Developmental Malformations
Barth syndrome
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Prader-Willi syndrome is a sporadic condition characterized by neonatal hypotonia, hypogonadism, and obesity. Small hands and feet, characteristic facies, developmental delays, growth hormone deficiency, and behavior problems are strongly associated with Prader-Willi syndrome. The explanation of how both Prader-Willi syndrome and Angelman syndrome could share the same deletion in chromosome 15q11-q13 established these disorders as human models of genomic imprinting and greatly enhanced our understanding of genetic diseases. Management of Prader-Willi syndrome requires a multidisciplinary team approach, including hormone therapies in combination with dietary, developmental, and behavioral interventions. There is no cure for Prader-Willi syndrome; however, clinical outcomes can be improved through targeted therapeutic interventions. Advances in our understanding of the complex genetic and molecular mechanisms underlying the pathophysiology of this condition may lead to new treatment options.
|
• Prader-Willi syndrome is a neurodevelopmental disorder characterized by infantile hypotonia, hypogonadism, obesity with hyperphagia, and characteristic facies. | |
|
• Prader-Willi syndrome is the most common genetic cause of severe obesity. | |
|
• Prader-Willi syndrome is an example of a genetic imprinting disorder resulting from lack of function in paternally inherited genes located at 15q11-q13. | |
|
• Management of Prader-Willi syndrome requires dietary, hormonal, developmental, and behavioral interventions. |
The first patient with Prader-Willi syndrome was described by John Langdon Down (who also described Down syndrome) in 1864. Prader, Labhart, and Willi published the first case series, describing nine children with infantile hypotonia, intellectual disability, hyperphagia with obesity, hypogonadism, and characteristic dysmorphic features in 1956 (137).
A quarter century later, the demonstration of a deletion in chromosome 15q11-q13 in 60% of patients with Prader-Willi syndrome suggested an etiology for this condition (99). However, the identification of patients without this deletion and the fact that Angelman syndrome, a distinct condition, shared the same deleted region (107) confused the issue.
In the mid 1980s, this dilemma was reconciled by the discovery of genomic imprinting–the concept that certain genes can be expressed differently depending on their parent of origin. Prader-Willi syndrome and Angelman syndrome became the first disorders identified as being caused by imprinting and have since been established as human models of the role of genomic imprinting in human disease (129). Whether or not a given patient has Prader-Willi syndrome or Angelman syndrome depends on the sex of the parent with whom the deletion originates or on uniparental disomy, the inheritance of two copies of a gene from one parent. The differential expression of alleles in the 15q11-q13 region is the result of modification of maternal and paternal contributions to the zygote during gametogenesis (130; 78).
|
• Prader-Willi syndrome has a characteristic clinical presentation consisting of severe neonatal hypotonia and feeding difficulties followed by insatiable hyperphagia and obesity, developmental delay, and behavioral disturbances after the age of 1. | |
|
• Neuroendocrinological abnormalities secondary to hypothalamic dysfunction are prominent, including hyperphagia, growth hormone deficiency, hypothyroidism, central adrenal insufficiency, and sleep disturbances. |
Prader-Willi syndrome is a sporadic condition characterized by neonatal hypotonia, hypogonadism, hyperphagia, obesity if diet is not restricted, developmental delay, abnormal behaviors, growth hormone deficiency, characteristic facies, and a cytogenetic or molecular abnormality of the long arm of chromosome 15. It is the most common genetic cause of marked obesity and obesity-related mortality (20). Historically, the course of Prader-Willi syndrome has been divided into a neonatal stage characterized by hypotonia and feeding difficulties, followed by an early childhood-onset stage of hyperphagia, obesity, and developmental delay that persists into adulthood. However, Prader-Willi syndrome is more complicated, encompassing a wide range of clinical manifestations that show variable expression on an individual level and in accordance with genetic subtype.
Prenatal period. Advanced maternal age is the only established risk factor for Prader-Willi syndrome (116). Prenatal characteristics include decreased fetal movements (reported in 82% to 89% of pregnancies), polyhydramnios (34% to 42%), small for gestational age (65%), asymmetric uterine growth (43%), preterm birth (15% to 31% of pregnancies), and high rates of Caesarian section (42% to 82%) (73; 69; 185). Birth asphyxia, in which newborns required assistive breathing, occurred in 23% to 32% of deliveries (177; 185).
Infancy. Prader-Willi syndrome commonly presents in the newborn period with profound hypotonia, which is central in origin. The infant is floppy with little spontaneous movement and a weak cry; dyskinetic movements are sometimes noted (114). Sucking and feeding difficulties have been reported in 96% to 99% of newborns, and 50% to 70% of children require tube feedings because of poor weight gain (ie, failure to thrive) (69; 185). Some infants have severe central hypoventilation and require mechanical ventilation for days to weeks. Many infants with Prader-Willi syndrome have swallowing dysfunction, including silent aspiration events. Hypogonadism is common in both sexes, presenting as genital hypoplasia in girls and micropenis and cryptorchidism in boys.
Starting around 9 months, infants begin to feed and grow more appropriately. The hypotonia starts to slowly improve between 9 to 12 months of age, though it typically never completely resolves. Delays in motor and language abilities are apparent by 6 to 18 months of age.
Early childhood. This period is characterized by weight gain starting at about 2 years of age. By 4.5 years, children show increased appetite and will become obese without intervention. Developmental delays persist, and cognitive impairments become evident. Characteristic behavioral challenges emerge, including anxiety, temper tantrums or outbursts, rigidity, and obsessive-compulsive behaviors (157). Most patients with Prader-Willi syndrome have growth hormone deficiency (52), which may cause short stature in early childhood if untreated.
Mid-childhood to adulthood. Hyperphagia with persistent uncontrolled hunger manifests at a median age of 8 years (119). Food- and non-food-related behavioral challenges escalate. Food foraging and hoarding, theft of food, pica, and other behaviors indicative of food obsession develop. Most patients have mild to moderate intellectual disabilities, multiple learning disorders, and academic difficulties (179). Endocrine abnormalities are common. Patients who are not treated with growth hormone will develop short stature in their second decade if they haven’t already done so. Delayed puberty is common for males and females. Other endocrine abnormalities, including hypothyroidism and type II diabetes mellitus, can occur in adulthood. Adults are at risk for developing psychiatric illnesses, including mood disorders and psychosis (157). Patients with Prader-Willi syndrome typically have a reduced lifespan.
Physical features. Physical features include dolichocephaly in infancy, small bifrontal diameter, a narrow face, almond-shaped eyes, strabismus, a narrow nasal bridge, and a small-appearing mouth with a thin upper lip and downturned corners (116). The distinct facial features of Prader-Willi syndrome may be attenuated by early treatment with growth hormone therapy. Thick saliva, crusted at the corners of the mouth, has been noted. Hypopigmentation with fair skin and hair is often seen in individuals with deletions. Small hands and feet are frequently associated with Prader-Willi syndrome but are not a universal feature. A typical metacarpophalangeal pattern profile has been noted (21). The hands are narrow with straight ulnar borders.
Obesity and hyperphagia. Classically, Prader-Willi syndrome has been divided into an infantile phase of poor weight gain and feeding followed by hyperphagia and obesity. More recent research has revealed a more subtle and complex progression of feeding behavior categorized into five distinct nutritional phases (and two additional subphases), which are summarized below in Table 1 (119). Of note, evidence supporting the existence of an adult phase involving restoration of satiety and normal appetite (phase 4) is weak and needs to be further validated (92).
|
Phase |
Age |
Description |
|
0 |
Prenatal |
Decreased fetal movements, intrauterine growth restriction |
|
1a |
0 to 9 months |
Hypotonia, poor feeding |
|
1b |
9 to 25 months |
Improved feeding, appropriate growth |
|
2a |
2.1 to 4.5 years |
Weight gain without appetite increase |
|
2b |
4.5 to 8 years |
Increased appetite and interest in food |
|
3 |
8 years to adult |
Hyperphagia, lack of satiety |
|
4 |
Some but not all adults (usually > 30 years) |
Return to normal appetite |
Hyperphagia with persistent uncontrolled hunger combined with low muscle mass leading to and further exacerbated by decreased physical activity causes obesity. Nearly all patients manifest marked obesity without intervention. Obesity can be prevented and weight controlled through dietary restriction and modification. Growth hormone therapy may also help prevent obesity through improved growth and muscle mass, but it does not reduce hyperphagia.
Developmental and intellectual delay. Nearly all patients (90% to 100%) with Prader-Willi syndrome will have delays in motor, language, and cognitive development (53; 179; 58). Motor milestones take approximately twice as long to achieve, with independent sitting occurring at 13 months, crawling at 16 months, and walking at 28 months (17; 83).
Patients show a range of intellectual and adaptive abilities, with approximately 40% of individuals with Prader-Willi syndrome having mild intellectual disability and 20% having moderate intellectual disability (58). The average intelligence quotient is in the 60s. Most children have multiple severe learning disabilities (179). Individuals with Prader-Willi syndrome have an unusual skill with jigsaw puzzles (31).
Expressive and receptive language function is significantly impaired in children with Prader-Willi syndrome, out of proportion to verbal intelligence levels (53). Speech abnormalities (articulation, nasality, intonation pitch, and volume) are common, and speech apraxia has been reported in 7% to 10% of children with Prader-Willi syndrome (58).
