






Neuromuscular Disorders
Viral and retroviral myositis
Jun. 16, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Walker-Warburg syndrome is a rare autosomal recessive disorder and the most severe presentation in the dystroglycanopathy spectrum. It combines congenital hydrocephalus; lissencephaly with cobblestone malformation; eye abnormalities, especially retinal dysplasia; and congenital muscular dystrophy. Walker-Warburg syndrome is genetic heterogeneous, with 14 genes identified to date. Gene functions converge on glycosylation of alpha-dystroglycan, a component of the dystrophin-glycoprotein complex mediating important interactions between cells and the extracellular matrix. The authors review the present knowledge of Walker-Warburg syndrome.
|
• Walker-Warburg syndrome is part of a spectrum of presentations within dystroglycanopathies. It is closely related to muscle-eye-brain disease and Fukuyama-type congenital muscular dystrophy as they all share many characteristics. | |
|
• Neuronal migration disorder with cobblestone malformation is the most typical expression. | |
|
• Eye anomalies affect anterior and posterior parts of the eye. | |
|
• Congenital muscular dystrophy with decreased staining for alpha dystroglycan glycosylation is a shared characteristic. | |
|
• Mutations in a number of genes involved in alpha-dystroglycan glycosylation account for more than half of cases. |
Walker-Warburg syndrome is named after the Danish ophthalmologist Mette Warburg and the American neuropathologist A. Earl Walker. These researchers pioneered detailed descriptions of the syndrome, with its highly characteristic ophthalmopathologic and neuropathologic components (68; 69). Warburg suggested autosomal recessive inheritance (69).
The phenotype is reflected in the different names applied. By 1983, all components then known appeared in the mnemonic: HARD±E, denoting hydrocephalus, agyria, retinal dysplasia, with or without encephalocele (48), but the proposal was changed to Warburg syndrome in honor of Warburg's pioneering contribution (49). Dambska and colleagues drew attention to the peculiar pattern of seeming architectural chaos with loss of neocortical stratification (17). The neuromuscular component manifesting as congenital muscular dystrophy was recognized through the work of Dambska and colleagues and Towfighi and colleagues (17; 62). An official subclassification of lissencephaly types, types 1 and 2, was initiated by Dobyns and colleagues, where type 2 belonged to Warburg syndrome or cerebro-oculo-muscular syndrome and type 1 to the regular 4-layered type of lissencephaly, typically found in the chromosomal Miller-Dieker syndrome and the isolated Norman-Roberts syndrome, presently known as LIS1 mutations (18). They also proposed to change the name of the disorder once more, this time to Walker-Warburg syndrome in equal recognition of Walker's early neuropathologic contribution. Following a suggestion by Haltia (20), Dobyns proposed to change the name to cobblestone lissencephaly (19). Thus, this malformation is referred to as either type 2 lissencephaly or cobblestone lissencephaly in the literature. The most recent classification guidelines for cortical malformations have coined the term “cobblestone malformation,” which is applicable to both cases with smooth or undersulcated brain with thin cortex in Walker-Warburg syndrome and cases with pachygyria and thicker cortex (57). These nomenclature changes reflect a better understanding of the shared neuronal migration deficits found in the cortex leading to different presentations based on severity. In classic lissencephaly, the neocortex is macroscopically smooth, whereas the neocortex in cobblestone malformation is “bumpy” and uneven, resembling the eponymous cobblestone.
Due to the heterogeneous presentation of dystroglycanopathies, when Walker Warburg syndrome has an identified genetic cause in the OMIM® database, it is also referred to as muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies) type A, or MDDGA, and labeled from MDDGA1 to MDDGA14.
Histological studies of postmortem tissue show that cortical lamination is severely disrupted, with neurons sometimes arranged in columns and whorls often surrounding penetrating vasculature.
Typically, the glia limitans is broken, and there are gaps in the pial basement membrane causing leptomeningeal, neuronal, and glial heterotopias.
Despite the nomenclature changes, the main constituents of the syndrome have been well established since the 1980s: (1) variable eye malformations with typical retinal dysplasia combined with anterior chamber abnormalities; (2) cobblestone malformation, hydrocephalus, hypoplasia, and malformation of the cerebellum; (3) congenital muscular dystrophy; and (4) autosomal recessive inheritance.
As gene discovery has progressed, revealing variable phenotypic presentations among mutations in the same gene, Walker-Warburg syndrome has become part of a spectrum of disorders with shared disruptions in alpha-dystroglycan glycosylation, termed “dystroglycanopathies.” Other dystroglycanopathies affecting the brain and eye are muscle-eye-brain disease and Fukuyama congenital muscular dystrophy. Additional less severe presentations include limb-girdle muscular dystrophy with or without intellectual disability.
Infants with Walker-Warburg syndrome are symptomatic from birth, and a diagnosis can be suspected if a routine ultrasound during the third trimester reveals hydrocephalus. Manifestations at birth include generalized hypotonia and severe generalized paresis, macrocephaly due to hydrocephalus (occasionally microcephaly), and eye abnormalities that encompass anterior as well as posterior chamber abnormalities.
