


Neuro-Oncology
Turcot syndrome
May. 27, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article reviews the diverse systemic manifestations of von Hippel-Lindau disease and discusses the historical context of the disease as well as current methods employed in the diagnosis and treatment of the disorder. Classic physical and radiographic manifestations are illustrated in a clinical vignette. New recommendations for the management of pancreatic lesions and for pheochromocytoma surveillance are reviewed.
|
• Von Hippel-Lindau disease is an autosomal dominant hereditary multisystem tumor syndrome with marked intra-and interfamilial variability. | |
|
• The tumors are highly vascular and mostly consist of hemangioblastomas of the CNS and retina, renal cell carcinoma, endolymphatic sac tumor of inner ear, and adrenal pheochromocytoma. | |
|
• There is correlation between the type of gene mutation and its phenotypic expression, particularly for pheochromocytoma and renal cell carcinoma. | |
|
• Molecular diagnostic testing for the von Hippel-Lindau gene (VHL gene) is available and can be performed on asymptomatic high-risk individuals and in high-risk pregnancies in the early prenatal period. | |
|
• Management mainly consists of excision of malignant lesions (eg, renal tumors) and surgical removal or ablation of symptomatic benign lesions (eg, hemangioblastomas), which are followed closely for possible recurrence or development of new lesions. |
Collins was the first investigator to recognize the inherited nature of what was called “retinal angiomatosis,” which we now call retinal hemangioblastoma (09). Von Hippel was the first to document the progressive nature of the retinal lesions (68).
The history of hemangioblastoma of the cerebellum was initiated by Hughlings Jackson (25). Several authors subsequently noted an association between cerebellar cysts and small cysts of the pancreas and kidneys (56; 69). These associated lesions later became known as the von Hippel-Lindau complex.
Tresling described the first family with an association between retinal and cerebellar tumors (66). Lindau combined, as a single entity, the clinical-pathological hemangioblastic alterations of the retina, cerebellum, spinal cord, and lesions of the pancreas, kidneys, adrenal medulla, liver, and epididymis (37). The term "von Hippel-Lindau disease" was first used in 1936 (14) and has been in common use since the 1970s (40). Melmon and Rosen reviewed the literature on the disease and suggested the clinical diagnostic criteria (46). In 1988, Seizinger and colleagues demonstrated the responsible gene at short arm of chromosome 3 (60). Latif and colleagues showed that the abnormal gene of von Hippel-Lindau disease behaves as a tumor suppressor gene (36). These molecular genetic findings established the definitive criteria for the disease to confirm the diagnostic viability of the clinical criteria (65).
In 2023, a newly formed International VHL Surveillance Guidelines Consortium met to create evidence-based, system-specific guidance around surveillance in the setting of von Hippel-Lindau disease (13).
|
• Von Hippel-Lindau disease is an autosomal dominant multisystem tumor syndrome with various benign and malignant neoplasms. | |
|
• The typical ocular lesion is the hemangioblastoma of the retina. |
The lesions are diverse and affect various organs, including the eye, central nervous system, kidney, pancreas, inner ear, adrenal gland, liver, and epididymis (male) or broad ligament (female). Given the diversity of the symptomatology corresponding to the involvement of the organs in each patient, the symptoms and clinical signs are individualized.
Melmón and Rosen established diagnostic criteria for von Hippel-Lindau disease (46). Since these criteria were established, the diagnostic criteria have been expanded to encompass molecular genetic confirmation.
|
Clinically actionable variant in the family? |
Clinical criteria |
|
Yes |
Patient carries the same variant AND at least one von Hippel-Lindau disease lesion |
|
No |
Absent first-degree relative with von Hippel-Lindau disease, then two von Hippel-Lindau disease lesions are required, one of which is hemangioblastoma |
|
Present first-degree relative with von Hippel-Lindau disease AND at least one von Hippel-Lindau disease lesion | |
|
Adapted from (03) | |
The following von Hippel-Lindau disease lesions are included in the criteria: hemangioblastoma in the retina or CNS; renal cell carcinoma; pheochromocytoma; pancreatic neuroendocrine tumor; and endolymphatic sac tumor.
The lesions excluded from the criteria were shown to occur in a high enough frequency in the general population to lose specificity but can still be supportive in making the diagnosis of von Hippel-Lindau disease. “Clinically actionable variant” refers to one that can be used to predict the risk of the phenotype in the affected patient. The presence of a pathogenic variant must be interpreted alongside the presenting phenotype as they are not always correlated.
