










Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Polymicrogyria is a complex malformation of cortical development due to disorder of neuronal organization, in which the process of normal cerebral cortical development is disturbed in an early stage of cortical organization. Macroscopically or grossly, including by MRI, it is defined by clusters of gyri that are too small and too numerous for the area. Microscopically, not only are there numerous gyri in clusters but also many of the gyri are fused to adjacent gyri with continuity of their molecular zones and synaptic activity without the normal intervening subarachnoid space with leptomeninges and small meningeal blood vessels. Lamination of the cortex of these gyri is also abnormal with many displaced and disoriented neurons of all types, but there are no dysplastic megalocytic neurons as seen in focal cortical dysplasia type II, hemimegalencephaly, or tuberous sclerosis, except in some cases associated with a somatic mutation in the mTOR signaling pathway. Discontinuities or gaps in the pial membrane and lamina limitans, including the fetal subpial granular glial layer of Brun, are morphological features that lead to gyral fusion. Synaptic continuity of fused molecular zones (of mostly excitatory synapses) of adjacent microgyria results in epileptogenic short-circuitry. Clinical manifestations of perisylvian polymicrogyria include epilepsy, speech and language disturbance, and cognitive deficits, but polymicrogyria can be associated with many genetic syndromes with additional neurologic impairments; the spectrum of genes identified in polymicrogyria is very large, hence, it is a heterogenous malformation.
|
• Polymicrogyria is a cerebral malformation of cortical development with excessive folding of small, numerous fused gyri and normal or abnormal cortical lamination. | |
|
• Fusion of gyri microscopically results in continuity of the molecular zone of adjacent small gyri without intervening pial membranes or leptomeninges; pathogenesis involves discontinuities in the pia of the cortex during gyration or sulcation after mid-gestation. | |
|
• The underlying etiologies are multiple but can be summarized into three categories: (1) genetic disorders in which a wide range of unrelated mutations are reported; (2) association with other malformations of the brain, such as schizencephaly and cerebellar dysplasia, or with recognized genetic syndromes; and (3) vascular infarcts or ischemic zones, resulting in a wide variety of neurologic signs and symptoms. | |
|
• The distribution of polymicrogyria most frequently is perisylvian; it always is associated with schizencephaly, but in other cases it may be focal in any lobe or can be generalized throughout the cortex of both hemispheres. | |
|
• High-resolution MRI is the most reliable imaging modality for diagnosis and evaluating the distribution and extent of the dysgenesis and association with other brain malformations; fMRI shows decreased activation; neuropathology provides microscopic details not revealed by imaging. | |
|
• Prenatal diagnosis by fetal MRI in the third trimester is possible. | |
|
• The most frequent cause of acquired fetal polymicrogyria due to prenatal infection is cytomegalovirus. | |
|
• Surgical resection of epileptogenic foci within polymicrogyria may provide good seizure control with better developmental outcomes in children but is not successful in all cases. Also, polymicrogyria is not always epileptogenic. | |
|
• Neuroanatomical features at the single neuron level determine epileptic potential by shifting the excitatory/inhibitory ratio: short circuitry of the molecular zones of adjacent fused gyri, paucity of keratan sulfate inhibition of glutamatergic synapses, and clustering of Cajal-Retzius neurons that cause dyslamination of the cortical plate in microgyria. In cases due to an AKT family or other gene affecting the mTOR pathway, megalocytic dysplastic neurons and glial cells are found. |
Originally coined in the early 1900s by Bielschowsky (121), the term, “polymicrogyria” was largely replaced by the term “microgyria” by the middle of the 20th century. Contemporary preference has seen a resurgence of the term “polymicrogyria,” which more accurately describes the findings in this disorder (122).
Classification. Several types with various manifestations and etiologies have been noted. Under the current classification of malformations of cortical development, the following types of polymicrogyria are defined by MRI (12; 11). They also can be so classified by gross neuropathology.
|
(1) Bilateral frontal polymicrogyria (BFP) |
Outdated names used for polymicrogyria include microgyria, micropolygyria, and status verrucosus deformis (39).
Polymicrogyria manifests as a spectrum of clinical signs and symptoms ranging from no manifestations to severe encephalopathy or cognitive impairment (74; 75). This disorder is associated with epilepsy in approximately 50% to 85% of patients (67; 72). Children often present with developmental delay, spasticity, or seizures; they are also often microcephalic. Some patients with polymicrogyria go undiagnosed until they have offspring with the disorder, who typically have more severe manifestations. Retrospectively, these patients will often report some difficulty in their medical or educational history (97).
The clinical presentation and course depend on localization and extent of the lesion, association with other brain malformations, and underlying genetic mutations or prenatally acquired lesions, but epilepsy and developmental delay are the most frequent symptoms in infancy. Seizure onset in the first year of life occurred in 44% in a Swedish study of 109 patients (89). Clinical diversity of polymicrogyria is largely determined by the extent of cortical involvement and localization of lesions with a topographic distribution of symptoms (74; 116).
(1) Frontal polymicrogyria typically have features of delayed motor and language milestones, spastic hemiparesis or quadriparesis, and mild to moderate mental retardation (29; 74).
(2) Bilateral frontoparietal polymicrogyria present with its most common symptoms of global developmental delay, dysconjugate gaze, pyramidal and cerebellar signs, and generalized seizures (29).
