLA-4 also block gadolinium enhancement, interrupting T cell-endothelial interaction. Rare patients can develop nephrogenic systemic fibrosis from certain preparations of gadolinium. Nephrogenic systemic fibrosis is more common with low glomerular filtration rate and diabetes, and it can be diagnosed with a skin biopsy.
Perivenular spaces are prominent due to inflammation (lymphocyte cuffing) (84). A “sand-like appearance” of the high convexity white matter is from dilated Virchow-Robin spaces (03). These dilated spaces correlate with Gd+ lesions (268), suggesting diffuse activation of multiple areas of the brain.
Spinal cord lesions are usually only one to two segments long, often multiple, and predominantly cervical (66%) (22). They are diffuse in only 13% and are most likely in chronic multiple sclerosis. Almost all are peripherally located, not in the central cord, perhaps related to inflammation near pial veins. In recently diagnosed multiple sclerosis, cord lesions help define dissemination in space (85% are positive with brain plus cord MRI versus 66% using brain MRI alone). The cord is often a target primary progressive multiple sclerosis, but over 90% of these patients also have brain lesions.
Normal-appearing white matter evinces slight abnormalities on MRI and MR spectroscopy and also more cerebral perfusion several months before overt MRI lesions appear (69). MR spectroscopy (MRS) shows biochemically abnormal lipid peaks in gray matter. Scans with 7 or 8 Tesla magnets reveal multiple lesions in gray matter and many in subpial and perivenous locations. PET scans show decreased cerebral glucose utilization in frontal and parietal cortex, and this correlates with low NAA/Cr ratios on magnetic resonance spectroscopy and low oxygen use on fMRI. Resting-state networks also have decreased functional connectivity on fMRI, affecting vision, working memory, and sensorimotor function. These measures decline over time in multiple sclerosis and suggest hypometabolism and mitochondrial respiratory dysfunction.
Magnetization transfer (MTR) values in normal-appearing white matter are lowest in chronic progressive multiple sclerosis and continue to decline. MTR values are slightly low in benign multiple sclerosis but do not decrease over time (69). Gray matter MTR loss correlates with progression on the EDSS and long-term disability. Of patients with clinically or lab-supported multiple sclerosis, 1.5% have negative conventional MRI scans yet low magnetization transfer values in pons, cerebellum, and periventricular areas (69). Changes are fastest in the corpus callosum. The mechanism for abnormal MTR may involve different immune or neurotrophic responses than in MRI-positive multiple sclerosis. T2+ plus T1+ MTR lesions have more demyelination and more axonal swelling and are often chronic inactive compared to T2+ only lesions (71). Gd+ lesions show some recovery on MTR. Diffusion tensor white matter tractography shows damage to the cingulum (executive function, attention, and speed) and U-fibers (attention, verbal and visuospatial speed, and memory) that correlate with cognitive impairment (166).
Lesions approach but stop before arcuate U fibers below gyri on T2. These arcs form an “open ring” and are a strong indicator of demyelinating disease. Antibody and complement in plaques (Lucchinetti type II) causes a T1 Gd-enhancing ring and hypointense T2 ring (130). Restricted apparent diffusion coefficient (ADC) on MRI is more common in demyelinating disease than in tumors or abscesses.
Brain atrophy is present in early multiple sclerosis and averages 0.9% per year, versus 0.1% to 0.2% in healthy controls. It evolves five times more rapidly than in normal brains, and up to 14 times faster during active secondary progressive multiple sclerosis. Atrophy is largely from axonal loss but also from demyelination and contraction of the neuropil. Axonal loss and T2 lesion volume are only partially correlated. White mater loss is best seen as thinning of the corpus callosum. Gray matter volume loss appears at first presentation in the thalamus and putamen, caudate, cerebellum, and cortex. White and gray matter atrophy correlate with cognitive decline and can be measured with third ventricular volume, a reflection of thalamic loss. CNS atrophy can also be measured with transcranial sonography of the third ventricle (width versus disability is inversely correlated at r = -0.6). Cerebellar gray atrophy correlates with loss of locomotion. Brain atrophy correlates better with changes in disability than T2 MRI lesions do and is most predictive in the cervical cord (274). Cervical cord atrophy is pronounced in primary and secondary progressive multiple sclerosis; C1 cross-sectional atrophy predicts, and thoracic gray matter atrophy correlates with, progression. Eventual atrophy is predicted by the presence of early T2 hypointensity and T1 Gd+ lesions (231), T1 “black holes,” and low brain volume in some studies. T2 hypointensity is likely from iron deposition (perhaps neurotoxic) and correlates with later brain atrophy. IFN-beta and glatiramer therapy do slow atrophy. Atrophy is slower with weekly IFN-beta than with high-dose, high-frequency interferon, possibly because the latter reduces inflammation (“pseudoatrophy”) or because there are differences in neurotrophin induction.