Behavior. A behavioral phenotype for Prader-Willi syndrome has been described consisting of hyperphagia, temper outbursts, anxiety, obsessive compulsive behaviors, rigidity, and social cognition deficits (157). Behaviors escalate with age and BMI in childhood and adolescence (58).
Many individuals with Prader-Willi syndrome show autism-like traits, including rigid behaviors and deficits in ability to recognize facial expressions, interpret emotions, awareness of personal space, and understanding other people’s perspectives and mental state (theory of mind) (63; 157). The prevalence of autism spectrum disorder amongst individuals with Prader-Willi syndrome is reported to be between 12.3% to 26.7% (05; 63). Discrepancy comes from differences in methodology and application of autism diagnostic criteria. Autistic features are more prevalent in individuals with uniparental disomy rather than deletions (63). Attention-deficit/hyperactivity disorder is common and occurs in approximately 26% of children with Prader-Willi syndrome (182).
Self-injury, especially skin picking, is commonly reported (50% to 60%) and may be secondary to pain insensitivity (83; 58). Rectal picking may lead to rectal ulceration and bleeding, and if unrecognized, unnecessary testing and interventions (151).
Obsessive compulsive behaviors occur in 37% to 88% of individuals with Prader-Willi syndrome (61; 157; 160), but only a minority (8% to 15%) of patients meet DSM-5 criteria for obsessive compulsive disorder (157; 160). Obsessive compulsive symptoms in Prader-Willi syndrome (eg, food obsessions, repetitive questioning) differ from typical preoccupations seen in obsessive compulsive disorder (ie, germs, illness, cleanliness), and individuals with Prader-Willi syndrome typically lack insight into their behaviors (157).
High rates of anxiety (46%) and depressive (18%), bipolar (approximately 5%), and psychotic (11% to 15%) symptoms are reported in Prader-Willi syndrome (175; 160). Psychotic features are typically associated with an affective mood disorder or occur as recurrent, rapid-cycling psychotic episodes, with onset typically in adolescence or early adulthood. Episodes of psychosis may be preceded by transient, acute somatic symptoms such as pain, flu-like symptoms, diarrhea, or urinary frequency. Psychotic features and bipolar symptoms are much more likely to develop in cases with uniparental disomy than in those with deletions (184). Later in life, satisfactory control of symptoms is often established with antipsychotic medications for those who have developed cyclical psychotic episodes, and the tendency to have further psychotic episodes markedly declines (98).
Endocrine. Nearly all patients with Prader-Willi syndrome will have some degree of hypogonadism, which is thought to originate from both central hypothalamic dysfunction and primary gonadal deficiency. Hypogonadism is typically evident at birth with cryptorchidism (93% to 100%), small testes (76%), scrotal hypoplasia (69%), and micropenis (78% to 90%) commonly observed in boys and clitoris or labia hypoplasia seen in 76% of girls (41; 161; 131). Hypogonadism can aggravate muscle weakness, obesity, osteoporosis, and fatigue.
Puberty is spontaneous in both sexes, with normal age of pubertal onset followed by pubertal arrest around 13 years of age in males (64; 161) or delayed but near complete sexual maturation in females (161). Adult males have reduced testicular volumes and low testosterone levels, and adult females often have low estradiol levels. Nearly all females show abnormal menstruation patterns. A minority (8% to 14%) of females with Prader-Willi syndrome achieve menarche, which occurs at an age range between 12 to 34 years and is typically followed by oligomenorrhea or secondary amenorrhea (161; 81). Precocious puberty can occur infrequently in males and females. No males have been reported to reproduce, but there is documentation of at least seven females with Prader-Willi syndrome reproducing (131).
The majority of patients with Prader-Willi syndrome show reduced growth hormone or insulin-like growth factor 1 (IGF-1) levels, or both. Prevalence studies suggest growth hormone deficiency is present in 75% to 85% of all children, and IGF-1 values are decreased in virtually all patients (52; 75). If untreated, growth hormone deficiency leads to short stature, which becomes manifest postnatally, and delayed growth of hands and feet. Short stature may not be a striking feature until the second decade when there is a failure of the pubertal growth spurt. Specific growth charts for infants and children with Prader-Willi syndrome, both treated and untreated with growth hormone, are available. Growth hormone deficiency has been shown to contribute to increased fat mass, poor muscle tone and strength, and reduced exercise tolerance (25).
Clinical hypothyroidism of central origin has also been reported in 24.4% of patients with Prader-Willi (52). Type II diabetes is reported in 25% to 35% of individuals with Prader-Willi syndrome, with average onset at 20 years of age or younger (16; 32).
Individuals with Prader-Willi syndrome may be at increased risk of adrenal insufficiency due to hypothalamic dysfunction, but the incidence of adrenal insufficiency is unknown. Multiple studies using different pharmacologic stimulation and assaying methods have reported discordant results, with some studies finding no significant increased risk compared to controls, whereas a number of studies report higher rates of adrenal insufficiency amongst both children and adults with Prader-Willi syndrome (96). Individuals with Prader-Willi syndrome should be closely monitored for signs of adrenal insufficiency.
Epilepsy. In comparison with Angelman syndrome, unprovoked epileptic seizures are relatively uncommon in Prader-Willi syndrome, but, nonetheless, are seen far more frequently than in the general population. Reported incidence rates have varied from 4% to 26% (67; 172; 68; 167; 160). Seizure patterns may be generalized or focal. Reported series have varied widely with respect to relative frequencies of generalized versus focal seizure patterns. It is likely that the ratio of generalized to focal seizure patterns in one of the larger cohort studies (n=38; 55.2% generalized, 44.8% focal) comes closer to representing a more accurate estimate for this disorder (173). In the series by Fan and colleagues, six of the 10 epileptic patients had generalized seizures (67). In contrast, focal-onset seizures predominated in the series published by Vendrame and colleagues, Takeshita and colleagues, and Gilboa and Gross-Tsur: 92%, 77.8%, and 80%, respectively (172; 68; 167). Epileptic seizures are seen more often in deletion patients than in those with uniparental disomy (67; 172; 68; 160). Febrile seizures also occur more frequently than in the general population (170; 172; 167; 173).
Sleep disorders. Excessive daytime sleepiness is common in Prader-Willi syndrome, with prevalence rates reported to be as high as 55% (160). Some patients have sleep-disordered breathing associated with severe obesity, whereas others have narcoleptic features with sleep-onset REM sleep. In the majority of individuals, sleep studies suggest that the hypersomnolence results from primary hypothalamic dysfunction (109). Central and obstructive sleep apnea are frequently encountered in persons with Prader-Will syndrome, occurring in over half (33; 136). Central apnea is more common in younger patients, whereas obstructive apnea is more common in older children (33). Although obesity undoubtedly plays a role in the pathogenesis of obstructive sleep apnea, overall, there is a poor correlation between obstructive sleep apnea and body mass index (136).
Orthopedic features. A variety of orthopedic problems have been reported; the most prominent are scoliosis (40% to 80%), kyphosis, flat feet, knock knees, and hip dysplasia (20% to 30%) (176; 58). Osteoporosis at all ages has been reported and can result in fractures following minor trauma (83). Bone mineral density is typically reduced, apparently due to increased bone turnover (174).
Additional clinical features. Individuals with Prader-Willi syndrome show reduced sensitivity to pain (83; 139). Temperature dysregulation, including temperature instability, hyperthermia, hypothermia, and cyclic asymptomatic temperature elevation (153), have been reported in individuals with Prader Will syndrome from infancy to adulthood. Although typically benign, temperature dysregulation can become life-threatening if not recognized. Delayed gastric emptying is very common (60% to 80%), whereas vomiting is typically reduced (58). This combination of reduced gastric motility and decreased vomiting, especially in the setting of hyperphagia, leads to an increased risk of gastric necrosis and perforation, which can be lethal and may be challenging to recognize in individuals with pain insensitivity and cognitive impairments. Asymptomatic dysphagia, including disordered swallowing with lack of attempt to cough or clear residue, is also common, leading to an increased risk of aspiration events (74).
|
• The average life expectancy for individuals with Prader-Willi syndrome is 29.4 years. |
Overall mortality rates in children and young adults are much higher in Prader-Willi syndrome compared to the general population or controls with intellectual disability. Butler and colleagues report that the average life expectancy for individuals with Prader-Willi syndrome is 29.4 years, with a 20% mortality rate by 20 years of age, 50% by 29 years of age, 75% by 42 years of age, and 99% by 60 years of age (20). Death in childhood was more likely due to respiratory failure or aspiration; in adults the most common causes of death were respiratory failure, cardiac disease, pulmonary thromboembolism, accidents, sepsis, and gastric causes (20; 133). Males with Prader-Willi syndrome are more likely to die of presumed hyperphagia-related accidents or cardiopulmonary factors than females (140).
Obesity is the major contributing factor to the morbidity and mortality of this condition, and there is a clear relationship between the degree of obesity and the frequency of medical complications, which may correlate with mortality (140). Deceased patients with Prader-Willi syndrome have greater BMI and more reported obesity-related comorbidities, including type 2 diabetes, cardiac problems, respiratory problems, and sleep apnea, compared to living individuals with Prader-Willi syndrome (140). Mortality due to cardiovascular and gastrointestinal-related causes, but not respiratory causes, has decreased since 2000, which is attributed to earlier diagnosis and treatment, including strategies to prevent obesity (112).