(3) Bilateral perisylvian polymicrogyria, the most frequent distribution, manifests as seizures that usually begin between 4 and 12 years of age as well as mental retardation, pyramidal signs, and pseudobulbar palsy that manifests as diplegia of the facial, pharyngeal, and masticatory muscles (facio-pharyngo-glosso-masticatory diplegia) (63). Dystonia occurs in some cases (05). Developmental delay may be observed, and malformations such as arthrogryposis (127), club feet, and micrognathia may be seen more commonly in the congenital form of the disorder. Perisylvian polymicrogyria is seen in all cases of schizencephaly, whether unilateral or bilateral. Some cases of bilateral perisylvian polymicrogyria also involve cerebellar dysplasia and ectopic neurohypophysis (166). Psychiatric symptoms such as mania and bipolar disorder may occur in young adults with polymicrogyria (26).
(4) Bilateral parasagittal parieto-occipital polymicrogyria patients present with seizures, cognitive slowing, and normal to mildly decreased intelligence (65).
(5) Bilateral generalized polymicrogyria not limited to the perisylvian region usually have variable degrees of cognitive and motor delay presentation, as well as spastic hemiparesis or quadriparesis and seizures. Most patients also have associated congenital abnormalities such as macrocephaly, scalp and limb defects, low-set ears, macrostomia, hypothyroidism, and sensorineural hearing loss (29; 127). Genetic mutations are the most frequent etiology.
(6) Unilateral polymicrogyria may be seen in various cortical locations, but manifesting signs and symptoms typically include spastic hemiparesis primarily of the upper extremity, variable degrees of mental retardation, and seizures. Contralateral body hemiatrophy and hemianopsia have also been reported in the unilateral forms (27; 133; 137). Unilateral perirolandic polymicrogyria may be accompanied by ipsilateral brainstem hypoplasia and contralateral hyperplasia (131). In unilateral cases, the corticospinal tract, as assessed by MRI and DTI tractography from the involved hemisphere, is asymmetrical and smaller, which may provide for less functional motor impairment if hemispherectomy is performed to control epilepsy because the tract on the other side may carry axons from both hemispheres (55; 06). Even if contralateral hemiparesis is present preoperatively, hemispherectomy may provide good results (100).
Two thirds of patients with polymicrogyria have involvement of the perisylvian region, whether unilateral or bilateral. The characteristic neurologic features reported in patients with perisylvian involvement are cognitive defects, dyspraxia consisting of nasal speech, dysarthria, dysphagia, drooling, and other signs of pseudobulbar palsy (75; 127). The reason for this perisylvian pattern is that about two thirds of the ventral surface of the early telencephalon after prosencephalic cleavage is buried in the frontal and temporal and insular lips of the operculum and, eventually, the Sylvian fissure; ventrodorsal genetic gradients in the vertical axis could be involved in the generation of this abnormal morphogenesis (145).
The spectrum of the conditions associated with polymicrogyria continues expanding. Polymicrogyria has been found part of the heterogeneous genetic presentation of the three forms of the megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome (MPPH1, MPPH2, and MMPH3). MPPH1 is caused by heterozygous mutation in the PIK3R2 gene on chromosome 19p13. MPPH2 is caused by mutation in the AKT3 gene on chromosome 1q43-q44; and MPPH3 is caused by mutation in the CCND2 gene on chromosome 12p13 (62; 01).
Polymicrogyria may occur as an isolated anomaly, but more often, it occurs in conjunction with other developmental abnormalities such as microcephaly, porencephaly, or cerebral encephalocele (39; 172). Fukuyama and colleagues showed thickened cortex with polymicrogyria in all cases of Fukuyama congenital muscular dystrophy with pachygyria/lissencephaly II that were studied neuropathologically (64). Others have classified the neuropathologic abnormalities seen in these patients as Lissencephaly type II (33).
The most frequent and constant association of polymicrogyria is with schizencephaly. Even if schizencephaly is unilateral, the associated polymicrogyria may be bilateral, including a similar topography in the contralateral nonschizencephalic hemisphere. Polymicrogyria occurs within or immediately adjacent to a schizencephalic cleft (64; 12) and involves not only the frontal and temporal lips of the cleft but also the “third lip” of the insula (145). Polymicrogyria may also occur in conjunction with, and in close proximity to focal cortical dysplasia type II (70). Both abnormalities are incited during approximately the same period of cortical organization during fetal development (68; 173). Polymicrogyria not limited to a perisylvian distribution; it rarely may be concurrent with subcortical laminar or “band” heterotopia and can be demonstrated by both prenatal and postnatal MRI (88; 112). Inconstant associations of polymicrogyria may be found in a wide variety of other cerebral malformations, including agenesis of the corpus callosum (71), lissencephalies and holoprosencephaly, and septo-optic-pituitary dysplasia with olfactory bulb hypoplasia (15).
A developmental vasculopathy, hereditary hemorrhagic telangiectasia, can be associated with cerebral aneurysms and ischemic infarcts of the brain; some cases also have bifrontal periventricular nodular heterotopia and perisylvian polymicrogyria (118). A strong coexistence of polymicrogyria is with the megalencephaly-capillary malformation syndrome (previously known as cutis marmorata telangiectasia congenita), associated with PIK3CA mutations of the mTOR signaling pathway (106; 99; 56; 119). Some of these cases can be further complicated by cerebral venous thrombosis (56) or cerebral arteriovenous malformations (87). The vascular disorder is one of angiogenesis during fetal development (87). Another ischemic cause of polymicrogyria during fetal life is twin-to-twin transfusion syndrome (82; 140).