T2 lesion volume has a weak correlation with disability (r = 0.2 to 0.3). T1 holes correlate more strongly, especially in secondary progressive multiple sclerosis (approximately 0.8). Without treatment, 56% of acute black holes will remain as permanent black holes. Callosal atrophy is a poor prognostic sign. Twenty-six percent of relapses appear without new or enhancing lesions on MRI (83). These are more likely with highly effective therapies, with long-duration disease, and with fatigue as part of the relapse and cause EDSS worsening of 0.45, versus 0.72 when lesions are Gd+.
Advanced MRI techniques aid in measuring CNS lesions. Three-dimensional FLAIR increases sensitivity and contrast. Shifts in magnetic resonance frequency define tissue architecture in new lesions. MRI texture analysis quantitates changes in local image intensity. Double inversion recovery reveals intracortical lesions not seen on FLAIR. 7-Tesla imaging shows the expanding inflammation as a hypointense rim and improves detection of cortical lesions. Injection of albumin-binding gadolinium with a 4-hour delay increases signal and decreases noise. [11C]PK11195 binding to a mitochondrial translator protein on PET scans correlates with disability, likely from microglial activation. Important concepts in MRI diagnosis are a visible central vein within a lesion (immune cells egress here; not seen in migraine and small vessel disease), subcortical lesions that spare the U-fibers, cortical lesions seen with 3D MRI sequences, slowly expanding lesions (in 92% of early disease), and lesions with paramagnetic rims (56% in early disease) due to iron-laden macrophages. A large choroid plexus plus paramagnetic rims is linked to poor cognition and more fatigue.
CSF in diagnosis. The CSF is the best non-MRI marker of multiple sclerosis. Analysis of CSF is helpful when patients have atypical clinical symptoms or MRI appearance, or early or late onset of disease. CSF reflects CNS inflammation and detects blood-brain barrier leaks (77). The CSF, in order of increasing frequency and importance for a diagnosis of multiple sclerosis, shows elevated protein, a moderate increase in white blood cells, increased IgG, IgG/albumin index, IgG synthesis rate (Tourtellotte or Link formulas), and oligoclonal bands or Ig kappa free light chains. Roughly equivalent as markers of multiple sclerosis, oligoclonal bands require matched serum to determine if the bands are unique to CSF; kappa free light chains do not, and are, thus, easier to use in clinic. IgA and IgM oligoclonal bands can provide additional information. Intrathecal IgM may be a high-risk marker for conversion from clinically isolated syndrome to multiple sclerosis (114).
Bands are present in 95% of multiple sclerosis patients but only 80% of primary progressive patients (233). The index, synthesis rate, and oligoclonal bands are increased in black compared to white patients by 30% to 40%, suggesting more active inflammation (211). The IgG index correlates with CSF IL-10 and IL6 levels. A positive MRI and CSF oligoclonal bands in clinically isolated syndromes predict conversion to definite multiple sclerosis in 90% of patients, at nine times the rate with negative tests, perhaps more predictive than dissemination in space on MRI.
When suspected cases of multiple sclerosis have no CSF IgG bands, a repeat study will show new bands in half of them (251). With only one CSF band, one in three later develops more oligoclonal bands, but one in 27 has cerebral lymphoma (49). Patients with diagnosed multiple sclerosis who have negative oligoclonal bands are less likely to develop more bands over time (233). Serum bands are increased in 44% of multiple sclerosis patients and are 10 times more common in women than in men (251). Inflammatory conditions such as SSPE, Lyme disease, neurosarcoid, CNS infections, Behçet disease, lupus, and adrenoleukodystrophy also often have unique CSF bands.