A significant number of adults (86%) continue to have behavior problems, with 23% requiring psychiatric hospitalization and 45% requiring medication for behavioral or psychiatric problems (27; 60). Most adults with this condition require some supervision.
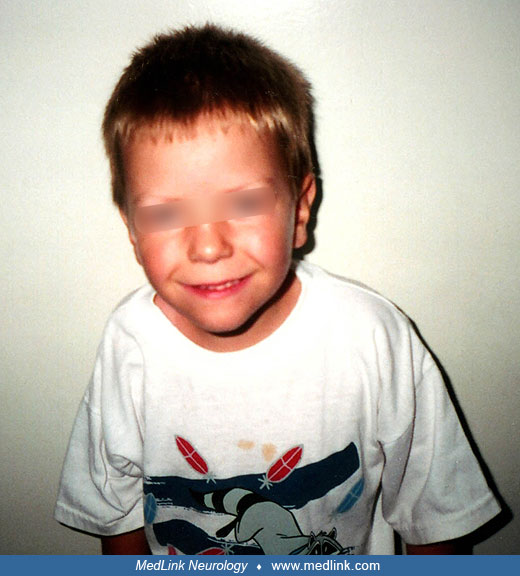
The patient, a boy aged 9, was the product of a term gestation that was uncomplicated except for the detection of oligohydramnios at 36 weeks. Cardiac decelerations occurred during labor, prompting an emergency cesarean section. Birth weight was 2.87 kg with Apgar scores of 6 and 7 at one minute and 5 minutes, respectively. The baby was given oxygen but required no further resuscitative efforts. He was noted to be markedly hypotonic and fed poorly. There was no muscle weakness or wasting, and tendon reflexes were brisk. The testes were undescended, and the scrotum was underdeveloped; otherwise, there were no obvious dysmorphic features.
Extensive blood work (including CBC, electrolytes, glucose, calcium, magnesium, creatinine, ALT, AST, ammonia, lactate, venous gases, and creatine kinase) was normal. LDH levels were increased at 1549U/L. A urine metabolic screen was negative, as was an organic acid spectrographic screen. Cranial imaging (head ultrasound and CT) was normal.
Family history for neonatal hypotonia, neuromuscular disease, and developmental delay was negative.
The baby’s feeding ability gradually improved to the point of allowing him to be discharged from the hospital. The working diagnosis at that time was central hypotonia of undetermined etiology.
At 8 months of age, the patient was reassessed at the Neurology Clinic. His development was delayed; he was unable to roll over, sit unaided, or crawl, and he had no interest in helping with feeding. Examination showed height, weight, and head circumference below the third percentile, whereas all had been previously normal. The boy was noted to have almond-shaped eyes and a downturned mouth. As before, he showed marked hypotonia with a head lag but normal limb movement against gravity and intact tendon reflexes.
The diagnosis of Prader-Willi syndrome was suspected on clinical grounds and was confirmed by the detection of a microdeletion at 15q11-q13, using methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) across the 15q11-q13 site.
The patient was referred to a developmental treatment program and had a carefully supervised diet. At the age of 6 years, he was significantly delayed from language and cognitive standpoints, functioning, in general, at about a 3- to 3.5-year-old level. Although his height was in the 98th percentile at 4 years of age, there was a subsequent gradual deceleration in growth velocity. At 8.5 years of age, his height and weight are in the region of the second percentile.
|
• Prader-Willi syndrome is caused by loss of expression of paternal genes in the 15q11-13 locus due to either deletions affecting the paternal chromosome, uniparental disomy, or imprinting errors. | |
|
• The type of mutation and size of deletion influences phenotypic expression of symptoms. | |
|
• The SNORD116 gene cluster appears to be critical to the pathogenesis of Prader-Willi syndrome. | |
|
• Hypothalamic dysfunction is implicated in many of the core endocrinologic and behavioral symptoms seen in Prader-Willi syndrome. |
The etiology of this condition is the lack of a paternally inherited gene or genes in the 15q11-13 locus. This occurs most commonly by deletion of the paternal copy (accounting for 60% of cases) but can also occur via maternal uniparental disomy (36% of cases) whereby both copies of the region are present but are of maternal origin and inactivated (19). A small number of patients (4%) have imprinting center microdeletions or epimutations (19). Rarely, other chromosome 15 abnormalities, including translocations, will cause Prader-Willi syndrome. In several reports, the relative percentages of patients having uniparental disomy has significantly increased relative to those with deletions.
Gene imprinting. The phenomenon of gene imprinting appears to be central to the biological mechanisms involved in the pathogenesis of Prader-Willi syndrome. Imprinted genes are active in each chromosome derived from one parent and inactive in the homologous chromosome from the other parent. This phenomenon appears to be common in genes involved in growth and development and has conferred an evolutionary advantage across species (130).
Imprinted genes are “marked” by methylation of DNA cytosine within the gene, changes in the configuration of chromatin, or histone modifications, which control transcription factor access to the DNA. Groups of imprinted genes are clustered in domains, and each domain is regulated by an independent cis-acting imprinting control region (ICR). The methylation state of the ICR determines the activity state of the genes: unmethylated ICRs activate the expression of genes within its domain, whereas genes in domains controlled by methylated ICRs are inactive. The mechanisms by which unmethylated ICRs control gene activity remain largely unknown. The proximal segment of the long arm of chromosome 15 harbors a number of imprinted genes and is the site of the deletions common to Prader-Willi and Angelman syndromes (78).
Genetic findings in Prader-Willi syndrome. Within the 15q11-q13 segment implicated in Prader-Willi and Angelman syndromes, there are a series of genes that are active in the chromosome derived from the father and inactive (methylated) in the maternally derived chromosome: MKRN3, MAGEL2, NECDIN (NDN), C15orf2, SNURF/SNRPN, and a group of small nucleolar RNA genes (snoRNAs) (130; 78; 09; 88). All of these genes are potentially involved in the pathogenesis of Prader-Willi syndrome. Just downstream from the snoRNAs are at least two genes (UBE3A and ATP10C) that are active in the maternally derived chromosome and inactive in the chromosome from the father. Mutations in the UBE3A gene have been shown to be associated with Angelman syndrome, a distinct disorder characterized by developmental delay, microcephaly, and seizures.
Prader-Willi syndrome results from the absence of activity of the paternally imprinted genes in the 15q11-q13 segment of the affected individual. This lack of active (unmethylated) genes occurs most frequently (approximately 60% to 70% of cases) due to a deletion in the 15q11-q13 region in the chromosome derived from the father. The maternally derived chromosome is intact, but the implicated genes have appeared to be completely inactive (methylated); however, this inactivation process is not always 100%. Similar sized deletions occur in both Prader-Willi and Angelman syndromes and are thought to be caused by low copy tandem repeats flanking the common break points, which are susceptible to recombination during meiosis (105).
There are five major breakpoints in the 15q11-q13 region (BP1-5). Most deletions in Prader-Willi syndrome occur between BP2-3 (type 2 deletion), whereas most of the remainder are between BP1-3 (type 1 deletion). In general, patients with type 1 deletions are more compromised than those with type 2. This difference may reflect absence of function in one or more unimprinted genes located between BP1 and BP2 (TUBGCP5, CYFIP1, NIPA1, NIPA2). Deletions confined to this region have been associated with intellectual impairment, speech and language delays, and autism spectrum disorder (23). Deletions that are smaller or larger than type 1 or type 2 deletions are termed “atypical deletions” and occur in 7% to 9% of those with deletions (89; 19). Larger deletions, especially BP2-BP5, include non-imprinted genes such as CHRNA7 (implicated in the so-called 15q13.3 microdeletion syndrome) and are associated with dysmorphic features not seen in most patients with Prader-Willi syndrome (89).
Of those patients with Prader-Willi lacking obvious deletions, the majority (approximately 36% of the total) have uniparental disomy, with both chromosome 15s derived from the mother (76; 19). This phenomenon is believed to occur most often from a combination of chromosome 15 nondisjunction in the egg and the subsequent loss of the paternal copy of chromosome 15 in the embryo, in which case the implicated genes are inactive in both chromosomes. If the nondisjunction event occurs during stage 1 meiosis, the egg will carry two nonidentical maternal chromosomes (ie, originating from each maternal grandparent), leading to uniparental heterodisomy. If the nondisjunction event occurs during stage 2 meiosis, the egg will carry two copies of the same maternal chromosome resulting in uniparental isodisomy. Uniparental isodisomy can be complete (accounting for 13% of disomy cases) or segmented (58% of disomy cases) and can be detected based on loss of heterozygosity over the 15q11-q13 region (19). Heterodisomy accounts for 30% of maternal disomy cases or 11% of all Prader-Willi cases (19).
In Western populations, the proportion of uniparental disomy cases in Prader-Willi syndrome appears to be increasing (178; 162; 19.) Possible explanations for this include advanced maternal age, which is thought to be a risk factor for nondisjunction events (147; 19) or a delay in the diagnosis of milder (ie, uniparental disomy) cases until later in life (69).