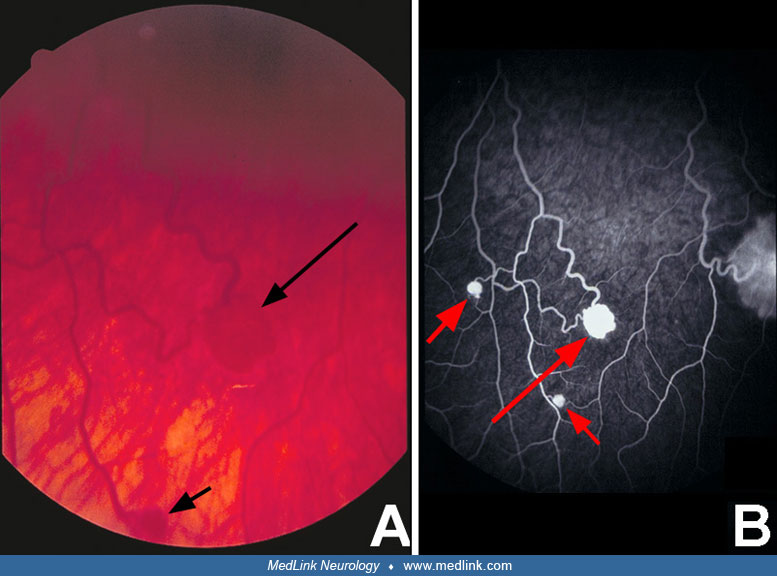
Eye. The typical ocular lesion is the hemangioblastoma of the retina, which was the first recognized pathological manifestation of von Hippel-Lindau disease, initially described as “nevi.” The retinal hemangioblastoma has been referred to as an angioma, hemangioma, or retinal angiomatosis and has been reported to occur in approximately 60% to 70% of patients; they are bilateral in 30% to 50% of cases with ocular involvement. Retinal lesions may become manifest at any age, including infancy. The principal symptoms or complications of retinal hemangioblastoma are glaucoma, cataracts, detachment of the retina, and sympathetic ophthalmitis, which results in unilateral or bilateral blindness in half the cases. Some tumors remain stationary and dormant for many years after diagnosis; however, the usual course is one of progressive growth at a moderate or rapid rate. Studies within the past several years indicate that the prevalence and biological progression of retinal hemangioblastoma and other lesions in von Hippel-Lindau disease are determined in part by the specific germline mutations within the VHL gene. When the optic disc is affected, the lesion can resemble papilledema, papillitis, or glioma of the optic nerve head (23).
Retinal vascular proliferation is a relatively uncommon fibrovascular lesion in patients with von Hippel-Lindau disease, and it is important to distinguish this from retinal hemangioblastoma.
Central nervous system. Hemangioblastomas of CNS are the main component of von Hippel-Lindau disease. They occur in 60% to 80% of cases, are multifocal in 42% of cases, and are the presenting feature in approximately 40% of cases (41; 40). The lesions are most commonly located in the cerebellum and spinal cord, less commonly in the brainstem (71), and rarely in the cerebral hemispheres (55), optic nerve (24), pituitary gland (12), pituitary stalk, and third ventricle (38).
The diagnosis is usually made in young adults, 24 to 40 years of age, with a mean age of 32 years; it occasionally has been seen in children less than 10 years of age (22). Typical of CNS lesions, presenting symptoms are dependent on the affected location. The most frequent symptoms are generalized headaches due to increased intracranial pressure and headaches localized to the occipital and upper cervical regions; less frequent symptoms include lethargy, anorexia, gait disturbances, dementia, and visual disturbances. The usual signs of hemangioblastoma of the cerebellum are ataxia, papilledema, nystagmus, bilateral convergent strabismus, and facial paralysis; less frequent findings are impaired consciousness, paresis of the lower extremities, diminished in visual acuity, and dysarthria.
The symptoms of posterior fossa involvement with hemangioblastoma appear approximately 15 years earlier in cases of von Hippel-Lindau disease than in sporadic cases of hemangioblastoma (10). The majority (80%) of cerebellar hemangioblastomas are located in the cerebellar hemispheres, 15% in the vermis, and only 5% in the floor of the fourth ventricle. Of these cerebellar tumors, 75% are cystic, usually with mural nodules (35). Approximately 10% are multiple and 10% are recurrent. Cerebellar hemangioblastomas usually produce erythropoietin, which may result in erythrocytosis (polycythemia).