Intrathecal synthesis of antibodies to measles in multiple sclerosis was described in 1962. Many other viruses, such as HHV-6, are also targets of a polyspecific B cell response. An index of CSF antibodies to measles, rubella, and herpes zoster (the MRZ reaction) improves sensitivity (66) and is low or absent in neuromyelitis optica. Antibodies to galactocerebroside, triose-phosphate isomerase, and glyceraldehyde-3-phosphate are elevated. These antibodies inhibit glycolytic enzyme activity and could disrupt neuroaxonal function. Reports of CSF antibodies to myelin oligodendrocyte glycoprotein and MBP have not been replicated, perhaps because of assay difficulty from conformational changes in the protein.
The CSF cell count is often slightly elevated at 5 to 10 per cubic um. Eighty percent of the white blood cells are T cells in stable multiple sclerosis and in healthy controls; T cells rise to 90% in active multiple sclerosis (205). CSF white blood cells are 90% CD3+ T cells (70% CD4, 20% CD8), 3% natural killer, 4% macrophages, and 5% B cells (35). The CD4/CD8 ratio reflects the blood (2/1) in relapsing-remitting multiple sclerosis, but CSF CD8 cells fall in progressive disease. Many CSF T and B lymphocytes are activated blasts (174). B cells are at lower levels than in blood. A high CSF B cell to monocyte ratio in CSF correlates with IgG levels and with rapid disease progression in relapsing and progressive multiple sclerosis.
Potential biomarkers in CSF, reflecting damage to brain cells and inflammation, include the following: proteins from axon cytoskeleton (neurofilament light chains, present in first multiple sclerosis attacks and peaking 8 weeks after an enhancing MRI lesion and higher with MRI T1 black holes, but increasing by 3% per year with age in healthy controls; neurofilament heavy chains, highest in secondary progressive multiple sclerosis, but present even in clinically isolated syndromes; actin; NAA; tau; and tubulin), neurons and axons (fetuin-A, NOGO receptor, parvalbumin), astrocytes (GFAP), membranes (24S-hydroxycholesterol, apoE4, NCAM-1), glia (GFAP—higher in nonrelapsing progressive disease/PIRA, S-100b), endothelial cells (endothelin, e-Selectin, PECAM-1, VCAM-1), immune cells (CD138, chemokines, chitinase, complement, cytokines, GM1 allotype of immunoglobulin, ICAM-1, MMP), and amyloid precursor protein (247). microRNAs, miR 155, miR 338 and miR 491, which regulate neurosteroid production in the brain are elevated in progressive multiple sclerosis brains; also miR922, miR181c, and miR633 are elevated in CSF and help differentiate between forms of multiple sclerosis. Neurofilament light chains currently have the most evidence to support their use as a biomarker, especially in studies of large groups.
N-acetyl aspartate (NAA) is abundant enough in neurons (10 mM) to be detectable with magnetic resonance spectroscopy. NAA decreases in CSF during secondary progressive multiple sclerosis as neurons die or lose function. Tau protein, a marker of axonal damage, increases 2-fold in progressive multiple sclerosis but also increases in other inflammatory diseases, and levels correlate with the IgG index. 14-3-3, a neuronal, axonal, and glial protein, is present in 10% of patients with transverse myelitis and multiple sclerosis (52). In clinically isolated syndromes, 14-3-3 predicts an earlier conversion to multiple sclerosis. It is also elevated in various dementias after extensive damage of the brain, especially in Creutzfeldt-Jacob disease where levels are high. Reduced ATP metabolites, perhaps reflecting high energy demand, correlate with more severe multiple sclerosis progression. Cystatin C may be uniquely cleaved by endogenous CSF proteases. A high level of myelin basic protein (MBP) in CSF and MBP-like material in the urine reflects damage to myelin and oligodendroglia in progressive multiple sclerosis (263) and correlates with the number of MRI T1 black holes. S-100b protein increases during flares. None of these is diagnostic in itself, but multiplex analysis coupled with reliable assays may be used in the future.
CSF neurotrophic factors also rise during different phases of multiple sclerosis. Neural cell adhesion molecule (N-CAM) and ciliary neurotrophic factor (CNTF) increase in serum during recovery from exacerbations. Nerve growth factor (NGF) sometimes increases during exacerbations, although levels fall as the disease becomes advanced. DJ-1 (PARK7) in astrocytes and neurons stabilizes neuroprotective Nrf2. DJ-1 is elevated 6-fold in relapsing-remitting Japanese multiple sclerosis versus non-inflammatory disease and correlates well with the multiple sclerosis severity scale (MSSS; r = 0.509) (108). Other growth factors are reduced. Growth hormone, which induces insulin-like growth factor-1, is neuroprotective and remyelinating and is at low levels in the CSF.