The third genetic mechanism for Prader-Willi syndrome is an imprinting center defect that is identified in the remaining 4% of patients. Some (< 0.5% of total patients) have been found to have microdeletions in the “bipartite imprinting center,” a region adjacent to the SNURF/SNRPN gene, with two components: AS-IC (Angelman syndrome imprinting center), located just upstream from SNURF/SNRPN, and PWS-IC (Prader-Willi syndrome imprinting center), which is embedded within exon1 of SNRPN (142; 58). The normal function of PWS-IC is to unmethylate the inactive genes on the 15th chromosome derived from the mother during gametogenesis in the male and to methylate (render inactive) the maternal UBE3A and ATP10C genes. Conversely, during oogenesis, the AS-IC inactivates the relevant genes on the paternally derived chromosome 15 and activates UBE3A and ATP10C (13). This mechanism allows for any of the 15th chromosomes in the gametes to function in the eventual zygote as appropriate male-derived or female-derived chromosomes, the ultimate raison-d’etre for the imprinting process. A deletion in the imprinting center of the maternally derived chromosome 15 in the sperm leads to a failure of conversion to the “male pattern” and ultimately results in 50% of the male’s offspring having two sets of inactive genes: the normally inactive genes from the mother and the nonactivated genes from the father. All these individuals have Prader-Willi syndrome and represent the only exception to the rule that Prader-Willi syndrome is normally a sporadic disorder (13). Imprinting defects can also be caused by epimutations, ie, an inherited change in gene expression not associated with DNA mutation. These individuals have maternal-only DNA methylation patterns but no detectable mutations (58).
The remaining identified mechanism for Prader-Willi syndrome is a chromosome 15 translocation (09). Isolated case reports of balanced translocations through the 15q11-q13 region, typically with no identifiable deletion, and one case report of an unbalanced translocation involving chromosomes 15 and 19 leading to a deletion in the 15q11-q13 locus have been published (42).
Possible roles of the imprinted genes. The individual roles of the inactivated paternally derived genes at the 15q11-q13 deletion site in the pathogenesis of Prader-Willi syndrome are far from clear. Although null mouse studies offer some insights into potential molecular mechanisms contributing to the somatic, behavioral, and metabolic symptoms of Prader-Willi syndrome, no mouse models are able to fully replicate the human Prader-Willi phenotype (145). Of the genes located within the Prader-Willi region, the SNORD116 cluster appears to be critically implicated in the pathogenesis of Prader-Willi syndrome; other genes may contribute to, but cannot fully account for, the Prader-Willi phenotype.
Individuals with microdeletions confined to the snoRNAs site in the 15q11-q13 region, particularly the SNORD116 cluster, show typical features of Prader-Willi syndrome (150; 59; 07). The SNORD116 gene locus produces a primary RNA transcript that is processed into two noncoding RNAs: Snord116 snoRNAs, which localize to the nucleolus of mature neurons, and the Snord116 host gene (37). Most snoRNAs function by methylating target rRNA or snRNA molecules; however, SNORD116 and SNORD115 snoRNAs have no identified rRNA targets. Instead, they may undergo further processing into smaller snoRNA molecules (psnoRNAs), which may perform a variety of regulatory functions (eg, splicing factors, activating proteins, stabilizing RNA) (66). In collaboration with SNORD115, the SNORD116 gene promotes production of at least 23 other genes, suggesting the presence of Prader-Will syndrome symptoms in individuals with deletions confined to the SNORD116 cluster may effectively result from downregulation of a number of other genes whose products are essential for normal brain development and function (65).
Snord116-null mice recapitulate the main features of Prader-Willi syndrome including low birth weight, hyperphagia, obesity, and endocrine abnormalities (141). The SNORD116 gene itself is expressed ubiquitously in mice but splicing and processing of the Snord116 transcript into snoRNAs only occurs in brain tissues (37). Using a selective SNORD116 knockout mouse model, Qi and colleagues demonstrated that elimination of Snord116 expression from neuropeptide Y cells of the arcuate nucleus in the hypothalamus, but not other brain tissues, reproduced the murine Prader-Willi condition, suggesting that Snord116 plays an important role in food and hunger control through regulation of neuropeptide Y neurons in the hypothalamus (141). Using a transgenic mouse model, Coulson and colleagues discovered that adding an exogenous Snord116 transgene to Snord116 knockout mice was not sufficient to rescue these mice from the Prader-Willi phenotype; additional cellular machinery present in wild type but not Snord116 knockout mice is necessary to process SNORD116 into functional snoRNAs (37). In addition, researchers have shown dysregulation of diurnal methylation patterns in the cortex of Snord116 knockout mice, including altered expression of diurnally methylated genes involved in neurogenesis, circadian entrainment, and metabolic pathways implicated in obesity (38). This work may provide new mechanistic insights into epigenetic influences on circadian rhythms and how alterations in regulation of circadian rhythm contribute to manifestations of Prader-Willi syndrome including sleep disorders, hyperphagia, and metabolic/endocrine dysfunction.
The SNORD115 gene produces an RNA transcript that binds to and potentially promotes alternative splicing or editing of the 5-HT2C serotonin receptor mRNA, which is expressed predominantly in the brain (66). A mouse model of altered serotonin 2C receptor RNA editing demonstrates many features of human Prader-Willi syndrome: failure to thrive, decreased somatic growth, neonatal hypotonia, and initially poor food consumption followed by post-weaning hyperphagia (123). However, both patients and knockout mice with SNORD115 gene cluster deletions lack typical Prader-Willi syndrome features (54; 149). It remains unclear whether downstream regulation of 5-HT2C via Snord115 (or perhaps other unidentified factors or cofactors) plays any significant role in the manifestation of Prader-Willi syndrome.
Nectin plays a role in axonal outgrowth, possibly through interactions with other neuronal proteins including Magel2, which help regulate cytoskeleton organization (100). Nectin appears to regulate respiratory rate. Most necdin-deficient mice (-/-) die in the neonatal period from respiratory insufficiency akin to the respiratory problems seen in neonates with Prader-Willi syndrome. Heterozygous necdin-deficient mice with the deletion only on the paternal allele have less severe respiratory difficulties and are much more likely to survive to adulthood (146). In theory, this improved outcome in heterozygotes should not be happening as the maternal allele is presumably silenced. As demonstrated by Rieusset and colleagues, however, deletion of the paternal origin necdin allele in mice is accompanied by varying degrees of residual expression of necdin mRNA derived from the maternal allele. This finding also appears to apply to human Prader-Willi syndrome as these authors also found NECDIN mRNA expression in the paraventricular and supraoptic nuclei of the hypothalamus in eight postmortem brains of Prader-Willi patients, whether of the deletion or isodisomy subtypes.
The respiratory insufficiency in necdin-null mice appears to be related to rhythm instability in the pre-Botzinger complex, the respiratory rhythm-generating center (143). Necdin-deficient mice surviving to adult life have reduced numbers of oxytocin-producing and gonadotropin-releasing hormone neurons in the hypothalamus (125; 121). Clinically, they demonstrate frequent skin-scraping activities and superior spatial learning, both of which are seen in humans with Prader-Willi syndrome (125). The former behavior may be related to increased neuronal apoptosis in sensory ganglia and a paucity of substance P-containing neurons in necdin-deficient mice (97); in turn, these findings may be relevant to the elevated pain thresholds seen in Prader-Willi subjects (139).
The MAGEL2 gene encodes a protein expressed in multiple brain areas including the hypothalamus and appears to be involved in brain structure and development, reproductive function and fertility, and multiple homeostatic processes including circadian rhythm, appetite and weight gain, and bone metabolism (08; 115; 03). A cohort of 78 patients with truncating mutations confined to the paternal allele of MAGEL2 have been described, and although they share similar early symptoms with Prader-Willi patients, they lack many of the typical morphological features and do not develop hyperphagia and obesity in later childhood and are classified as a distinct syndrome, Schaaf-Yang syndrome (113). Autism spectrum disorder (ASD) has a much higher prevalence in patients with Schaaf-Yang syndrome, suggesting Magel2 may play a role in the development of autism spectrum disorder symptoms (113).
Magel2-null mice demonstrate different but complementary features to those seen in Prader-Willi syndrome: neonatal growth retardation, impaired reproductive function with infertility, altered bone homeostasis, changes in circadian rhythm, and dysregulation of feeding and weight leading to excessive weight gain after weaning and increased adiposity in adult life (08; 115; 03). With respect to regulation of food intake and weight gain, Magel2-null mice more closely follow a Prader-Willi trajectory than that of patients with Schaaf-Yang and, thus, have provided insights into the feeding impairments seen in Prader-Willi syndrome. Magel2-null mice show progressive leptin insensitivity and reduced volumes of anorexigenic axons in the arcuate nucleus of the hypothalamus, a crucial center for appetite control (138; 108).
MKRN3 encodes a protein (makorin ring finger protein 3) involved in the regulation of puberty and may play a role in the hypogonadism and infertility seen in Prader-Willi syndrome (35).
Kanber and colleagues describe two patients with atypical deletions in the Prader-Willi region on the paternal chromosome 15 that include C15orf2 and the SNURF-SNRPN locus (which encompasses the SNORD116 gene cluster), but not MKRN3, MAGEL2, and NDN genes (88). Both patients show classic Prader-Willi syndrome, indicating the MKRN3, MAGEL2, and NDN genes are not sufficient to cause Prader-Willi syndrome.