Spinal cord hemangioblastomas present with a frequency three times greater than those of the brainstem (35). Symptoms of spinal cord compression occur early, and pain often precedes the loss of sensation. The localization of spinal cord hemangioblastoma is intramedullary in almost two thirds of cases, extramedullary intradural in one fourth, and extradural in the rest. Half the tumors occur in the thoracic region, 40% in the cervical region, and the remainder in the lumbar segments. Rarely is the tumor encountered in the sacral region or the cauda equina. The CSF may be xanthochromic, with elevated protein and occasionally subarachnoid hemorrhage. Erythrocytosis has not been described in spinal hemangioblastoma as it has been with cerebellar lesions.
Hemangioblastomas of the brainstem in von Hippel-Lindau disease are generally rare and typically localize to the medulla oblongata.
Screening guidelines include regular imaging to identify lesions prior to their clinical presentation and have improved clinical outcomes.
Systemic lesions. Lesions of the kidneys, pancreas, and epididymis are frequently asymptomatic, and their detection may be incidental. Patients with adrenal tumors (pheochromocytomas) are more often symptomatic.
Kidney. The renal lesions of von Hippel-Lindau may be benign cysts or clear cell carcinomas with a partial or predominant cystic component; both present with a similar incidence. Renal cysts may be solitary or multiple. The risk of developing renal cell carcinoma varies in different clinical forms of von Hippel-Lindau disease (see classification below). In the most common forms (types 1 and 2B) this risk approaches 70% (41; 52); it is the cause of death in 20% to 50% of affected patients (35). Malignant renal cell tumors in von Hippel-Lindau disease are multiple and bilateral in 60%, an important difference from sporadic cases (53). The detection of renal cell carcinoma among multiple cystic lesions can be difficult, though new methods of imaging are making early and correct diagnosis much easier.
Pancreas. Lesions of the pancreas in von Hippel-Lindau disease are present in over 60% of patients and are usually benign and cystic. Pancreatic neuroendocrine tumors (pNET) are seen in 11% of patients with von Hippel-Lindau disease and are often non-functioning (03). Like the renal, CNS, and retinal lesions in von Hippel-Lindau disease, pNETs are substantially vascular. Most of these lesions grow slowly and are asymptomatic, although up to 8% can undergo malignant transformation (11). Familial cystic or solid lesions of the pancreas should raise suspicion for von Hippel-Lindau disease or multiple endocrine neoplasia. Most pancreatic lesions are asymptomatic, but some may obstruct the biliary and pancreatic ducts and result in pancreatitis, fibrosis, and atrophy of the pancreas. If there is significant loss of the cells populating the islets of Langerhans, diabetes mellitus may result. When symptomatic, patients may present with abdominal pain, mass effect in the abdomen, and endocrine and exocrine insufficiency of the pancreas. In select patients with resultant pancreatitis, pancreatectomy may be performed.
Adrenal gland. Pheochromocytomas are usually unilateral, but bilateral tumors are not uncommon. The risk of developing this tumor varies in different forms of von Hippel-Lindau disease (see classification below). The mean age of onset is 20 to 29 years old, though some present prior to 18 years of age. The tumor may result in hypertension, unstable blood pressure, and paroxysmal headaches. High urinary concentrations of catecholamines and metanephrine are suggestive of the diagnosis. The diagnosis can be confirmed by an abnormal pressor response. Radiological study with ultrasonography, MRI, and MRA discloses the tumor.
Epididymis and broad ligament. Epididymal lesions consist of simple cysts or papillary cystadenomas; they are found in 10% to 60% of male patients with von Hippel-Lindau disease and present at an average age of 23 years (35; 06). They may be bilateral and rarely impair fertility. It can be found by routine physical examination but is often an incidental finding at autopsy. These lesions generally are not painful. Although often clinically insignificant, the presence of this tumor in a patient with a close relative with von Hippel-Lindau disease is often instrumental in performing additional studies to establish the diagnosis of that disease. Papillary cystadenoma of the broad ligament is an analogous lesion that may occur in women with von Hippel-Lindau disease. The diagnosis of this lesion can be reliably made by ultrasonography.
Inner ear endolymphatic sac tumor. These are slow-growing, low-grade papillary adenocarcinomas involving the temporal bone. Such lesions occur in an estimated 10% of patients with von Hippel-Lindau and are frequently bilateral (39). Symptoms of disequilibrium or aural fullness affecting patients with known von Hippel-Lindau disease may be an early indication of endolymphatic dysfunction. Abrupt hearing loss and tinnitus are other symptoms that may suggest the presence of an endolymphatic sac tumor (05; 32). Audiometry can reveal low-frequency hearing loss before patients report noticing changes in their hearing and is the recommended form of screening for children with von Hippel-Lindau disease. Thin-slice MRIs of the inner ear can begin in adulthood. Early diagnosis and surgical resection of these tumors can prevent permanent loss of hearing.