Evoked potentials in diagnosis. Evoked potentials are occasionally needed to confirm multiple sclerosis (eg, when MRI and CSF are normal), but they should not be used for the routine diagnosis of multiple sclerosis. The frequency of abnormal evoked potentials in definite multiple sclerosis is visual=90%, auditory=80%, and somatosensory=70%. Visual evoked potentials become less variable 4 weeks after the onset of optic neuritis and are now quite standardized compared to the pre-MRI era. Visual evoked potentials latency reflects demyelination and has less spontaneous recovery than other visual measures such as fields and acuity. Auditory evoked potentials are seldom helpful in making the diagnosis. Neurophysiological studies such as vestibular evoked myogenic potentials, multifocal visual evoked potentials, motor (magnetic) evoked potentials, and the P300 event-related potential could also provide information about central nervous system function and prognosis (149).
Serum tests in diagnosis. Erythrocyte sedimentation rate or C-reactive protein, antinuclear and anticardiolipin antibodies (44), Sjögren syndrome A and B antibodies, angiotensin converting enzyme, vitamin B12 and methylmalonate, and vitamin D levels should be ordered when appropriate. Antinuclear antibodies are present at low levels in over 20% of patients. Because vitamin D is low in many multiple sclerosis patients and because low levels are linked to more exacerbations and faster progression, correction of deficits is likely to have clinical benefit. CRP, a marker for inflammation from many etiologies, has a modest correlation with relapses, progression, and MRI activity.
DNA transcription from many genes is controlled by methylation. Circulating methylated DNA profiles are highly abnormal in multiple sclerosis plasma (151).
Ophthalmologic diagnosis. The optic neuritis workup is funduscopy, visual acuity, perhaps optical coherence tomography (below), a neurologic examination, MRI, and possibly lumbar puncture. Perimetry shows a scotoma that is typically central or diffuse but sometimes is peripheral. Low-contrast Sloan letter charts are more sensitive than the standard Snellen measure of visual acuity. A loss of one line of low-contrast acuity correlates with a 3 mm2 increase in MRI T2 lesion volume; one line of high-contrast loss correlates with a 6 mm2 increase in MRI T2 lesions (267). Loss of acuity can also reflect post-geniculate white matter damage.
Visual evoked potentials are often abnormal in the affected eye but return to normal within 2 years in one third of eyes. Visual evoked potentials are more sensitive than optical coherence tomography (170). Multiple sclerosis patients with normal visual acuity but no history of optic neuritis will have subclinical optic tract lesions detected with visual evoked potentials (82%), contrast sensitivity (73%), OCT (60%), pupillary light reflex (52%), flight of colors (36%), and color vision (Ishihara plates) (32%) (255; 170). Low contrast visual evoked potentials stimulation (just as with visual acuity tests) and multifocal visual evoked potentials (which correlate with retinal nerve fiber layer thickness) are more sensitive than conventional tests. Electroretinograms measure macular function. The optic nerve head component of the ERG measures the transition from membrane to saltatory conduction at the optic nerve head, and it is abnormal in multiple sclerosis.
Retinal tomographs (OCT) reproducibly measure retinal nerve fiber layer thickness, retinal ganglion cells, and macular volume. One third of macular volume is from neurons, and macular edema correlates with visual function. OCT can define acute optic neuritis, demonstrate a second lesion in clinically isolated syndromes, and monitor atrophy and progression as a surrogate marker to complement the neurologic examination. OCT usually shows retinal nerve fiber layer thinning 3 to 6 months after optic neuritis. OCT is abnormal in half of the “normal” fellow eyes in multiple sclerosis patients after an episode of optic neuritis. Thinning of retinal nerve fiber layer (RNFL) is faster in optic neuritis eye (69 µm) than in “normal fellow” eye (partially affected, at 95 µm) and healthy control eyes (103 µm) (254). RNFL thickness is affected most in the temporal quadrant. Retinal nerve fiber layer loss occurs over time in some patients, even without symptoms of optic neuritis. In some studies, retinal nerve fiber layer and total macular volume is lower in progressive multiple sclerosis than in relapsing-remitting multiple sclerosis. The decline is faster in patients with more active disease. Loss is even worse in neuromyelitis optica, where it is more diffuse than in multiple sclerosis and affects the superior and inferior quadrants. However, change over time in multiple sclerosis may be too slow to use OCT as a measure in trials in relapsing/remitting multiple sclerosis.