Other associations of polymicrogyria have been demonstrated with neonatal adrenoleukodystrophy (12) and a case of bilateral perisylvian polymicrogyria occurring in a young girl with a Chiari I malformation has been reported. Initially, Robin and colleagues reported 32 patients with deletion of chromosome 22q11.2 and found that there was a striking predilection for right-sided polymicrogyria (136; 24), most common in the perisylvian region and often with mega-cisterna magna or mild cerebellar vermis hypoplasia (124). Nguyen and colleagues reported association of duplication and triplication of chromosome 18q22.2 abnormalities. Focal small foci of polymicrogyria have been identified incidentally within the insular cortex of asymptomatic patients, and focal polymicrogyria has been found associated with chromosome rearrangements detected by CGH microarray analysis (130; 113).
The complications of polymicrogyria are primarily related to epilepsy and cognitive and motor deficits. Neonatal seizures can be severe, refractory to medications, and even life-threatening (21). Prognosis is determined by the underlying etiology as well as distribution and severity of the disease. Poor prognostic indicators include involvement of more than half of one cerebral hemisphere and bilateral brain involvement (09). However, early identification of surgical candidates so they can undergo early epilepsy surgery can improve significantly their developmental outcome (86; 162). In addition to well-known clinical manifestations of epilepsy and cognitive delay, specific disturbances of speech, language, and oral functions may be prominent in perisylvian polymicrogyria (19).
A 14-year-old female presented with seizures refractory to medical therapy. At 5 months of age, she was diagnosed with epilepsy consisting of jerking movements, rolling back of her eyes, and a blank stare. Since then, her seizures had occurred at random, but often during the process of awakening from sleep. During the seizures, she also would make a clicking noise with her mouth, and her head would drop forward. The seizures typically occurred two to three times per day and would last 1 to 5 minutes and then subside. Despite various medication regimens, her seizures were poorly controlled. She had moderate mental retardation and developmentally was at the level of a 6- or 7-year-old child. Physical examination revealed slight hypotonia of the upper and lower extremities with decreased deep tendon reflexes and a mildly unsteady gait. MRI with and without gadolinium showed distortion of the frontal right gray-white matter junction with right frontal sulcal and gyral thickening that extended into the right insula.
An EEG showed mild generalized slowing, continuous polymorphic slowing in the right frontal region, and frequent epileptiform sharp-wave discharges over the right superior frontal region. Surgical intervention ensued, with resection of the right frontal epileptogenic focus. The resection specimen grossly had a nodular cortical surface with multiple, small coalescent gyri. Microscopically, there was cortical thinning with a simplified lamination pattern and excessive folding of the gyri.
A diagnosis of polymicrogyria was rendered. After surgery, the patient continued to have refractory seizures; however, the frequency of her seizures was mildly decreased, and the patient was more verbal and interactive than she previously had been.
Until recently, the majority of cases of unilateral and bilateral polymicrogyria were described to be of unknown causes (122; 123). However, advances in the genetic etiologies of brain malformations, including exon sequencing, have revealed multiple specific genes that cause polymicrogyria (48; 158; 03). In addition to the genetic etiologies, environmental etiologies, such as intrauterine infections (eg, cytomegalovirus, toxoplasmosis, syphilis, and varicella-zoster), as well as fetal cerebral ischemia from various causes, such as twin-twin transfusion syndrome, still remain as significant risk factors (39).
Multiple syndromes such as Aicardi syndrome, Foix-Chavany-Marie syndrome, glutaric academia type II, histidinemia, Leigh syndrome, maple syrup urine disease, mitochondrial respiratory chain deficiency, neonatal adrenoleukodystrophy, Pelizaeus-Merzbacher disease, thanatophoric dysplasia, Walker-Warburg and Zellweger syndrome, as well as familial occurrences of polymicrogyria have been confirmed to be due to specific genes that lead to the development of polymicrogyria (74; 102; 151; 174; 25; 115; 128; 36). Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis in many malformations of cortical development including polymicrogyria (96). Hemidystonia with polymicrogyria has been reported with ATP1A3 mutations (92). An association of polymicrogyria with megalencephaly-capillary malformation, which can result in cerebral venous infarctions, has been described (56; 142).
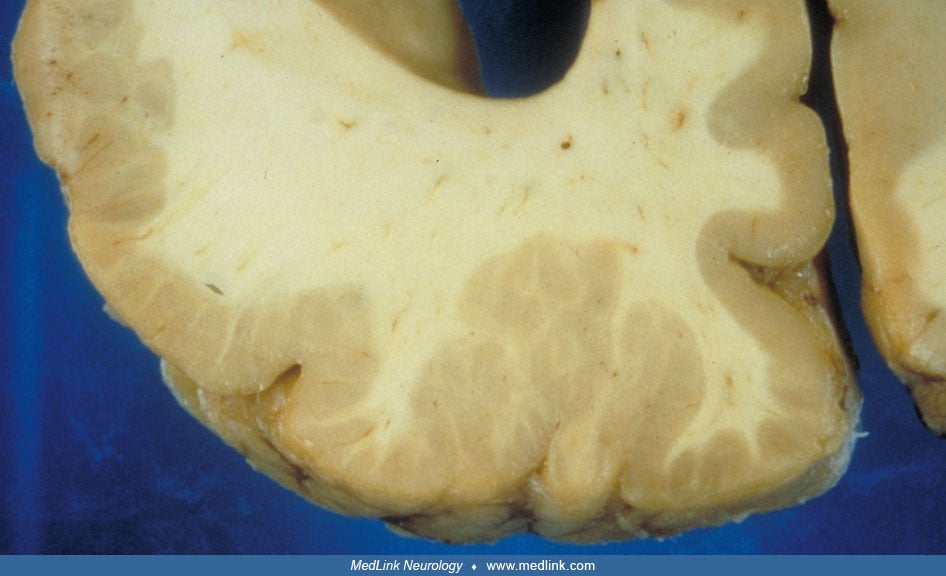
Typically noted at autopsy, the surface of the brain has increased convolutional complexity with miniature gyri that are fused to one another, or piled on top of one another, forming an irregular, bumpy surface.