The proposed role of SNURF/SNRPN in Prader-Willi syndrome comes largely from human evidence, particularly the production of the classic Prader-Willi phenotype by the disruption of exon 2 and exon 3 of the SNURF component in balanced translocations (95). Contradictory evidence comes from mouse models in which neither snurf-deficient nor snrpn-deficient mice demonstrate features in common with Prader-Willi syndrome (125).
Finally, there are reports of cases with typical features of Prader-Willi syndrome related to microdeletions confined to the snoRNAs site in the 15q11-q13 region, particularly the SNORD116 cluster (150; 59). The snoRNAs include SNORD64 (one copy), SNORD107 (one copy), SNORD108 (one copy), SNORD109 (two copies), SNORD116 (29 copies), and SNORD115 (48 copies) (141). An in vitro study by Falaleeva and colleagues revealed that SNORD116, in collaboration with SNORD115, promotes the production of mRNAs from at least 23 other genes, suggesting that the presence of Prader-Will syndrome symptoms in individuals with deletions confined to the SNORD116 cluster may result from, effectively, downregulation of a number of other genes whose products are essential for normal brain development and function (65).
Snord116-null mice recapitulate key features of Prader-Willi syndrome including low birth weight, hyperphagia, and obesity (141). Interestingly, Qi and colleagues demonstrated that selective deletion of Snord116 from neuropeptide Y (NPY) neurons resulted in the same phenotype as the global deletion (141). In addition, researchers have shown dysregulation of diurnal methylation patterns in the cortex of Snord116 knockout mice, including altered expression of diurnally methylated genes involved in neurogenesis, circadian entertainment, and metabolic pathways implicated in obesity (38). This work may provide new mechanistic insights and understanding how genetic factors, through effects on methylation, contribute to manifestations of Prader-Willi syndrome including sleep disorders, hyperphagia, and metabolic/endocrine dysfunction.
Genotype-phenotype correlations. Larger deletions in the 15q11-q14 region have been associated with additional clinical manifestations. Deletion of the human P gene, downstream from UBE3A, results in a decrease in pigmentation. This phenomenon can be seen in both Prader-Willi and Angelman syndromes as the P gene is not imprinted (128). Such patients typically have central visual impairment, in part related to retinal hypopigmentation, as is seen in albinism (40). Inclusion in a deletion of one or more of the three nonimprinted GABA-receptor genes at the 15q11-q13 site may account for some of the cases of febrile seizure and epilepsy in Prader-Willi syndrome (67; 172). Windpassinger and colleagues described a 2-year-old boy with Prader-Willi syndrome caused by a large 15q11-q14 deletion resulting from an unbalanced translocation t(3; 15). In addition to depigmentation and central visual impairment, this patient also demonstrated relative macrocephaly, retrognathia, preauricular tags, and bilateral club feet, presumably related to the absence of other nonimprinted genes in the 15q13-q14 region (183).
There are cognitive and behavioral differences between individuals with uniparental disomy and those with 15q deletions: those with uniparental disomy are significantly more likely to develop psychotic and bipolar features, whereas individuals with 15q deletions have significantly lower full-scale IQ and verbal IQ (184) and are more likely to have speech apraxia (58).
Individuals with uniparental disomy have difficulty recognizing faces and emotional states (180; 63) and are twice as likely to meet DSM-5 criteria for autism spectrum disorder (05). These differences may be related to overexpression of UBE3A and ATP10C genes, which are significantly increased in disomy lines compared to controls and 15q11-q13 deletion cases (10).
Epigenetic considerations. Evidence of a second mechanism for the autistic features seen in Prader-Willi syndrome has emerged, a mechanism that does not directly involve the paternally imprinted genes in the 15q11-q13 region. Nagarajan and colleagues found that MeCP2 expression was significantly reduced not only in patients with Rett syndrome but also in subjects with Prader-Willi syndrome, Angelman syndrome, Down syndrome, and idiopathic autistic spectrum disorder (126). These results suggest the possibility that the autistic symptoms seen in Prader-Willi, Angelman, Down, and Rett syndromes may relate to MeCP2 deficiency, the mechanism in the non-Rett cases being an epigenetic phenomenon.
It is, thus, possible that other clinical features of Prader-Willi syndrome are secondary to relatively remote downstream effects on other genes or genetic mechanisms. A mouse model of altered serotonin 2C receptor RNA editing (a process mediated by snoRNA genes) demonstrates many features of human Prader-Willi syndrome: failure to thrive; decreased somatic growth; neonatal hypotonia; and initially poor food consumption followed by post-weaning hyperphagia (123).
The hypothalamus in Prader-Willi syndrome. From the clinical perspective, there are grounds for suspecting hypothalamic dysfunction in Prader-Willi syndrome. The hypogonadism is associated with low gonadotropin levels, low gonadal steroid levels, the presence of Sertoli cells, and the variable numbers of Leydig and germinal cells in the testes (17). Growth hormone secretion is low in both obese and nonobese patients and has shown diminished response to provocative tests (36). The temperature instability problems sometimes seen in the first stage of the disorder also presumably stem from hypothalamic dysfunction (153). Finally, the characteristic insatiable hyperphagia suggests a defect in the hypothalamic satiety center, a hypothesis supported by the findings of multiple studies involving Prader-Willi mouse models (44; 108; 141). Interestingly, excess food consumption in these mice does not appear to be determined by the sweetness of food items, but by their caloric content (44).
Neuropathological studies. Neuropathologic studies in Prader-Willi syndrome are few, and the results are inconsistent (79; 144). There are no characteristic gross or microscopic abnormalities in the hypothalamic region or elsewhere. Swaab reported a specific reduction in oxytocin-containing and vasopressin-containing neurons in the paraventricular nucleus in comparison with controls (165); oxytocin neurons are important in the regulation of food intake. A similar study of the infundibular nucleus by the same group showed that growth hormone-releasing hormone neurons were not reduced in Prader-Willi syndrome in comparison with obese controls (72).
Swaab’s observations concerning oxytocin neuron depletion in Prader-Willi syndrome are interesting given the accumulating evidence that oxytocin is an important neuromodulator in facilitating facial recognition of emotional states, peer recognition, and interpersonal bonding behavior (56; 55).
Down-regulation of oxytocinergic activity has, in fact, been demonstrated in Magel2-deficient mice (117). Interestingly, daily administration of subcutaneous oxytocin to these mice in the first postnatal week permanently protects the mice against the later evolution of deficits in social behavior and learning (117).
Hayashi and colleagues reported a pathological study of a deletional Prader-Willi patient who had a selective loss of cholinergic neurons in the pedunculopontine tegmental nucleus, a finding that could account for the patient’s hypotonia and REM sleep abnormalities (80). Cholinergic neurons in the nucleus basalis of Meynert, in contrast, were normal, as were GABAergic interneurons in the cerebral cortex and catecholaminergic neurons in the brainstem. These are intriguing findings that have yet to be replicated.
Postmortem transcriptional analysis of hypothalamic tissue in patients with Prader-Willi reported by Bochukova and colleagues identified increased expression of genes that signal hunger and proinflammatory genes (11). Upregulated genes were located predominantly in microglial cells. There was downregulation of neuronal genes, including genes that signal fed state and genes associated with neurogenesis, synaptic plasticity, and neurotransmitter release. Specifically, the authors noted decreased levels of oxytocin mRNA (supporting Swaab’s 1997 neuropathological study) and decreased levels of brain derived neurotrophic factor (BDNF) and its receptor, trkB (NTRK2), mutations to which have previously been associated with symptoms that overlap with the Prader-Willi phenotype. Addition of BDNF to a SNORD116 deficient human neuroblastoma cell line partially improved neuronal growth, suggesting decreased BDNF/NTRK2 expression may contribute to the neurodysgenesis or neurodegeneration associated with Prader-Willi syndrome (11).
MRI-based studies. MRI studies of subjects with Prader-Willi syndrome published to date are limited by small sample populations studied at different stages of brain maturation and by variations in imaging methodology and analysis. Nonetheless, there is a growing body of evidence showing a diffuse range of brain abnormalities in Prader-Willi syndrome.
Conventional structural MRI studies have identified a variety of abnormalities, including cortical atrophy, ventriculomegaly, cerebellar abnormalities, corpus callosum dysgenesis, incomplete closure of the insula, and pituitary hypoplasia (87). Similar findings are commonly seen in other genetic and neurodevelopmental conditions and are not specific to Prader-Willi syndrome.
Morphologic MRI studies have shown heterogenous results. Reduced brain volumes have been found in the orbitofrontal cortex (an area associated with the limbic dopamine system involving motivation and reward learning), hypothalamus (associated with homeostasis and hunger control), regions within the dorsolateral and medial prefrontal cortices (associated with inhibitory regulation and emotional control), and the posterior inferior lobule of the cerebellum (132; 87). In contrast, increased brain volumes and cortical thickness have been reported in grey matter diffusely, in sensorimotor and subcortical regions, and in dentate nuclei of the cerebellum (110; 87). Widespread reductions in the local gyrification index (a quantitative measure of cortical complexity comparing the ratio between the total surface area of the brain, including sulcal regions, to the surface perimeter area) have been reported in frontal, temporal, and parietal lobes (102; 24). Contradictory results regarding basal ganglia and anterior cingulate cortex volume changes have been published.