Thickness of the ganglion cell layer (GCL), in contrast, does not correlate with a history of optic neuritis. However, GCL thickness is reduced in multiple sclerosis patients and correlates with brain gray matter atrophy on MRI.
Optic nerve cross-sectional area on MRI correlates with retinal nerve fiber layer thickness (r = 0.66). OCT measures do correlate with visual evoked potential amplitude but not with latency. Retinal nerve fiber layer thickness atrophy is associated with motor disability (r = 0.2-0.4) and cognitive problems (r = 0.5) (252). Peripapillary retinal nerve fiber layer thickness, and also a composite of [ganglion cells + inner plexiform layers], correlate with cortical gray and caudate atrophy (223). However, correlations of RNFL with atrophy of the visual cortex that suggested trans-synaptic degeneration were not compared to atrophy of other cortical areas. Inner nuclear layer thickness correlates with FLAIR lesion volume. With current technology, however, OCT is not a replacement for visual evoked potentials and MRI, especially in clinically isolated syndromes, because it is less sensitive.
MRI studies show multiple cerebral white matter lesions in one fourth to three fourths of optic neuritis patients, depending on the series; most of the MRI lesions involve the visual radiations.
The spinal fluid in idiopathic optic neuritis contains elevated protein, mild lymphocytosis, elevated IgG index, and oligoclonal bands (50% to 70% vs. 95% in multiple sclerosis).
Quality of life scales in diagnosis. Quality of life scales are inexpensive, simple to administer, and measure a wide range of the problems seen in multiple sclerosis (34; 33). They predict changes in disability and are objective measures of therapeutic outcomes. However, present scales are insensitive and have not contributed to trial monitoring.
Confusion in diagnosis and misdiagnosis. In some patients with clinically definite multiple sclerosis but negative MRI, other techniques such as evoked potentials or magnetic transfer imaging will show damage. When CSF has no oligoclonal bands, as in approximately 3% of patients, prognosis is better and brain MRI lesions are fewer. Four years after a diagnosis of multiple sclerosis, only half of these oligoclonal band-negative patients become positive (272). Similarly, one third of patients with one oligoclonal band will eventually develop multiple oligoclonal bands on follow-up (49).
Incidental, unexpected multiple sclerosis-like lesions on an MRI scan, without symptoms or signs of multiple sclerosis, are often referred to neurologists (radiologically isolated syndrome, RIS). Autopsy studies mentioned above show a significant reservoir of undetected multiple sclerosis. However, multiple sclerosis-like MRI lesions could be from vascular disease, tumor, and possibly migraine headache; strokes without overt symptoms are five times as common as symptomatic strokes—the same ratio as in multiple sclerosis. Nonetheless, in 30 patients with incidental brain MRI lesions, 23 (77%) developed new MRI lesions by 6 months, and 11 (37%) had clinical conversion to multiple sclerosis (148). In 41 patients (seven treated) followed for 2.7 years, 59% had new MRI lesions; 30% had one or more clinical attacks at a median time of 5.4 years (178; 179). Ten percent develop primary progressive multiple sclerosis over 5 years. Male sex doubles the chance of a second attack; (asymptomatic) cervical or thoracic cord lesions triple the risk, and infratentorial MRI lesions, CSF oligoclonal IgG bands, and younger age increase risk. A quarter of patients with radiologically isolated syndrome have cognitive impairment, 40% have frontotemporal cortical MRI lesions, and N-acetylaspartate levels are two standard deviations below normal in normal-appearing white matter (in 44% of radiologically isolated syndrome) and cortex (61%) (245). Asymptomatic cord lesions, present in one third of patients with brain lesions, predict a future attack or progression in 80%. Thus, early treatment is reasonable for some cases with MRI lesions only, and several drugs are highly effective in this case, but commitment to a therapy after a misdiagnosis is a danger.
MRI lesions can lead to MRI-supported misdiagnosis of multiple sclerosis. Patients may be treated with expensive and dangerous drugs, “therapeutic mislabeling.” Reluctance to reverse the misdiagnosis by neurologists raises ethical issues because continued therapy does not provide full disclosure of a patient’s condition.