This appearance is often described as that of cobblestones or Moroccan leather (66; 67). The heaping up of the mini-gyri accounts for the apparent cortical thickening that is often seen radiographically (67). The distribution of the lesions often corresponds to a particular arterial territory, most commonly of the middle cerebral artery. Polymicrogyria is often seen at the border of porencephaly or schizencephaly (68).
Microscopically, small, fused gyri are noted with shallow sulci. In the unlayered form of polymicrogyria, which is the more common of the two types, there is cortical thinning with the presence of only a molecular layer and a neuronal layer without further laminar organization. In the layered form, four distinct layers of the cortex are seen corresponding to a molecular layer and two layers of neurons separated by an intermediate layer of a few cells and myelinated fibers (66).
Additional histologic findings include an abnormal structure of cortical neurons and presence of heterotopic neurons in the white matter (80).
In acquired forms of the disorder, the more commonly accepted hypothesis is that hypoxic-ischemic insults occur during gestational weeks 20 and 24 (172; 121). Williams and colleagues demonstrated the presence of six layers of the cortex within polymicrogyric regions using Golgi-impregnated tissue. Each layer was continuous with the analogous layer in the adjacent unaffected cortex. However, it was the gyral pattern that was topographically anomalous, inferring that insult had occurred to a normally developed cortex (172). This postmigratory destruction is further supported by the findings that 4-layered polymicrogyria shows normal positioning of neurons in layers II, III, IV, and VI with very few layer V pyramidal cells identified in the sparse intermediate layer. The implication here is that neuronal migration was completed prior to occurrence of the cortical insult (67).
The pattern of distribution of polymicrogyria in the cerebrum and cerebellum tends to correspond to distal regions of perfusion by the major cerebral arteries. Of note, the vasculature of the brain is already established during the first half of gestation, further supporting the post-migrational theory (172). A preference for involvement of the perisylvian may predominantly be due to the fact that this region is more susceptible to intrauterine ischemic insults (164).
In cases resulting from infectious etiologies, neither the degree nor distribution of infection dictates the distribution or extent of polymicrogyria. It is, therefore, likely that the pathogenesis of the disorder relies more heavily on perfusion failure as a result of intrauterine infection rather than direct cytopathic effects of the causative infectious agent (172; 121). Fetal cytomegalovirus infection is the most frequent cause of ischemic polymicrogyria induced by prenatal infections (94).
Anatomic observations made by Hallervorden in 1949 and Bankl and Jellinger in 1967 demonstrated cases of classic polymicrogyria in women who had been exposed to large doses of carbon monoxide at 20 and 24 weeks of gestation, respectively (40; 172). This transient perfusion failure occurred in the fetuses at a time when neuronal migration should have been completed.
Animal models. Rat models developed by Dvorak and colleagues showed evidence of a 6-layered cortex in the four layers of polymicrogyric cortex. These findings were similar to those seen by Williams and colleagues just a few years prior. However, the rat models also showed that this phenomenon could occur in the very late stages of neuroblastic migration (47). Dvorak and colleagues also discovered that neuroblasts could migrate through regions of partial necrosis and come to rest in a predetermined layer of the cortex. This meant that intrauterine insult during neuroblastic migration did not necessarily lead to complete migrational arrest (46). Some authors now believe that the intrauterine insult occurs during late neuroblast migration in the unlayered type and post-migration in the layered type of polymicrogyria (64; 121). Focal transcranial cortical freeze lesions in neonatal mice can produce schizencephalic cortex and polymicrogyria, the former yielding increased excitatory synapses with increased dendritic spine density, whereas polymicrogyria produce decreased numbers of inhibitory synapses; both conditions are conducive to epilepsy (45). Induced malformation of neocortex resembling human polymicrogyric cortex in neonatal rats is associated with alterations in dendritic structure and decreased calbindin expression in GABAergic interneurons, but also preservation of glutamate receptors in these cells (84).
Familial cases. Familial cases have been documented with both autosomal dominant and autosomal recessive heredity. Brothers with Joubert syndrome and polymicrogyria have been reported (59). Bilateral frontoparietal polymicrogyria, bilateral perisylvian polymicrogyria, and bilateral generalized polymicrogyria have all shown familial occurrences (91). Bilateral frontal polymicrogyria and bilateral parasagittal parieto-occipital polymicrogyria have only been reported in sporadic cases (74). Bilateral perisylvian polymicrogyria shows a tendency towards occurrence in males. This led to the discovery of a genetic abnormality localized to Xq28. Both unilateral and bilateral perisylvian polymicrogyria have been associated with several chromosomal aneuploidy syndromes. Perhaps the most common genetic abnormality seen with polymicrogyria is deletion of chromosome 22q11.2 (12; 136). It has been reported that deletion of 1p36 is also one of the most common chromosomal abnormalities associated with polymicrogyria (74). An unbalanced translocation resulting in 1q44qter monosomy and 12p13.3pter trisomy has also been identified in some patients with unilateral polymicrogyria (27). Bilateral frontoparietal polymicrogyria has been mapped to 16q12.2-21 and is associated with mutations of the GPR56 gene (12; 74). This gene encodes for a G protein coupled receptor. This regulates cell cycle signaling in neuronal progenitor cells, which is essential in cortical regional patterning (167). Mutations of the PAX6 gene on chromosome 11p13 were seen in a family with unilateral polymicrogyria as well as in mice (74). Xq22 mutations in the SRPX2 gene have been associated with bilateral perisylvian polymicrogyria. This gene is involved the development of the speech cortex through proteolytic remodeling of the extracellular matrix (156; 167). Robin and colleagues have hypothesized that haploinsufficiency of certain genes expressed in vascular tissue that supplies the embryonic brain act in concert with genetic modifying factors, thus, causing hypoperfusion of the developing brain (136). Other mutations reported in polymicrogyria are duplications of 2p16 (04) or of 17p13.3-p13.2 (158). In a Finnish cohort of bilateral perisylvian polymicrogyria, five of 21 (24%) cases were confirmed as de novo with pathological variants of five genes: DDX23, NUS1, SCN3A, TIBA1A, and TUBB2B (77). Genetic mutations were similarly confirmed in one-third of Swedish patients with bilateral polymicrogyria (89).