There is some evidence that changes in brain volumes may differ with age and stage of brain maturation as well as mutation subtype, with 15q deletion individuals appearing to show reductions in grey matter volume without signs of cortical atrophy, whereas those with the maternal disomy subtype have more severe early brain atrophy and increased cortical thickness (85; 104; 87).
This diffuse range of structural brain abnormalities and changes in cortical complexity suggest there may be early systemic dysregulation of neurodevelopmental processes leading to widespread alterations in brain structure, which provide the biological basis for the cognitive impairment, developmental delay, and other symptoms associated with Prader-Willi syndrome (110). Areas of increased cortical thickness may reflect impaired neuronal migration and reduced synaptic pruning, whereas areas of volume loss without cortical atrophy could be consistent with a degree of simplification or relative lack of complexity. More studies and new models, such as machine learning, are needed to clarify inconsistencies and better elucidate the nature and functional implications of grey matter volumetric differences in Prader-Willi syndrome.
To date, only a handful of small diffusion tensor MRI imaging studies have been published suggesting white matter dysfunction. Possible delays in axonal maturation or damaged white matter tracts may play a role in the pathogenicity of Prader-Willi syndrome; however, consistency across these studies is poor, and larger longitudinal studies are needed to better characterize white matter changes and their relationship to the Prader-Willi phenotype (87).
Functional MRI experiments examining eating behaviors in Prader-Willi syndrome report that individuals with Prader-Willi syndrome show hyperactivation in frontal (including prefrontal cortex) and limbic (including hypothalamus, amygdala, hippocampus, anterior cingulate cortex) regions associated with food motivation, both before and after meals, as well as functional dysconnectivity in neuronal networks associated with eating and reward in a resting state (158; 84; 103; 87). Preliminary data suggest possible functional connectivity differences between genetic subtypes. Individuals with 15q11-q13 deletions appear to show greater hyperactivation of the food motivation circuit pre-and post-meals, and individuals with maternal uniparental disomy appear to show hyperactivation of inhibitory control networks (dorsolateral cortex, parahippocampal gyrus) associated with cognitive control of food-seeking behaviors (84; 87). Although these fMRI results do not clearly localize the origin of hyperphagia in Prader-Willi syndrome, they do suggest a fundamental alteration in neural mechanisms of satiety.
Endocrinological aspects. There has been speculation that the hyperphagia in Prader-Willi syndrome might be linked to aberrations in levels of two closely related circulating peptides, ghrelin and obestatin. Ghrelin is a stomach-derived orexigenic compound that induces fat deposition by increasing food intake and decreasing fat utilization, particularly in conditions of starvation and cachexia. In contrast, obestatin decreases food intake and decelerates gastric emptying, reducing body weight. Both compounds are derived from the posttranslational cleavage of the same gene product, preproghrelin. In principle, either excessive levels of ghrelin or inadequate levels of obestatin might lead to hyperphagia and obesity.
In fact, plasma ghrelin levels are consistently elevated in older subjects with Prader-Willi syndrome in comparison with obese controls (50; 168; 18); ghrelin levels are also elevated in a mouse model of Prader-Will syndrome (44). Beauloye and colleagues showed that, in children and adults with Prader-Will syndrome, the elevated ghrelin levels are primarily in the acetylated form (04). In contrast, infants with the disorder have low levels of acetylated ghrelin and high levels of unacetylated ghrelin, suggesting that the latter form may be anorexigenic (04). Contrary to expectation, obestatin levels in Prader-Willi subjects were not found to differ from those in obese controls, regardless of age, either in the fasting state or following a glucose load (18; 134).
These somewhat conflicting findings suggest that ghrelin may not be the direct cause of hyperphagia in Prader-Will syndrome but that its elevated plasma concentrations may be secondary to a more fundamental defect that has not yet been identified. This conclusion is supported by a controlled trial of octreotide, a potent suppressor of ghrelin production. As expected, Prader-Willi subjects taking octreotide had a significant and sustained reduction in ghrelin levels; there were no significant changes, however, in food-seeking behaviors and body mass index (51).
Hyperphagia in Prader-Willi syndrome has also been linked to low circulating levels of brain-derived neurotrophic factor (BDNF) and to a failure of BDNF blood levels to rise following a glucose load (77; 12). BDNF knockout mice also demonstrate hyperphagia and obesity (77). Bueno and colleagues also demonstrated that plasma leptin levels are elevated in Prader-Willi syndrome, as they are in obese typical adults (12). Leptin is a peptide produced by adipocytes, and it normally stimulates anorexigenic neurons in the hypothalamus. Again, these BDNF and leptin results may be secondary to a more fundamental hypothalamic disturbance (12).
Also, indicative of a fundamental disturbance in hypothalamic regulation in Prader-Willi syndrome is the finding that orexin A levels are also elevated in this disorder (111). Orexin A (hypocretin1) is involved in arousal, regulation of the sleep/wake cycle, metabolism and energy expenditure, and reward seeking behavior (111). Finally, there is also a suggestion that the obesity in Prader-Willi syndrome may not result exclusively from overeating. Those patients who manage to lose weight require lower caloric intakes, approximately 60% of the norm for their age, to maintain reduced weight (17). Energy expenditure appears to be below normal, but studies measuring metabolic rates have been inconclusive. Thyroid hormone, lipid profiles, insulin, and glucocorticoid levels are comparable to those of obese individuals (17). Fat cells are larger than in controls, but the number of fat cells and the uptake of fat and fatty acid composition are not increased (06; 17).
An alternative mechanism for fat production in Prader-Willi syndrome was suggested in a study of dispersed cell cultures of pre-adipocytes (15). Prompted by extensive previous work in Wevrick’s laboratory on the necdin null mouse, these authors found that necdin overexpression inhibited the conversion of pre-adipocytes to mature fat cells, whereas reduced necdin levels in tissue culture enhanced this conversion.
Dysregulation of the growth hormone/insulin-like growth factor 1 axis is prominent in most patients with Prader-Willi syndrome. Pituitary growth hormone reserve levels decrease with age, which may, at least in part, contribute to the changes in growth velocity observed in older children and adolescents (75).
|
• Prader-Willi syndrome affects approximately 1 in 10,000 to 1 in 30,000 individuals in North America. | |
|
• Both sexes are affected equally. |
Prader-Willi syndrome has an estimated prevalence of 1 in 10,000 to 30,000. There are no differences in prevalence between sexes or ethnicities (58). Prevalence rates vary by study and location. Large-scale epidemiological surveys have reported a prevalence of 1 per 16,602 in North Dakota (14) and 1 per 8000 in rural Sweden (01). A population-based newborn screening study in Australia identified a birth prevalence of 1 in 8290 (70). Prevalence in the United Kingdom has been estimated at 1 in 45,000 to 1 in 52,000 (181).
• Prader-Willi syndrome is considered a sporadic genetic syndrome with low recurrence rates. | |
• In rare cases in which inheritable genetic mutations are identified, genetic counseling can help families make informed decisions for future pregnancies. | |
• Prenatal testing to detect Prader-Willi syndrome can be conducted for certain high-risk pregnancies. |
A small recurrence risk (less than one) has been cited for families with one child with Prader-Willi syndrome (71). In order to ascertain the risk of recurrence, it is essential to characterize the molecular genetic basis for Prader-Willi syndrome in each patient. For those with deletions, uniparental disomy, and de novo balanced translocations, the recurrence risk is low. Where the proband is found to have an imprinting center mutation, however, the recurrence risk is 50% (13). In such situations, further cases can be prevented via prenatal diagnosis.
A second scenario exists in which accurate prenatal diagnosis can prevent the birth of a child with Prader-Willi syndrome. On occasion, chorionic villus sampling for advanced maternal age has revealed trisomy 15, whereas amniocentesis has revealed a fetus with 46 apparently normal chromosomes, a phenomenon referred to as confined placental mosaicism (26; 124). Such pregnancies may produce a child with Prader-Willi syndrome secondary to maternal disomy, with loss of the paternal chromosome 15 present in the trisomic chorionic villus cells. In pregnancies where trisomy 15 is identified on chorionic villus sampling, the fetal cells should be evaluated for the presence of maternal disomy for chromosome 15.
Routine second trimester karyotyping of amniocytes in high-risk mothers (for advanced maternal age, abnormal maternal serum screening, or abnormal fetal ultrasound findings) may reveal absence of the normal band at 15q12 (27 out of 26,041 samples) (28). This suggests the possibility of identifying both Prader-Willi and Angelman syndromes prior to the achievement of potential fetal viability; however, only a small fraction (less than 5%) of samples missing the 15q12 band subsequently tested positive via methylation analysis for Prader-Willi or Angelman syndromes (28).
The differential diagnosis varies depending on the age of presentation. For a newborn with hypotonia, it should include those disorders that can give rise to profound hypotonia and both central and peripheral causes. Hypoxic-ischemic encephalopathy, prematurity, intraventricular hemorrhage, congenital brain anomalies, spinal muscular atrophy, myasthenia gravis, myotonic dystrophy, congenital myopathies, Zellweger syndrome, other chromosomal defects, and other conditions must be considered.
Normal electromyograms and nerve conduction velocities, creatine phosphokinase levels, muscle biopsy, metabolic screens, and the absence of cerebral pathology on imaging studies are seen in Prader-Willi and will exclude most of these other conditions.