Imaging is expensive and can lead to decisions that are not evidence-based. MRI-based decisions to change treatment to another partially effective therapy should be made with caution in patients with definite multiple sclerosis (eg, one new lesion after years of clinically stable disease). Premature termination of a quite effective therapy in this highly variable disease can potentially harm patients. Baseline activity can’t be ignored as it predicts future multiple sclerosis activity. For example, a patient who had 20 new lesions per year before therapy and who then has one new lesion per year and no clinical attacks should not be called a “non-responder” to therapy. Personalized medicine versus evidence-based decisions are discussed by Caplan (31).
A diagnosis of multiple sclerosis should be judiciously questioned when there are “red flags” such as:
|
• No eye findings (optic nerve or motility) or conversely, prominent uveitis or retinitis | |
|
• No remissions | |
|
• Localized disease | |
|
• Repeated episodes in the same part of the CNS | |
|
• Atypical clinical features: aphasia, altered consciousness, connective tissue disease symptoms, extrapyramidal symptoms, fevers, homonymous visual field defects, third nerve palsy, no fatigue or heat sensitivity, no long tract findings, no sensory or bladder symptoms, no constipation, progressive myelopathy without bladder involvement, peripheral neuropathy, late (older than 60 years) or early age of onset | |
|
• Very high sedimentation rate or CRP | |
|
• Normal CSF and no oligoclonal bands (217); high white count greater than 50 cells/µl or protein higher than 100 mg/dl | |
|
• Normal MRI of brain and spinal cord, or atypical MRI with small lesions (less than 3 mm), basal ganglia or internal capsule involvement, bilateral “mirror image” lesions, diffuse confluent white mater lesions, or longitudinal cord lesions spanning more than two vertebral segments. |
These red flags should prompt confirmatory tests for multiple sclerosis, such as CSF analysis, evoked potentials, and eye OCT. In the absence of objective evidence for multiple sclerosis or other disease, follow-up investigations should be kept to a minimum.
The treatment of multiple sclerosis is discussed in the following MedLink articles: Multiple sclerosis: management; Clinical trials in multiple sclerosis; Personal approach to multiple sclerosis treatment; and Multiple sclerosis: treatment of its symptoms.
Pregnancy as a treatment for multiple sclerosis is as potent as some approved drug therapies. The exacerbation rate during pregnancy at 0.14 per year is less than baseline at 0.36 per year and is approximately 70% lower in the third trimester. The severity and rate of attacks increase during the 3 to 6 months postpartum (to 1.00 attacks per year in the “fourth trimester”) (48). The average exacerbation rate during the entire year (pregnancy and postpartum period) is equivalent to the baseline rate. Mothers who smoke while pregnant have a 3-fold increased risk of having children who will develop multiple sclerosis (168); sun exposure in the second trimester reduces risk of childhood relapse by one third.
Multiple sclerosis is unlikely to appear de novo during pregnancy. Pregnancy decreases the later risk of a progressive course (219). Each baby and each further birth reduce the risk of multiple sclerosis by 50% (192). Furthermore, nulliparity is associated with an earlier onset of progressive disease, but there is a delay of the onset of progressive multiple sclerosis with each subsequent pregnancy (273).
A decline in family size and later age at first birth may be linked to evolving demographic factors and to the increasing frequency of multiple sclerosis in women.
The decline in exacerbations during pregnancy is presumably due to a shift from Th1 to Th2 type immunity and to immunosuppressive factors such as IL-10 that prevent rejection of the placenta and fetus. Treg cells decline during pregnancy, suggesting that they are of little consequence in multiple sclerosis. Estrogens are also important, and include estrone (E1, with one OH group), estradiol (E2), and estriol (E3), which is being studied as a treatment for multiple sclerosis. Estriol has 1/50th the estrogenic effect of estradiol but appears to have stronger effects on immunity. Estriol progressively increases during pregnancy. The progesterone/17-beta-estradiol ratio falls during the third trimester of pregnancy, when clinical activity is low. A rebound in immune function after delivery exacerbates disease activity. In support, high estradiol and low progesterone or oophorectomy in women, and low testosterone in men, are linked to more Gd+ MRI lesions; male to female sex change with hormonal therapy increases the risk of developing multiple sclerosis by 7-fold (89). Additionally, chimerism of circulating cells from the fetus may induce immune tolerance and neuroprotection. These fetal cells tend to reside in the mother’s bone marrow and brain.