Other rare genetic mutations that cause familial polymicrogyria and micrencephaly include RTTN (31), COL18A1 (30; 35), NF1A (14), and KIF5C mutations (104). A rare familial autosomal recessive trait is a defect in occludin (OCLN), an important component of the tight junction complex providing apical intercellular connections between adjacent cells in endothelial and epithelial tissues; it is expressed as polymicrogyria and pachygyria, band-like calcifications in basal ganglia and cerebral cortex (probably due to ischemia from endothelial involvement of parenchymal capillaries), and progressive acquired microcephaly; clinical neurologic features are severe global developmental delay and epilepsy (18; 78). Knobloch syndrome is an autosomal recessive disorder consisting of polymicrogyria with early onset of retinal detachment due to vitreoretinal degeneration and occipital cranial defects clinically expressed with refractory severe epilepsy (30; 171). Homozygous mutation of the COL18A1 gene is demonstrated in siblings (30). More details of the diverse genetic profile in polymicrogyria is found below in this review in the Genetics section.
Epileptogenesis in regions of polymicrogyria has received a significant amount of attention in several studies. Some animal studies on polymicrogyric brains have shown hyperexcitability due to a decrease in GABA-A receptors and an increase in postsynaptic glutamate receptors in the microgyric cortex (66). Others have shown that that the hyperexcitability is found in the histologically normal cortex surrounding the polymicrogyria (72; 152). This supports the findings that thalamic sensory afferents are lacking in the microgyric regions and that these afferents are redirected to adjacent areas that become hyperexcitable (66). The gaps in the pial membrane in human polymicrogyria lead to gyral fusion and synaptic short-circuiting of the continuous molecular zones of cortex of adjacent gyri (157; 42); the molecular zone contains almost exclusively glutamaterigc axodendritic synapses, thus, shifting the balance of the excitatory/inhibitory ratio in favor of excitation (143). By contrast, there is dendritic loss in the molecular zone in Down syndrome and epilepsy is uncommon (163). GPR56 promoter of gene expression in GABAergic neurons (and axosomatic synapses) in marmosets also might be altered (110). Corticothalamic circuits also are altered if the thalamus is hypoplastic with polymicrogyria (13).
Neuropathological studies disclose that one of the primary developmental defects in the pathogenesis of polymicrogyria, regardless of etiology, is discontinuity of the pia mater and the glia limitans at the surface of the cortex. Neuropathological series confirm that polymicrogyria is a common endpoint of many different etiological processes (76; 165; 49).
Historically, an overwhelming number of cases were from patients in psychiatric hospitals. But even in the mid-1900s it was postulated that the incidence of polymicrogyria in patients without seizure disorders or psychiatric disturbances must be higher than what was recognized (39). Today, with increased use of high-resolution imaging modalities, the recognition of this disorder is increasing (67; 74). However, the incidence is still largely undetermined at this time. There are no reports of ethnic preference. However, some X-linked forms are more common in males (32; 28; 74; 09). Overall prevalence of polymicrogyria in the Stockholm, Sweden region of 109 patients was .3 per 10,000 children (89).
No definite preventative measures are known at this time. Careful prenatal monitoring with avoidance of risk factors for infectious agents in pregnant women may reduce the risks of events that may lead to polymicrogyria. Genetic counseling of patients with hereditary polymicrogyria syndromes may also be warranted. A patient with bilateral polymicrogyria and intractable infantile spasms was treated with vigabatrin, a reasonable therapy, but developed white matter spongiosus that was suspected to be due to this medication, and he died with SUDEP (125). Neuroimaging monitoring of patients on vigabatrin might detect early such cases so that the treatment can be changed.
One of the most important and often difficult distinctions is between polymicrogyria as a developmental malformation and chronic fetal ischemic encephalopathy that results in gyral atrophy during development and ulegyria (85; 90). These two entities usually are easily distinguished neuropathologically at the microscopic level but may be a more difficult distinction by neuroimaging. Lissencephaly type II can be distinguished from polymicrogyria by obliteration of the subarachnoid space, complete disorganization of the laminar pattern, and deep-seated nodular heterotopia (60).
Other malformations of cortical development are differentiated by imaging and histopathologic characteristics. Polymicrogyria may be only one component of multiple malformations of brain and periphery such as polydactyly (117; 83). Unilateral polymicrogyria may be associated with ipsilateral brainstem atrophy with deficient corticospinal tract fibers and contralateral motor deficits (137).