Metastasis. Of the tumors frequently seen in von Hippel-Lindau disease, renal cell carcinoma, pancreatic neuroendocrine tumors, and pheochromocytomas have the potential to metastasize. Renal cell carcinomas are the most common and can metastasize to the CNS. Tumor-to-tumor metastasis is well documented.
Metastatic involvement of the nervous system occurs relatively rarely (48), and the origin of the tumor can be any other organ (most commonly kidney). Complications of CNS disease, including metastasis, is the most common cause of death.
Classification. Based on the likelihood of developing pheochromocytoma, von Hippel-Lindau disease phenotypes may be divided into types:
Type 1: Low risk for pheochromocytoma
Type 2: High risk for pheochromocytoma
Type 2A: Pheo+ CNS hemangioblastoma, low risk for renal cell carcinoma
Type 2B: Pheo +CNS hemangioblastoma, high risk for renal cell carcinoma
Type 2C: Pheochromocytoma only
Type 3: Chuvash polycythemia
This classification is helpful in research for correlating the effect of specific mutation with pVHL function and phenotype but is less useful for clinical management due to its intrafamilial variability (40).
Many factors influence the prognosis of von Hippel-Lindau disease. The most important factors include localization, size, number, and biological nature of the tumors. CNS hemangioblastomas and renal cell carcinoma metastasis to the CNS are the most frequent causes of death.
Early detection of the disease and the associated lesions and management of the tumors of various organs by physicians with special expertise in these organs have markedly improved the prognosis in patients with von Hippel-Lindau disease. The general life expectancy for women is 50 years; it is approximately 60 years for men (73).
A 25-year-old Caucasian woman first presented for medical care at the age of 18 years following a 2-month history of progressive difficulty with ambulating, handwriting, thinking, and blurring of vision with headache. She acutely experienced severe nausea and headache in the days preceding her admission.
Her neurologic examination was notable at that time for papilledema, right dysmetria, and right Babinski. CT scan revealed a left cerebellar 5 cm x 5 cm enhancing lesion with a cystic component compressing the fourth ventricle with early hydrocephalus.
The lesion was surgically excised without complication. The tumor was diagnosed as a cerebellar hemangioblastoma.
Given the young age of presentation and a similar tumor previously diagnosed in her mother, she received extensive medical surveillance for systemic manifestations of von Hippel-Lindau disease. MR of C-spine revealed multiple heterogenously enhancing nodular masses throughout the posterior fossa as well as cervical cord consistent with multiple hemangioblastomas.
Subsequent fundoscopic examination revealed multiple bilateral retinal hemangioblastomas.
Additional evaluations in the succeeding months revealed the presence of multiple cystic lesions within the head of the pancreas as well as a solid tumor within the right mid-kidney. The renal lesion was surgically excised and diagnosed as renal cell carcinoma.
Von Hippel-Lindau disease is inherited as an autosomal dominant trait with high penetrance (over 90% by 65 years of age). The associated neoplasms are age-dependent and show allelic-specific expressivity. The disease is caused by germline mutations in the VHL gene (a tumor suppressor gene), and the associated tumors are initiated by biallelic inactivation of this gene (36; 65). Over 500 gene variants have been described to date (59).
VHL gene and pVHL. The gene is located at chromosome 3p25-26 and contains three exons and a coding sequence of 639 nucleotides. It is expressed in all tissues and encodes two protein products: a full-length form (p30, 213 amino acids) and a shorter form (p19, 160 amino acids). The VHL gene products, particularly the p19 isoforms, have been conserved throughout evolution and can be detected in all multicellular organisms examined to date (Kaelen 2007; 49). Both protein isoforms behave similarly in most functional assays, so they are referred to as pVHL. This protein has been implicated in a variety of cellular functions, including transcriptional and posttranscriptional gene expression, extracellular matrix formation, apoptosis, and ubiquitylation (28; 58; 51).