In utero hormones could affect offspring. The right-hand finger 2D:4D ratio is increased in men with multiple sclerosis, suggesting fetal exposure to lower androgen/estrogen balance. In vitro fertilization increases risk of exacerbation 7-fold and MRI activity 9-fold (41). Gonadotrophin-releasing hormone induced IL-8, IL-12, TGF-beta, and IFN-gamma and facilitated transmigration across a blood-brain barrier model. It also induced 17-beta-estradiol, which increased anti-MOG antibody titers but did not affect cytokines. Interferon-beta therapy enhances fertility, perhaps by enhancing implantation (208).
Female hormones affect disease activity; 82% of women report worse symptoms before menses (235). The progesterone/17-beta-estradiol ratio increases during the luteal phase of the menstrual cycle, and this corresponds to higher MRI activity (196). MRI activity increases during ovulation when estradiol is high and progesterone is low (13). Catamenial symptoms improve with aspirin, and this does not appear to be from effects on body temperature. Birth control pills tend to reduce the incidence of multiple sclerosis in some studies.
Fifty-four percent of women have worse symptoms during menopause, and 75% feel symptoms improve with hormone replacement therapy of menopausal gonadotropin at very levels; effects may parallel those of in vitro fertilization. Progression may be more rapid at menopause. Menopause and removal of ovaries cause low-grade systemic inflammation, which can be prevented with low-dose estrogen replacement (01). A small clinical trial suggests that estriol decreases relapses and MRI lesions.
Breastfeeding, which elevates prolactin, was believed to have no effect on the exacerbation rate (172). However, one study shows women who breastfeed for more than 15 months have half the chance of developing multiple sclerosis (144), and a meta-analysis suggests that breastfeeding may be protective (137). A potential confounder in earlier studies was more smokers in the group who developed multiple sclerosis. Also, some women breastfeed for several days after delivery and then stop in order to begin an immunomodulatory drug; these women are more likely to have had active disease before or during pregnancy.
Mothers with multiple sclerosis in Norway had babies with slightly lower birth weights (46). They also needed more frequent induction and interventions during delivery but had no increase in birth defects or mortality. Perhaps confounding the data, these mothers were 2 years older than the control group. Other studies show similar trends for adverse delivery outcomes in women with severe disability (76), but duration of hospitalization is not longer.
Historically, multiple sclerosis therapies were classified as pregnancy category B (glatiramer), C (fingolimod, fumarate, interferons, natalizumab, alemtuzumab), X (teriflunomide), and D (mitoxantrone). Ocrelizumab has not been assigned a pregnancy category as it was developed after that drug labeling system was phased out by the U.S. Food and Drug Administration, and studies of its use during pregnancy are ongoing. The narrative structure now in use by U.S. Food and Drug Administration is more useful in guiding treatment decisions on an individual basis. Glucocorticoids rarely cause orofacial malformations. Interferons and glatiramer do not affect risk of fetal loss, although IFN-beta causes slightly smaller babies in some studies, but not others. IFN-beta and glatiramer should not cross the placenta; natalizumab does not cross in the early trimesters. However, fingolimod can cause cardiac malformations and teriflunomide is teratogenic for many organs. Both have long half-lives and cross the placenta. Dimethyl fumarate crosses the placenta but has a shorter half-life and causes small babies with delayed bone formation (142).
In the rare event of a relapse during pregnancy, corticosteroids should be avoided during the first trimester as they are linked to fetal malformations, such as orofacial clefts. Steroids are generally considered safe in the second and third trimester. Prednisone, prednisolone, and methylprednisolone cross the placenta at minimal concentrations. IVIG has also been used for relapses in pregnancy. Note, there is no long-term benefit of steroid use in multiple sclerosis. There is no contraindication to an MRI without contrast during pregnancy.
General anesthesia has no effect on multiple sclerosis (12). Direct trauma to the spinal cord or brain, barbotage, or intracerebral electrodes may predispose to lesions (194), but there are also reports of multiple sclerosis patients with direct brain trauma and surgery that did not cause any plaque formation (209). Experience suggests that brain biopsy and thalamic stimulation do not induce plaques.