Neuroimaging. MRI has been demonstrated to be the most useful and common modality for diagnosing polymicrogyria, as it is more sensitive than any other neuroimaging technique. MRI appearances may vary, revealing multiple small gyri, an apparently thickened cortex with an irregular border between the gray and white matter junction, or even a paradoxically smooth cortex in which a smooth molecular layer fuses over underlying microsulci (68; 10; 160). A reduction in white matter is also common, as are other white matter abnormalities. Patients with deletion of chromosome 22q11.2 may also demonstrate agenesis of the corpus callosum and cerebellar hypoplasia (161). By contrast, some patients have an excessively large corpus callosum with polymicrogyria (22). Large corpus callosum may be a maturational delay or arrest of retraction of commissural collateral axons during fetal and early postnatal life (147). Three-dimensional magnetic resonance imaging is currently the recommended imaging modality as it allows a better appreciation of the true extent of brain involvement. Functional fMRI also is useful in showing decreased reactivity in areas of polymicrogyria (95). Prenatal MRI in the third trimester often can detect polymicrogyria (134; 94).
CT scans have very limited utility in the diagnosis of polymicrogyria. They are helpful to visualize better calcifications due to intrauterine infections and associated band-like calcifications associated with recessive mutations (20; 114).
Diffusion tensor imaging (DTI) and functional MRI in unilateral polymicrogyria in nine patients demonstrated asymmetry in the corticospinal tract at the level of the midbrain cerebral peduncles; pronounced asymmetry predicted preserved motor function after hemispherectomy because of bilateral projections of the CST from the preserved hemisphere that arose during fetal life (55).
Neurophysiology. Electroencephalography, including routine and prolonged video-EEG monitoring, is used to characterize the background and the interictal and ictal patterns that define the features of each of the structural epileptic syndromes due to the various forms of malformations of cortical development seen in this condition. In a study, 10 of 16 patients with polymicrogyria had normal EEG activity. The remaining six had at least focal abnormalities, and three of those also had generalized EEG abnormalities (164). Teixeira and colleagues also showed that EEG abnormalities were most often seen in patients with generalized, hemispheric, and holosylvian polymicrogyria. There was a good correlation between extent of cortical involvement and severity of clinical features and EEG abnormalities. There is a positive correlation between a slow background on EEG and the presence of cognitive delay, motor deficit, and epilepsy. However, a majority of the patients they studied (55%) had no EEG abnormalities whatsoever (164). In patients with perisylvian polymicrogyria, epileptiform activity, when present, is typically localized to the frontotemporal and temporal regions, and non-epileptiform abnormalities are more often seen in the cortico-temporal regions (164). It should be taken in consideration that many children could present initially with generalized onset ictal patterns, such as diffuse electrodecremental response or generalized paroxysmal fast activity, but subsequently, these children can be identified later to have focal epileptogenic zones that could make them amenable to potentially successful epilepsy surgery, with the help of techniques such as SISCOM guided depth electrodes implantation prior to resection of an epileptogenic zone. Continuous spike-wave complexes during sleep in polymicrogyria are likely related to cortico-thalamic circuitry (13). Electroclinical changes from infancy to adulthood after corpus callosotomy were described in three siblings with bilateral frontoparietal polymicrogyria (91). fMRI studies suggest that polymicrogyric cortex may predispose to seizures via abnormal network topology (149; 70; 34). Combined EEG and invasive epileptogenicity mapping in pediatric patients with refractory epilepsy and focal polymicrogyria may be useful to surgical decisions of brain resection (148).
Neuropathology. Excellent gross and microscopic neuropathological studies of polymicrogyria are available (157; 42). Gross inspection at autopsy or by neuroimaging, especially MRI, disclose polymicrogyria as focal clusters of excessively small and too numerous gyri within a given region of neocortex; the distribution is perisylvian, focal in other regions, or generalized throughout the cortex.
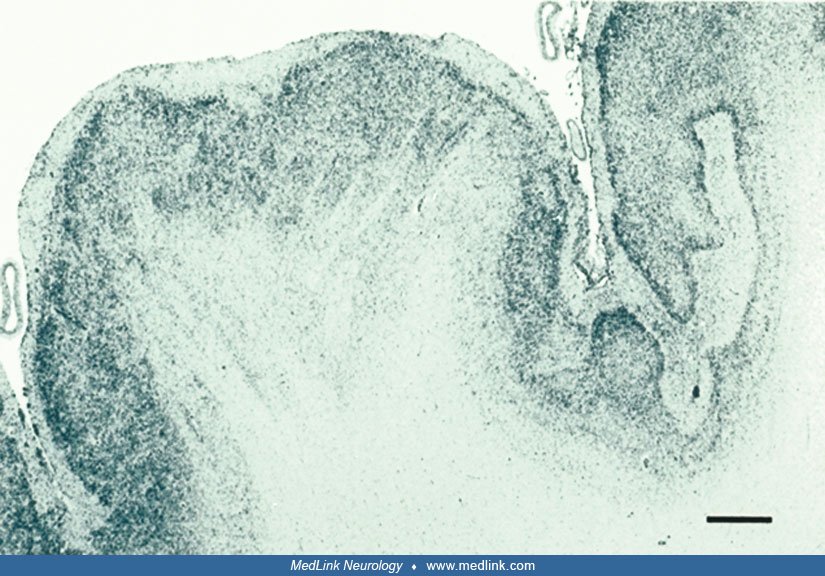
But microscopic examination reveals a great deal more, particularly if histological stains are supplemented by immunocytochemical reactivities. The laminar structure of the microgyria may be preserved, but the number of neurons reduced (79), or there may be dyslamination with unlayered architecture or disorganized and disoriented neurons (52; 53). Radial micro-columnar architecture, such as that which occurs in focal cortical dysplasia type I and in normal cortical histogenesis during the first half of gestation, is found occasionally, especially in perisylvian polymicrogyria associated with schizencephaly (17).