Gene mutations. Approximately 23% of cases of von Hippel-Lindau disease result from de novo mutation of the VHL gene (61). Germline mutations of the VHL gene are spread throughout the three exons. Missense mutations are the most common, but other forms of mutation and large deletions can also occur. Biallelic inactivation may occur through one of several pathways, including intragenic mutations, mitotic recombination events, and hypermethylation of the promoter region. Alteration of the VHL gene is typically due to insertion, deletions, or missense mutations (36). Mutations of the VHL gene are also common in sporadic hemangioblastomas and renal cell carcinomas.
There is a correlation between certain germline mutations and particular clinical phenotypes (62). In families with von Hippel-Lindau disease type 1 (high risk for hemangioblastoma and renal cell carcinoma but low risk for developing pheochromocytoma), mutations are nonsense or truncating, which markedly disrupt the folding of pVHL (64; 40). Individuals with von Hippel-Lindau disease type 2 have a higher risk of developing pheochromocytoma, and nearly all have a missense germline mutation. This type is further divided into three subtypes; 2A (high risk for pheochromocytoma and hemangioblastoma but low risk for renal cell carcinoma); 2B (high risk for pheochromocytoma, hemangioblastoma, and renal cell carcinoma); 2C (pheochromocytoma only). This subtype shows a specific missense germline mutation in the VHL gene (76).
Pathogenesis. The tumors in von Hippel-Lindau disease are initiated by random somatic cell mutations, resulting in bialleic loss of the tumor suppressor VHL gene and its protein product pVHL. Among the various functions of this protein, the pVHL-deficient cells’ inability for ubiquitylation (and subsequent degradation) of hypoxia-inducible factor (HIF-1alpha and HIF-2alpha) has been implicated in the development of highly vascular tumors in von Hippel-Lindau disease (43; 08; 28; 52).
Normal pVHL binds to elongin C, which forms a complex with elongin B, Cul 2, and Rbx1. This complex, due to its ubiquitin ligase activity, interacts with the hydroxylated alpha subunits of HIF and targets them for rapid degradation by proteasomes. Under normoxic conditions and in the presence of functional pVHL, hydroxylated alpha subunits are rapidly degraded. In hypoxia, alpha subunits of HIF cannot be hydroxylated for binding to functional pVHL; this results in transient stabilization of the HIF-alpha, causing their accumulation in the cytoplasm and subsequent translocation to the nucleus, where they result in activation of the transcription factors for a variety of hypoxia-inducible genes such as vascular endothelial growth factor, erythropoietin, and transforming growth factors TGF-alpha and TGF-beta. Likewise, in von Hippel-Lindau disease, where pVHL is abnormal or absent, accumulation of HIF results in abnormal signaling, even in the presence of oxygen (29; 42; 28; 49). These chains of interactions appear to play a key role in the formation of the vascular tumors characteristic of von Hippel-Lindau disease. Studies suggest that targeted inhibition of some of these factors or their receptors may suppress the growth of tumor cells in advanced metastatic renal cell carcinoma and other von Hippel-Lindau disease-associated tumors (07). The broader concept of the role of hypoxia in inflammation and tumor progression in the context of von Hippel-Lindau disease is reviewed by Bader and Hsu (02).
Histopathology. The two most common tumors of von Hippel-Lindau disease hemangioblastoma and renal cell carcinoma are discussed here.
Hemangioblastoma is a fine vascular network of endothelial-lined channels or caverns, generally without the presence of neurons or glia. In the cerebellum, it may be cystic and contain xanthochromic fluid. A small, highly vascular nodule is generally found in the wall of the cyst. The nodule is usually in contact with the leptomeninges at the surface of the cerebellum. The "pseudoxanthomatous" stroma consists of polygonal cells with foamy lipid-laden cytoplasm. The histogenesis of the stromal cells is not known. Studies suggest that stromal cells share protein expression and topographic distribution with hemangioblast progenitor cells (17; 54). Extramedullary hematopoiesis may be present within cerebellar hemangioblastoma, and concomitant paraneoplastic erythrocytosis has been reported (78). Hemangioblastomas are well-demarcated and generally are not locally invasive, nor do they tend to metastasize.
Renal lesions may be cystic or solid. The tumors are highly vascular and consist of clear cells. The cysts are small (range from several millimeters to 2 cm in diameter), have a gray appearance, and are filled with clear liquid. They are lined by a layer of atypical cuboidal cells that are similar to those covering solid and cystic renal cell carcinomas. Renal tumors may invade neighboring zones and metastasize through venous channels to the adrenal glands, spinal cord, and other sites of the CNS.