It is important to recognize that infants with Prader-Willi syndrome may not manifest the characteristic dysmorphic features of the disorder, including hypogonadism (122). For this reason, it is worthwhile to consider DNA methylation screening for all neonates with undiagnosed central hypotonia.
Patients with Schaaf-Yang syndrome, which is caused by truncating mutations to the MAGEL2 gene located within the paternally imprinted Prader-Willi 15q11-q13 locus, show similar early symptoms, including neonatal hypotonia, developmental delays, and poor feeding and growth, but patients typically do not develop hyperphagia and often lack the dysmorphic features associated with Prader-Willi syndrome. Schaaf-Yang syndrome was previously described as Prader-Willi-like syndrome; however, the characterization of this syndrome has established distinct clinical features justifying its classification as its own distinct syndrome (113).
In the older child, the differential diagnosis for the untreated syndrome will focus on other causes of marked obesity as well as developmental delay and intellectual disability. Laurence-Moon syndrome, Bardet-Biedl syndrome, Cohen syndrome, and Börjeson-Forssman-Lehman syndrome as other genetic causes of obesity should be considered, but these usually can be eliminated by history and physical features.
In addition, at least three other chromosomal deletion syndromes present with neonatal hypotonia, hyperphagia, obesity, mental deficiency, and dysmorphic features: 6q16.2 (SIM1 gene) (171), 1p36 (43), and 9q34 (34). As well, a Prader-Willi syndrome-like phenotype has been described in individuals with maternal uniparental disomy of chromosome 14 (86). These syndromes should be considered in patients with Prader-Willi-like features but lacking a 15q11 methylation defect.
Prader-Willi syndrome is associated with obesity; multiple endocrinologic disorders [growth hormone deficiency (40% to 100%), type 2 diabetes (25%), hypothyroidism (25%), central adrenal insufficiency, sleep disturbances]; orthopedic complications (scoliosis, kyphosis, hip dysplasia, osteoporosis); and increased rates of behavioral conditions, including autism spectrum disorder (12% to 27%), obsessive compulsive disorder, attention-deficit/hyperactivity disorder, and psychosis.
|
• Prader-Willi syndrome is diagnosed via genetic testing. |
The diagnosis of Prader-Willi syndrome is suspected based on clinical presentation and is confirmed via genetic analysis. As Prader-Willi syndrome can be caused by multiple genetic mechanisms, a combination of molecular genetic testing techniques is needed to confirm the diagnosis and identify the genetic subtype, which is essential for both clinical monitoring and prognostication and determining recurrence risks.
Currently, most testing paradigms start with DNA methylation analysis to screen for lack of the male methylation pattern in the paternal chromosome (or maternal-only imprinting) at the 15q11-13 site, followed by one or more confirmatory tests to identify the genetic mechanism. DNA methylation testing can be accomplished using either methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) across the 15q11-q13 site or methylation-specific polymerase chain reaction (MS-PCR) at the SNRPN region of the 15q11-q13 locus (166; 58). DNA methylation analysis can identify more than 99% of Prader-Willi syndrome cases but cannot differentiate subtypes (MS-MLPA can identify different deletions and often mosaicism but cannot distinguish between uniparental disomy and imprinting center defects, whereas MS-PCR fails to distinguish between any of the genetic mechanisms) (166). Thus, if a maternal-only methylation pattern is detected via methylation analysis (indicative of Prader-Willi syndrome), additional testing is needed to determine the genetic mechanism.
A variety of confirmatory testing strategies can be used to determine the genetic subtype, each with its own advantages and disadvantages:
Oligo-small nucleotide polymorphism (SNP) combination array (OSA). OSA uses oligonucleotide and SNP probes to detect deletions, chromosomal rearrangements, uniparental disomy due to isodisomy or segmental isodisomy, and most small imprinting center defects and can detect rare deletions in the SNORD116 cluster that are negative on MS-PCR methylation analysis (58). This technique can be done at the same time as methylation analysis and will identify the molecular cause in most (approximately 90%) individuals, except those with uniparental heterodisomy (accounting for 8% to 11% of Prader-Willi syndrome cases) or epigenetic imprinting defects (2% to 4%) (58). If methylation is abnormal but OSA testing is negative at the 15q11-q13 locus, DNA polymorphism testing (DNA sequencing) of proband and parents is recommended (58).
Chromosomal microarray analysis. Chromosomal microarray analysis is a combination of comparative genetic hybridization with SNP array. It offers a similar diagnostic yield as OSA testing and will screen the entire genome for copy number variant changes. By itself, it cannot differentiate Prader-Willi syndrome from Angelman syndrome (cannot determine methylation pattern), thus, chromosomal microarray testing is typically conducted sequentially following an abnormal DNA methylation test result. Chromosomal array testing cannot identify individuals with uniparental heterodisomy or imprinting defects. Additional testing, such as trio (proband and parents) DNA sequencing, would be necessary to detect these subtypes.
DNA methylation analysis. Traditionally, DNA methylation analysis has been followed by DNA fluorescence in situ hybridization (FISH) testing, which can detect deletions (covering approximately 60% of mutations) and chromosomal rearrangements but does not identify size of deletion, uniparental disomy, or imprinting defects (58). Thus, additional molecular testing is necessary if FISH analysis is negative or information about deletion subtype is desired.
Whole exome sequencing (WES). WES with sequence variant analysis of SNRPN can be combined with MS-MLPA methylation analysis to offer a highly sensitive, streamlined molecular testing strategy that is able to detect all genetic subtypes in a single analysis. WES can identify rare imprinting center deletions without the need for parental DNA analysis and can detect other genetic disorders and cytogenic abnormalities (164); however, it remains a costly test with limited availability.
Other. Muscle biopsy, electromyograms, and nerve conduction studies are typically normal or show nonspecific findings (58) and are not recommended in the diagnostic work-up for Prader-Willi syndrome. Neuroimaging, likewise, has no specific role in the diagnosis of Prader-Willi syndrome.
|
• Management of Prader-Willi syndrome requires a multidisciplinary team approach involving geneticists, developmental pediatricians, endocrinologists, dieticians, neurologists, and behavioral specialists. | |
|
• Prevention of obesity is vital to the long-term health of patients and is managed through restriction of caloric intake and behavioral interventions; there are no effective pharmacologic interventions to control weight. | |
|
• Growth hormone supplementation starting in infancy is recommended for patients with Prader-Willi syndrome. |
Introduction. At the present time, there are no identified treatment modalities that can address the fundamental genetic defect in individuals with Prader-Willi syndrome: the lack of key functional gene expression in the Prader-Willi region of chromosome 15q11-q13. The various “treatment” modalities described below only address some of the symptoms of Prader-Willi syndrome, with greater or lesser degrees of success.
Neonatal and infant care. The floppy newborn with feeding problems will often require gavage feeding. Ventilatory support may be necessary, sometimes for several weeks. Cryptorchidism and other genital anomalies may be corrected surgically. All infants with Prader-Willi syndrome should undergo comprehensive feeding and swallowing evaluations (152).
Dietary management. Children and adults require behavioral and nutritional management to control the hyperphagia and subsequent obesity. The goal when possible is the prevention of obesity before it occurs, but weight reduction in the already overweight individual may result in a significant improvement in associated medical conditions (83).
In children, food intake must be closely monitored to prevent excessive weight gain. The actual caloric needs of the individual appear to be lower than those of children of the same age. Caloric intake should be set at 60% to 80% of the age-appropriate daily allowance, with vitamin supplementation (120). Control of the environment by locking food away, an increase in activity, and management of food-related behaviors such as tantrums and food stealing are important components of treatment. Exercise programs should accompany dietary therapy.
Normative growth charts for children with Prader-Willi syndrome not receiving growth hormone therapy have been published. Because these individuals are short in stature, weight should be adjusted for height rather than age to determine ideal body weight (82; 22).
Surgical management of obesity (eg, gastric bypass, gastroplasty, biliopancreatic diversion) is associated with poorer outcomes in patients with Prader-Willi than normal obese patients and is not routinely recommended (155).
Hormone treatment. Growth hormone therapy has been used in children and adults with Prader-Willi syndrome since the early 2000s. Consensus guidelines published by the Growth Hormone Research Society recommend considering recombinant human growth hormone therapy for patients with genetically confirmed Prader-Willi Syndrome before onset of obesity, which can occur as early as 2 years of age, in conjunction with dietary, environmental, and lifestyle interventions (45).
Most endocrinologists start growth hormone supplementation during infancy. A meta-analysis of growth hormone treatment in children with Prader-Willi syndrome reported high-quality evidence for improvements in stature, growth velocity, body composition (lean body mass and % fat mass), and short-term decreases to BMI with better long-term control of BMI, as well as improvements in cognitive and motor abilities at a young age (135; 29). Growth hormone treatment in infants with Prader-Willi syndrome has been shown to prevent deterioration of cognitive skills and improves total IQ measured in later childhood, likely through its interactions with insulin-like growth factor which in turn effects brain growth and development (169). Positive effects of growth hormone therapy are seen in patients with or without measured deficiencies in growth hormone levels, thus, from a medical perspective growth hormone level testing is not necessary prior to starting therapy (45). There is no established duration of treatment. Most published trials involve treatment regimens of 1 to 4 years, and the most current recommendation from the Growth Hormone Research Society is to continue growth hormone administration for as long as benefits outweigh the risks (45). Growth hormone therapy requires close surveillance and monitoring. Growth hormone therapy is contraindicated in all patients with severe obesity, uncontrolled diabetes mellitus, untreated severe obstructive sleep apnea, active cancer, or psychosis (45).