One of the most striking and important neuropathological features that is not appreciated in neuroimaging is the gaps or discontinuities in the pia mater that enables fusion of adjacent microgyria with continuity of their molecular zones (79; 157; 76; 42). Synaptophysin immunoreactivity shows synaptic activity across this abnormal continuity of the molecular zone between gyri at abnormal sites (17). Short-circuitry of mainly excitatory synapses of the fused molecular zones of adjacent microgyria is an explanation of why polymicrogyria often is epileptogenic (143). Recurrent interactions in local circuits are a neurophysiological basis of seizure generation (126).
This pattern of fused gyri is seen in polymicrogyria of all etiologies, and there is no correlation with lamination patterns of the cortex of the gyri (76). However, gyral fusion is more extensive and more frequent in genetic forms of polymicrogyria than in acquired forms such as ischemic encephalopathy (Sarnat unpublished data). This may explain why some cases are much less epileptogenic than others. Gaps in the pia limitans enable overmigration of neuroblasts and glial cells into the leptomeninges within the shallow sulci above the areas of fusion of the molecular zones of adjacent gyri (157). Physical properties of the leptomeninges are also altered by thickening and reduplication of the pial collagen layer and increased meningeal vascularity.
In addition, there is clustering of Cajal-Retzius neurons in the molecular zones of microgyria with persistent Reelin production, which contributes to the dyslamination of the cortex and also may favor epilepsy (50; 103; 76; 42). Another factor to explain epilepsy in polymicrogyria is that the expression of keratan sulfate, a normal extracellular proteoglycan that binds to neuronal membranes during development, but not to dendritic spines, and selectively repels the formation of glutamatergic (excitatory) synapses while enabling GABAergic (inhibitory) synapses is deficient in cortical zones of polymicrogyria (144). Excessive heterotopic neurons and synaptic plexi in the U-fiber layer beneath polymicrogyria also may contribute to the epileptogenic malformation (146).
In fetuses of 18 to 29 weeks gestational age who had suffered ischemic infarcts of the cortex with focal necrosis, adjacent cortex often had polymicrogyria with pial defects and also an inflammatory response of astroglia and microglia (42). Cajal-Retzius neurons of layer one (molecular zone) were hyperplastic, clustered, and sometimes displaced deep within the cortical plate; subplate neurons also were hypertrophic, particularly in young fetuses of less than 20 weeks gestation. Radial glial fibers by 18 weeks showed altered growth patterns with abnormal lateral branching (42).
Genetics. Nearly all cases of polymicrogyria confirmed by neuroimaging merit genetic investigation unless another cause, such as a congenital infection, is demonstrated. Multiple specific genes can cause polymicrogyria (44; 48; 158; 49; 03). The most frequent mutations in a deep sequencing study of 123 patients were TUB1A and PIK3R2, but others included PIK2CA, NEDD4L, COL4A1, GPPSM2, GRIN2B, WDR62, TUBB2B, TUBB3, ACTG1, and FH (158). Some, such as the PIK family of genes, are part of the mTOR signaling pathway that causes megalocytic dysmorphic neurons. Other candidate genes, such as de novo missense variants, are PANX1, QRICH1, SCN2A, and compound heterozygous variants in TMEM161A, KIF26A, and MAN2C1, each with consistent genotype-phenotype relationships in multiple families (03). Other isolated case reports associate polymicrogyria with ATP1A3 (92), ADGRG1 (91; 98), SCN2A (58), and the channelopathy gene KCNMA1 (61).
Germline homozygous missense variants of DEPDC5 cause bilateral polymicrogyria with refractory early-onset epilepsy, developmental delay and macrocephaly (168). Compound heterozygous pathogenic variants of CEP83 are another basis of bilateral perisylvian polymicrogyria with cognitive impairment, epilepsy, and renal disease (120). X-linked bilateral polymicrogyria in a novel KIF4A variant is another cause of cognitive deficit, epilepsy, and complex other brain malformations of the hippocampi, dysgenesis of the corpus callosum, and cerebellar heterotopia and aplasia (93). Monoallelic missense mutation variants involving the Reelin gene, which is important in neuroblast migration and organization/lamination of both cerebral and cerebellar cortices, result in diverse abnormalities in gyration/foliation, including pachygyria and polymicrogyria (135).
Novel human polymicrogyria loci are demonstrated at 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2 (44). Genetic techniques such as comparative genome hybridization and whole-exome sequencing are important in diagnosis (16; 81). Deletion of chromosome 22q11.2 in patients with polymicrogyria, especially perisylvian polymicrogyri, was reported by Robin and colleagues to be the most common genetic abnormality associated with polymicrogyria (136). Testing for the GPR56 gene should be pursued in all patients with bilateral frontoparietal polymicrogyria (12). Copy number variants (CNVs) also play an important role in the etiology spectrum of the malformations seen in polymicrogyria, either as direct cause or as genetic risk factors. Mutations of the phosphoinositide-3-kinase regulatory subunit PIK3R2 can result in perisylvian polymicrogyria (105). The SCN3A gene encodes sodium channel Nav1.3 that is highly expressed in the brain, and missense variants of this gene cause severe infantile epileptic encephalopathy and impaired oral motor development, profound intellectual disability and diffuse generalized polymicrogyria (175; 154). The closely related SCN2A gene can produce Ohtahara syndrome with seizure onset within a day after birth, perisylvian polymicrogyria, and white matter heterotopia (169).