Approximately one in 36,000 children are born with von Hippel-Lindau disease (18).
|
• Surveillance screening for new tumors is central to the management of patients with von Hippel-Lindau disease. |
There are no specific preventive measures for any of the neoplasms associated with von Hippel-Lindau disease. However, early diagnosis of this disease in individuals at risk results in timely management of the tumors and reduces morbidity and mortality among these patients. Also, genetic counseling of patients with von Hippel-Lindau disease and genetic testing of at-risk individuals prior to pregnancy can be utilized. Fetal genetic studies can be performed, as can preimplantation genetic diagnosis, in pregnancies at risk for von Hippel-Lindau disease.
The differential diagnosis of von Hippel-Lindau disease includes sporadic tumor cases with the same types of lesions as in von Hippel-Lindau disease but not associated with this disease. The differential diagnosis is not important with regard to therapy, which is the same in both isolated lesions and those that present in a patient with von Hippel-Lindau disease. However, the occurrence of such tumors should prompt consideration for evaluation of von Hippel-Lindau disease.
The principal differential diagnosis of hemangioblastoma of the cerebellum is astrocytoma, which may be cystic or solid. Those of the brainstem must be differentiated from syringobulbia and those of the spinal cord from cystic astrocytoma and syringomyelia.
Renal cell carcinoma in patients with von Hippel-Lindau disease is differentiated from sporadic cases by an earlier presentation, its multifocal nature, and its frequent cystic component. Pancreatic neuroendocrine tumors are also seen in multiple endocrine neoplasia and should illicit a genetic work-up to identify a possible underlying tumor syndrome.
Chuvash polycythemia (also called von Hippel-Lindau disease–associated polycythemia or familial erythrocytosis type 2) is an autosomal recessive blood disorder caused by biallelic pathogenic variants in the VHL gene that cause a congenital increase in the number of red blood cells.
Minimal diagnostic criteria (Table 2) for patients with a known family history of von Hippel-Lindau disease include the presence of a single retinal hemangioblastoma or cerebellar hemangioblastoma, renal cell carcinoma, or pheochromocytoma (11). Multiple pancreatic cysts are sufficiently rare within the general population such that their presence in an at-risk patient is a strong indication that they have inherited the mutation. In contrast, renal and epididymal cysts are generally not reliable indicators that an at-risk person carries a von Hippel-Lindau disease mutation due to the fact that they occur with a fairly high frequency within the population. However, the presence of multiple renal cysts at an early age is strongly suspicious for a diagnosis of von Hippel-Lindau disease as such a presentation is rare within the general population (11).
|
Clinically actionable variant in the family? |
Clinical criteria |
|
Yes |
Patient carries the same variant AND at least one von Hippel-Lindau disease lesion |
|
No |
Absent first-degree relative with von Hippel-Lindau disease, then two von Hippel-Lindau disease lesions are required, one of which is hemangioblastoma |
|
Present first-degree relative with von Hippel-Lindau disease AND at least one von Hippel-Lindau disease lesion | |
|
Adapted from (03) | |
The following von Hippel-Lindau disease lesions are included in the criteria: hemangioblastoma in the retina or CNS; renal cell carcinoma; pheochromocytoma; pancreatic neuroendocrine tumor; and endolymphatic sac tumor.
The lesions excluded from the criteria were shown to occur in a high enough frequency in the general population to lose specificity but can still be supportive in making the diagnosis of von Hippel-Lindau disease. “Clinically actionable variant” refers to one that can be used to predict the risk of the phenotype in the affected patient. The presence of a pathogenic variant must be interpreted alongside the presenting phenotype as they are not always correlated. Dilated binocular funduscopic examination is the recommended mode of surveillance.
Fluorescein angiography can detect lesions before they are evident by unenhanced funduscopic examination and is the test of choice in questionable lesions (27).
The diagnosis of cerebellar hemangioblastoma is easily established following MRI or MRA, which provide precise identification of the size, solid or cystic structure, site of the nodule, feeding vessels and draining veins, and relations with surrounding structures. Contrast-enhanced MRI is capable of demonstrating a larger number of lesions, can separate the tumor from the surrounding zone of edema, and can determine whether the lesion is cystic or solid. In addition, MR angiography is capable of evaluating vascular lesions anywhere in the body including the retina. However, a detailed examination of the ocular fundus is still essential to detect retinal hemangioblastomas.
Given the limitations of CT in studying the posterior fossa and spinal cord, this technique is not a reasonable alternative to MRI. It may, however, complement the MRI for the study of lesions in the abdominal organs, especially if enhanced by contrast.