Prior to starting growth hormone, all patients should undergo comprehensive multidisciplinary evaluation including endocrine assessment; referral to sleep clinic for sleep oximetry, or polysomnogram, or both; ear nose throat assessment for causes of upper airway obstruction; and scoliosis evaluation if indicated (45). Growth hormone therapy can exacerbate underlying endocrine disorders, including central adrenal insufficiency and worsening of diabetic symptoms due to insulin antagonism (30). At least 28 unexpected deaths have been reported in patients receiving growth hormone, predominantly in morbidly obese individuals with coexisting upper airway obstruction and sleep apnea or respiratory infections or both (39; 02). Growth hormone has not been directly linked to any deaths (02). Further study is needed to better understand the role of growth hormone treatment in adult populations, as well as long-term effects of mortality and morbidity, including effects on bone health, lipid profiles, and cardiovascular risk.
Testosterone in males and estrogen in females has been successfully used to induce sexual development during adolescence. This can lead to fertility in female cases; thus, sex education is essential for patients receiving estrogen. Symptomatic glucocorticoid and thyroid hormone treatment are indicated for adrenal and thyroid deficiencies, respectively.
There are no consensus guidelines on screening, investigation, and management of congenital adrenal insufficiency. Diagnostic testing should be conducted in any individual with symptoms and can be considered in all (asymptomatic) individuals with Prader-Willi syndrome at least once as routine screening or prior to major surgery (96). Caregivers should be educated in recognizing signs and symptoms of adrenal insufficiency. Individuals with confirmed adrenal insufficiency should be treated with hydrocortisone during times of stress, including illness or major surgery.
Pharmacologic interventions. The use of medication to control appetite or improve satiety in this condition has been, in general, unsatisfactory (30). Currently, there are no approved pharmacologic therapies for obesity or hyperphagia in Prader-Willi syndrome; however, there is optimism that the recent success of glucagon-like peptide-1 receptor agonists in promoting weight loss and appetite reduction in non-Prader-Willi populations could potentially translate to individuals with Prader-Willi syndrome (116). Multiple small trials and case series involving the use of glucagon-like peptide-1 receptor agonists in individuals with Prader-Willi syndrome have been published, which show positive results in terms of safety, tolerability, and potential weight gain and appetite (127), with one case report showing a new glucagon-like peptide-1 receptor agonist (semaglutide) that was effective even when initial (older) glucagon-like peptide-1 receptor agonists were not (154). A proof-of-concept pilot trial involving 13 patients with Prader-Willi indicated safety and efficacy for diazine choline-controlled release tablets in reducing hyperphagia and aggressive behaviors (91). Diazine choline-controlled release is a KATP channel agonist used to treat hyperinsulinemic hypoglycemia that enhances the effects of leptin in the hypothalamus to decrease hyperphagia signals (91). Larger controlled trials are needed to further study the roles of glucagon-like peptide-1 receptor agonists and other potential weight loss medications like diazine in individuals with Prader-Willi syndrome.
Other behaviors such as aggression and obsessive behaviors have responded to pharmacological and behavioral therapies. Topiramate shows efficacy in decreasing self-injurious and impulsive behaviors, including skin-picking, but does not suppress appetite (159). N-acetylcysteine has also been shown to successfully treat and, in some cases, completely resolve skin-picking, likely either by modulation of NMDA receptors or by elevating levels of glutathione (118). Sertraline was effective in significantly reducing temper outbursts in 13 of 14 patients with Prader-Willi syndrome (48), whereas aripiprazole reduced outbursts in seven of 10 patients (49). Risperidone is indicated for episodes of psychosis associated with uniparental disomy. Serotonin reuptake inhibitors have shown some efficacy in treating obsessive compulsive behaviors.
Based on animal research, there is a possibility that oxytocin may be useful as a therapeutic intervention to improve social interaction skills as well as behavioral difficulties shown by individuals with Prader-Willi syndrome. In a randomized, double-blind, placebo-controlled cross-over study of 25 children with Prader-Willi syndrome, Kuppens and colleagues found that, for children less than 11 years (n=17) of age, twice daily intranasal oxytocin for 4 weeks resulted in significant improvement in social and food-related behaviors (93). A similar improvement was not seen in children aged 11 or older.
With respect to the management of excessive daytime sleepiness, De Cock and colleagues demonstrated a significant improvement in this symptom in nine Prader-Willi subjects treated with modafinil, an agent used to treat narcolepsy (47). The authors recommended that the drug not be used in patients with severe obstructive sleep apnea. Central sleep apnea in infants with Prader-Willi syndrome can be markedly diminished with the use of nocturnal oxygen therapy at relatively low rates of flow (0.25 to 1 L per minute) (33). Why oxygen supplementation diminishes central sleep apnea is unclear. Cohen and colleagues speculated that chemosensitivity to oxygen and carbon dioxide levels in Prader-Willi syndrome are diminished and that mild degrees of low oxygen saturation may be sufficient to dysregulate control of breathing (33).
Developmental and educational interventions. Children with Prader-Willi syndrome require appropriate developmental surveillance and intervention. Most children will benefit from early childhood intervention programs and often need speech language, occupational, and other therapies. Children will also require evaluation for educational placement paired with psychoeducational testing to assess for intellectual and learning disabilities and attention issues. Vocational training is recommended for the older child.
Treatments on the horizon. Advances in DNA targeting technologies have led to new research strategies focused on activating silenced Prader Willi syndrome associated genes on the maternal allele. In 2017, Kim and colleagues demonstrated successful restoration of expression of silenced genes at the maternal 15q11-q13 site using two epigenetic modifying transcription factors in human fibroblast tissue culture and a mouse model (90). One of the disadvantages of small molecules regulators, such as transcription factors, is that they are not specific and could potentially target numerous loci leading to off-target effects. Targeted epigenome editing using CRIPSR-based techniques could solve this problem. Rohm and colleagues utilized a targeted CRISPER-dCas9-based epigenetic editing approach in a human pluripotent cell model to identify regulatory elements at the 15q11-q13 locus controlling expression of Prader Willi associated genes and then activate the imprinted genes via both targeted transcriptional activation and DNA demethylation (148). Interestingly, dCAS9-based demethylation led to stable long-term gene expression of Prader Willi genes, suggesting one-time treatments could be possible.
Although much work remains to be done to translate this work safely in vivo to human subjects, these results provide preliminary evidence for further exploration of epigenetic-based regulatory factors (in this case activation of the maternally suppressed genes at the 15q11-q13 locus) as a treatment strategy for Prader-Willi syndrome.
Dietary measures and effective weight management play an essential role in preventing complications of obesity, including type 2 diabetes, cardiac disease, respiratory problems such as sleep apnea, fatty liver, and gastric paresis. Implementation of balanced restricted-calorie diets have shown positive effects in body composition and weight in children with Prader-Willi syndrome (120). Schmidt and colleagues showed that children with Prader-Willi syndrome who received a well-balanced, restrictive diet starting at 14 months of age had normal BMIs at 10 years of age (156).
There are now a multitude (more than 30) of published, small, randomized control trials suggesting that growth hormone therapy provides a range of physical benefits, including linear growth, reduced body fat, increased muscle mass, improved resting energy expenditure and exercise tolerance, and improvements in motor abilities (45). There is an increasing body of evidence suggesting that growth hormone therapy may improve cognition and adaptive functioning, especially if started before the age of 1 (101; 62; 57; 29). In a large cohort study of 173 children with Prader-Willi syndrome, Dykens and colleagues reported that those receiving growth hormone treatment had significantly higher verbal and composite IQs as well as adaptive communication and daily living skills compared to those not receiving hormone therapy (62).
Growth hormone therapy may also be useful in adults with Prader-Willi syndrome. In a relatively large, randomized, placebo-controlled crossover study (n=39), Sode-Carlsen and colleagues found that growth hormone treatment was accompanied by a significant increase in muscle volume, a reduction of abdominal subcutaneous fat, and an increase in lean body mass (163). Beneficial effects on cognition have been shown to persist in adults even after growth hormone treatment has been discontinued (94).
Testosterone supplementation can exacerbate behavioral problems, including aggression. Estrogen supplementation can lead to osteoporosis and possible fertility in female patients.
Pregnancy with a Prader-Willi syndrome child may result in breech presentation and decreased fetal movements.
There is no information available concerning pregnancy in women with this condition; it rarely occurs, as fertility is probably decreased due to hypogonadism.
Patients with Prader-Willi syndrome are placed at risk during anesthesia by problems including disturbances in body temperature, preoperative diabetes, intraoperative arrhythmias, cor pulmonale, and reduced pulmonary reserves secondary to obesity. There may be problems with venous access and apnea during the postoperative period (46). No cases of malignant hyperthermia have been reported, but newborns with hypotonia are at risk for this condition. Patients may be at increased risk of aspiration of gastric contents.
Careful medical assessment, including pulmonary evaluation and attention to glucose control and body temperature intraoperatively, is advised (106).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Kim Smyth MD
Dr. Smyth of the Alberta Children's Hospital Research Institute has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026