The genetic involvement leading to polymicrogyria can be divided in two types: one due to ischemia-related genes and another due structural function-related genes. Mutations that lead to ischemia where polymicrogyria has been found include alterations of the Occludin gene (OCLN), the COL4A1/COL4A2 gene, the ALX4 and MSX2 gene, deletions in the 22Q11, and the somatic mutation in GNAQ that causes the Sturge-Weber syndrome. CCND2 mutation results not only in perisylvian polymicrogyria but also obstructive hydrocephalus and somatic anomalies, including postaxial polydactyly (101). Brain malformations in CCND2 not only involve the cortex but may also include dysplasia and hypoplasia of brainstem structures and cerebellum, and hypomyelination (23). Multisystemic developmental anomalies also occur in Pallister-Killian syndrome caused by mosaic tetrasomy of chromosome 12p; the brain malformations are bilateral perisylvian polymicrogyria, ventriculomegaly, and hypoplasia of the corpus callosum (129). GRIN1 is another now established genetic association with polymicrogyria (38).
Genetic mutations that primarily cause disruption of the neuronal structural organization include multiple new genes that encode proteins that participate in the integrity of the centrosome, cilia, and microtubule, as well as proteins that participate in the integrity of the basement membrane. The more important genes are the GPR56 and the tubulin genes, which are mutated in many malformations of cerebral cortical development including in polymicrogyria (138).
In generalized bihemispheric polymicrogyria, a strong association is seen in the tubulinopathies (51; 139). Polymicrogyria has been reported in patients with mutations in TUBA1A, TUBB2B, TUBB3, and TUBA8. Mutations of the TUBB2B are known to cause asymmetric polymicrogyria (73; 158). The association of TUBA1A mutation was reported to cause eye abnormalities polymicrogyria, agenesis of the corpus callosum, microcephaly, and refractory epilepsy (111). Extensive bilateral polymicrogyria also is seen with de novo mutations in the gene GRIN1, which encodes GluN1, the essential subunit of the N-methyl-D-aspartate (NMDA) receptor (57). Mutation of the LAMC3 gene also causes generalized polymicrogyria (176).
Mutations of the PIK3CA and other PI3K-AKT-mTOR pathway genes have been identified in several overgrowth syndromes associated with polymicrogyria, such as hemimegalencephaly (41; 107; 158; 159). Pathogenic variants in PTEN also can be associated with polymicrogyria (150).
Other genes found to be associated with polymicrogyria include mutations of the WDR62 (WD Repeat Domain 62) gene, as well as mutations of the EOMES (Eomesodermin) gene, which encodes a transcription factor that influences regional gene expression and is involved in implementing regional identity in the cortex. Finally, mutations of the LAMC3 (laminin, gamma 3) gene are associated with occipital polymicrogyria. ATP1A3 variants were found in 124 patients with polymicrogyria by whole-exome sequencing (108).
The autosomal dominantly inherited BICD2 gene is associated with a form of congenital-onset spinal muscular atrophy and arthrogryposis multiplex congenita, but the phenotype is now expanded to also include microcephaly, bilateral perisylvian polymicrogyria, micrognathia, and respiratory insufficiency in the neonatal period (132).
Pallister-Killian syndrome, a rare disorder caused by mosaic tetrasomy of chromosome 12p and characterized by facial dysmorphism, hypotonia, developmental delay, and epilepsy, also exhibits polymicrogyria and may include cerebral calcifications (69).
Given the diversity of genes found to be involved in the etiology of this condition, the availability of next-generation sequencing could make the detection of multiple genes that cause polymicrogyria more cost effective (48).
Laboratory. There is no specific biochemical testing for the diagnosis of polymicrogyria; however, very long chain fatty acids should be obtained if there is a strong suspicion of Zellweger syndrome (67).
Montenegro and colleagues reported a lower frequency of epilepsy in patients with polymicrogyria compared to patients with other malformations of cortical development, and they reported that seizures in polymicrogyric patients are more easily controlled with antiepileptic drugs (109).
Surgical resection of refractory epileptic foci or hemispherectomy may provide good seizure control and developmental outcome in children with polymicrogyria (86; 170; 100). Using diffusion tensor imaging and fiber tractography, Isik and colleagues were able to evaluate white-matter tracts in vivo, thus, localizing the epileptogenic foci surrounding the polymicrogyric cortex. They report a case of a child who had a 90% reduction in seizures 12 months postoperatively (72). In some cases of diffuse bilateral polymicrogyria with intractable epilepsy, total corpus callosotomy is effective (08). However, some authors report that complete removal of polymicrogyric cortex identified on MRI does not always result in seizure control (07).
Intrauterine infections, such as cytomegalovirus and toxoplasmosis, remain significant risk factors for polymicrogyria (74). A fetus identified to be affected with parvovirus B19 was found to have polymicrogyria on a prenatal MRI (37).
Intrauterine hypoxia and ischemia were found in pathological studies of fetuses with polymicrogyria and skeletal muscle delay, which suggested the pathogenesis was due to an intrauterine hypoxic ischemic event (141). Correlation of prenatal and post-natal MRI have also demonstrated the association between prenatal stroke and polymicrogyria. Amniotic bands in rare cases of polymicrogyria also suggest an ischemic etiology (54).
Despite modern preventive obstetric care, lack of prenatal care, alcohol use, and young maternal age remain significant risk factors for polymicrogyria and schizencephaly (43).
Unsuspected congenital perisylvian polymicrogyria was discovered because of postpartum seizures with preeclampsia (02).
There is no contraindication for anesthetic administration in patients with polymicrogyria. Potential autonomic dysfunction can be anticipated in patients having epilepsy surgery involving the insular region. Careful monitoring in the intensive care unit should prevent complications such as hypotension, hypertension, tachycardia, nausea, and vomiting (153). Physiologic differences in the pediatric population, such as lower mean arterial pressure and lower autoregulatory reserves, should be carefully evaluated when administering anesthesia (155). Otherwise, precautions should be taken as with any patient with a seizure disorder, severe intellectual disability, or metabolic disease.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026