Ultrasonography remains a recommended test for the study of lesions in the epididymis and may play a role in the evaluation of intra-abdominal lesions. However, it is not recommended as a screening tool for pancreatic lesions (34).
Ultrasound offers the advantage of real-time visualization and facilitates sampling of the tumor by fine needle aspiration or core biopsy. The diagnosis of pheochromocytoma is suggested by specific tests for this tumor, specifically the urinary elevation of catecholamines and metanephrine; biopsy or fine needle aspiration then confirms the diagnosis.
Guidelines for the surveillance and management of an individual with von Hippel-Lindau disease include an annual medical history and physical examination for manifestation of symptoms attributable to the various components of the disease. It is also generally recommended that a patient with von Hippel-Lindau disease be managed by a von Hippel-Lindau disease clinical care center when possible, so a holistic, multidisciplinary approach case be used to best support the patient. System specific guidance is as follows.
|
1) CNS hemangioblastoma: Baseline MRI of the neuroaxis at the age of 11, then continued every 2 years (21). | |
|
a. Screen between the ages of 11 and 65 or at onset of neurologic symptoms | |
|
2) ELT: Diagnostic audiograms every other year. Establish a baseline MRI of internal auditory canals between the ages of 15 and 20 or at the onset of symptoms (45). | |
|
a. Screening between the ages of 11 and 65 or at the onset of audiovestibular signs and symptoms. | |
|
3) Pancreatic neuroendocrine tumor (PNET): Screen every 2 years if there are no lesions on imaging. | |
|
a. Screening is to start no later than the age of 15, until the age of 65. | |
|
b. If PNET is found, then the patient should be followed in a von Hippel-Lindau disease clinical care center when possible. | |
|
c. If routine surveillance is not possible, refer to a von Hippel-Lindau disease clinical care center if a tumor with a diameter greater than 1.5 cm is identified, if there is any tumor growth between two scans, or in cases of suspected PNET metastases (34). | |
|
4) Retinal hemangioblastomas: Dilated eye exam every 6 to 12 months starting before the age of 1 (13). | |
|
5) Pheochromcytoma/paraganglioma: Annual metanephrines starting at the age of 5. Blood pressure and pulse measurements annually starting at the age of 2 (13). | |
|
6) All intraabdominal lesions: MRI abdomen with/without contrast every 2 years starting at the age of 15 (13). | |
Retinal hemangioblastoma may be diagnosed by careful examination of the ocular fundi. Dilated binocular funduscopic examination is the screening test of choice to detect this lesion. Intracranial, spinal, intraabdominal, and epididymal tumors can be detected with MRI and MRA. Depending on the size, location, and nature of the tumor and the presence or absence of clinical symptomatology, the indications for surgical or other types of treatment may be established. Mathematical models to determine the optimal age for initial screening and frequency of screening for various Von Hippel-Lindau disease-associated lesions were published that propose earlier screening for adrenal lesions and delayed screening for retinal lesions (33). Revised screening criteria for children have been proposed (57), with the latest update by the International VHL Surveillance Guidelines Consortium and VHL Alliance released in 2023 (13).
Eyes. The management of retinal hemangioblastomas is through a combination of surveillance and both ablative and nonablative treatment. Surveillance should start as early as possible as it is the most frequent presentation diagnosed in childhood (03). The tumors in the region of the optic disc should be kept under surveillance unless they show evidence of progression due to potential visual loss through ablative treatments (74).
Central nervous system. Hemangioblastomas of the cerebellum are more easily approached than those of the brainstem or spinal cord. Although the resection of the solitary tumor is successful in a high percentage of cases, the final result depends on several factors, such as the nature of the hemangioblastoma, cystic versus solid (better results in cystic than in solid lesions), localization, and size. Microsurgical techniques and stereotactic radiosurgical treatment with preoperative embolization of vessels that feed the hemangioblastoma have considerably improved the prognosis of these tumors in symptomatic patients with nonresectable tumors (47; 01). The result of a study suggests infratentorial craniospinal irradiation as a potential therapeutic option for hemangioblastomas in von Hippel-Lindau disease (63). Nevertheless, the strong tendency of multifocality and recurrence remain serious problems that are incompletely resolved. Erythrocytosis, probably provoked by erythropoietin stimulation from secretions of hemangioblastoma cells, resolves after resection of the tumor and reappears in recurrent cases.
Hemangioblastomas of the brainstem are more challenging surgically, and their excision should be limited to the symptomatic tumors (75). Radiosurgery also may be used to treat symptomatic tumors that are not surgically resectable.