Between 2019 and the present, significant research has evaluated the relationship between multiple sclerosis and COVID-19 infection, including mortality, risk factors, effects of disease-modifying therapies, and vaccination.
Some studies have concluded that multiple sclerosis does not significantly increase the mortality rate from COVID-19 infection (15). However, others have observed up to a 24% increased risk of death from COVID-19 in patients with multiple sclerosis relative to the general population (197). These higher estimates may at least partly be attributed to age and comorbid disease, which could lower the estimate to closer to that of the general population. Obesity, cardiovascular and pulmonary diseases, and diabetes are risk factors for worse outcomes and are equivalent in people with and without multiple sclerosis.
Multiple sclerosis–specific risk for severe or fatal COVID-19 is increased by progressive disease course and worse disability (198). Male sex has variably been identified as a risk factor. Importantly, untreated patients have more severe COVID-19 relative to patients who were receiving disease-modifying therapies.
Although disease-modifying therapies appear to protect against severe or lethal COVID-19 infection, there is heterogeneity among the effects of specific drugs. Anti-CD20 therapies have the most consistently observed adverse relationship for severe COVID-19 infection. Conversely, interferons are protective (238). Occasional studies show no increased risk with anti-CD20 therapies and additionally that patients on high-efficacy therapies are less likely to contract COVID-19 compared with those with no disease-modifying therapy (15). The commonality across studies is that untreated patients have more risk.
Infectious symptoms of COVID-19 in patients with multiple sclerosis are similar to those in the general population. Fever and dyspnea were more common in hospitalized patients, and headache and anosmia were more common in ambulatory forms. High fever was associated with the development of acute respiratory distress syndrome (153).
Does COVID-19 lead to progression of multiple sclerosis disability? A single study reported that the occurrence and severity of COVID-19 are both associated with clinical disability worsening in patients with multiple sclerosis (187). In one other study, COVID-19 infection was associated with exacerbation of multiple sclerosis in 57% (n=404) of the patients; however, pseudo flares were included in that percentage and disease-modifying therapies reduced the chance of developing frankly new multiple sclerosis symptoms during the infection (81). There may be publication and authorship bias against negative studies. Follow-up studies have not demonstrated multiple sclerosis worsening with COVID-19 infection (63).
The possibility that COVID-19 infection incites demyelination is being explored. The eSARS-CoV-2 nucleocapsid exhibits significant homology with 22 multiple sclerosis-associated proteins including PLP, raising the possibility of molecular mimicry (140).
A 2023 review of 19 observational studies concluded that COVID-19 vaccination does not increase the risk of relapse and serious adverse events in multiple sclerosis. The minimal risk of vaccination, weighed against the risks of SARS-CoV-2-related complications and possibly multiple sclerosis exacerbations, indicates that vaccination should be encouraged in most patients (242). Vaccine response is altered by disease-modifying therapies. The longevity of postvaccination SARS-COV-2 IgG humoral immune response was lower in fingolimod, ocrelizumab, and rituximab treated multiple sclerosis patients. No significant differences were found in postvaccination SARS-COV-2 IgG titers between healthy subjects and untreated multiple sclerosis patients or patients treated with interferons, glatiramer, alemtuzumab, cladribine, dimethyl fumarate, natalizumab, and teriflunomide (04). Vaccination, nonetheless, protects against a more severe course of COVID-19 in people with multiple sclerosis, and moreover, T cell response to vaccines appears to be unaffected or even increased with ocrelizumab. The ideal time for vaccination may be 3 weeks before the next anti-CD20 infusion (75).
Investigation on the short-term and long-term effects of COVID-19 infection on people with multiple sclerosis is ongoing. In summation, there is some concern about increased mortality and disease activity associated with COVID-19 infection, but robust evidence exists for the protective effects of disease-modifying therapy and vaccination.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Anthony T Reder MD
Dr. Reder of the University of Chicago received honorariums from Genentech, Genzyme, and TG Therapeutics for service on advisory boards and as a consultant and stock options from NKMax America for advisory work and an unrestricted lab research grant from BMS.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuroimmunology
Jul. 02, 2026
Neuromuscular Disorders
Jun. 16, 2026
Neuropharmacology & Neurotherapeutics
May. 14, 2026
Neuroimmunology
Apr. 26, 2026
General Child Neurology
Apr. 24, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuromuscular Disorders
Apr. 23, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026