Spinal cord hemangioblastomas should be treated only if they produce progressive symptoms. The long-term results are considerably better in cases treated surgically than in those not operated (44). Moreover, diverse techniques of modern surgery, such as microsurgery, tumoral embolization, laser therapy, radiotherapy, and radiosurgery, also have provided more options for treatment.
Renal lesions. Surgical excision of benign renal cysts is usually not recommended even though excision may prevent malignant transformation. For renal cell carcinoma, surgery is deferred until the largest lesion measures greater than 3 cm as lesions smaller than 3 cm have very low rates of metastasis (70). Nephron-sparing surgery is performed to preserve renal function and delay the need for dialysis and renal transplant (04). To minimize future procedures, removal of all feasible lesions is recommended with the use of intraoperative ultrasound to help identify smaller lesions. In the United States, lesions smaller than 3 cm are followed with active surveillance. Radiofrequency ablation and cryotherapy ablation of renal lesions can also be utilized; however, surgeries following thermal ablation techniques have higher rates of adverse events (30).
An HIF-2alpha inhibitor called belzutifan has become available to treat tumors associated with von Hippel-Lindau disease (26).
Pancreas. Recommendations for the management of pancreatic lesions have been suggested; cystic lesions can generally be managed by observation, whereas solid lesions are of greater concern due to risk of malignancy (31). Biochemical markers are not recommended as a screening tool for PNET, though plasma pancreatic polypeptide may be used in patients with von Hippel-Lindau disease–related PNET (34). Pancreatectomy is usually not indicated, even in the presence of a completely cystic pancreas, if the patient is asymptomatic and there is no evidence of malignancy. Complete excision is indicated for mucinous cystadenoma and for carcinoma.
Pheochromocytoma. This tumor should be excised as soon as it is diagnosed and localized if the patient is otherwise stable. A few reports have advocated for adrenal-sparing surgery in pediatric patients so as to postpone steroid replacement therapy (15; 67).
Epididymis and broad ligament. In general, it is not necessary to excise cystadenomas.
Inner ear endolymphatic sac tumor. Early detection and surgical treatment of small lesions, when hearing is still present, may reduce the incidence and severity of hearing loss and vestibular dysfunction associated with these tumors (32).
Tumors of the brainstem and spinal cord carry a high risk, including damage to neural tissue during surgical excision. The size of the lesion is important for selection of operative technique. A small retinal tumor may be resolved with photocoagulation or cryotherapy, whereas larger tumors often associated with secondary changes, such as retinal detachment, exudation, or hemorrhage, carry a high risk of visual loss. The cumulative probability of visual loss in all VHL gene carriers by age of 50 years was estimated to be 35% (72). Small, solitary tumors confined to the kidney may be excised by a glomerular sparing surgery or laser ablation, whereas a large mass may require total nephrectomy. The prognosis, in spite of surgery, continues to be poor in cases where the tumor has spread outside of the kidney and in cases of bilateral renal cell carcinomas.
In select cases, pregnancy may influence the course of von Hippel-Lindau disease; additionally, it can constitute a complication in cases requiring surgical intervention (50; 20). Patients with known cerebellar hemangioblastoma may develop intracranial hypertension late in pregnancy (19), and growth of cerebellar hemangioblastoma during pregnancy has been demonstrated (16). However, another group has now shown that hemangioblastoma progression is not altered by pregnancy, which has sparked debate in the field (77). The general consensus appears to be that small patient populations are limitations in all of the studies and that a larger cohort will need to be utilized in order to fully explore the effect of pregnancy on hemangioblastoma progression in patients with von Hippel-Lindau disease.
Additional reported complications include the onset of eclampsia late in pregnancy due to inoperable pheochromocytoma (19). Improved methods of nonradiological imaging have reduced the risk to the fetus and the mother during diagnostic studies. Fundamental problems result from proximity of some structures (pancreas, adrenals, and kidneys) to the uterus and of the necessity of using general anesthesia with tumors in any location except in most ocular lesions, which can be treated under local anesthesia.
Any type of anesthesia may be used in patients with von Hippel-Lindau disease at any age. Standard precautions should be taken for pregnant women and hypertensive patients with pheochromocytoma.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Yelena Wilson DO
Dr. Wilson of Akron Children's Hospital has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 27, 2026
Neuro-Oncology
May. 23, 2026
General Child Neurology
May. 12, 2026
Neuropharmacology & Neurotherapeutics
May. 11, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026