Epilepsy is not a complication in all patients, but at least one third develop seizures during infancy or early childhood. Seizures may be generalized major motor, partial, myoclonic, or a mixture of types.
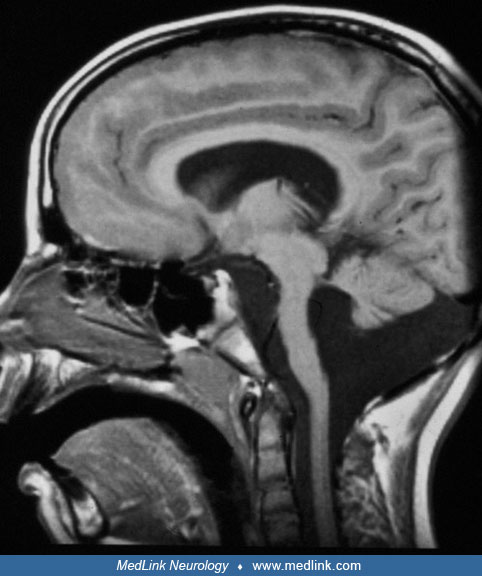
Ultrasonography, CT, or MRI will reveal gross hydrocephalus, together with partial aplasia of the cerebellar vermis, hypoplasia of the cerebellar hemispheres, and abnormal structure of the brainstem. In one third of the patients, posterior encephalocele or occipital meningocele is encountered. MRI is superior in demonstrating all the cerebral abnormalities of which the disease is composed (16; 57). MRI findings include a smooth neocortical surface, an irregular inferior cortical margin, hypoplasia or partial aplasia of the cerebellar vermis, and hypoplasia of the pons and brainstem. Jissendi-Tchofo and colleagues emphasized characteristic MRI abnormalities of the midbrain and hindbrain (32). Kinking of the brainstem in sagittal images is a characteristic finding; however, this abnormality is also observed in tubulinopathies (27). Rarely, cerebellar cysts may be found (52).
T1-weighted inversion recovery MRI of a 4-month-old male with severe hydrocephalus due to Walker-Warburg syndrome. In this section, the border between cortex and adjacent white matter is irregular because of the irregular struc...
Ophthalmological presentation includes microphthalmia and both anterior chamber (cataracts, colobomas, Peters anomaly) and retinal findings, such as retinal dysplasia and retinal detachment.
Serum creatine kinase is usually elevated. Muscle abnormalities consist of increased variability in diameters, affecting both fiber types: small fibers, with regenerated (basophilic fibers), internal nuclei and rounded fibers, with increase of endomysial connective tissue and increase of fat tissue. Diagnosis of dystroglycanopathy is usually confirmed by finding negative staining for alpha-dystroglycan glycosylation using a glycoepitope-specific antibody in muscle biopsies (45). It is most common to reach a molecular diagnosis using next-generation sequencing via either a neuromuscular disease panel or exome sequencing (74). POMT1, POMGNT1, FKTN, and FKRP mutations are the most common in different populations (24; 58; 59; 34).
Overall, presentation will be variable, with some combination of findings in the brain, eye, and muscle listed in Table 1. Most patients die before their second birthday, but survival may be more prolonged (69).
|
Eye findings | ||
|
• Anterior chamber | ||
|
- Corneal opacities | ||
|
• Posterior chamber | ||
|
- Persistent hyperplastic primary vitreous | ||
|
Brain findings | ||
|
• Agyria with cobblestone aspect of the neocortex | ||
|
Muscle findings | ||
|
• Creatine kinase elevated in serum | ||
|
Other associations (variable) | ||
|
• Polyhydramnios | ||
|
Facial dysmorphia | ||
|
• Low set ears | ||
Prognosis is uniformly lethal at an early age in typical Walker-Warburg syndrome. Although some patients may reach childhood, most will die before their second birthday, and some will not survive the neonatal period because of respiratory problems. Survival in muscle-eye-brain disease and Fukuyama-type congenital muscular dystrophy is longer, and puberty may be reached.
A female patient was born as the third of nonconsanguineous parents who previously had two healthy children. She presented at birth with macrocephaly, 42 cm (+ 6 standard deviation), and normal 3530 g body weight. Advanced hydrocephalus was demonstrated by immediate ultrasonography. She had no other outward stigmata. She had mild flexion contractures of elbows, hips, and knees without deformations. The muscles felt tense, she had areflexia, and her creatine kinase was 3460 IU at 4 days. MRI showed a serrated inferior margin of the neocortex suggesting type 2 or cobblestone lissencephaly, apparently fused frontal lobes, hypoplastic ventral pons, and a hypoplastic cerebellar vermis. Ophthalmoscopy revealed ablatio retinae on the right side. A muscle biopsy from the femoral rectus muscle revealed increased diameter variation, acting on both types, endomysial fibrosis, and some regenerating fibers. Immunohistochemistry showed absence of staining of alpha-dystroglycan glycosylation. Electron microscopy revealed a loss of normal sarcomere structure in the majority of the fibers. A diagnosis of Walker-Warburg syndrome was made. She received ventriculoperitoneal drainage that had to be changed twice because of obstruction. She deteriorated progressively, needing constant gavage feeding from 3 months. She had bouts of aspiration pneumonia and died at 4 months.
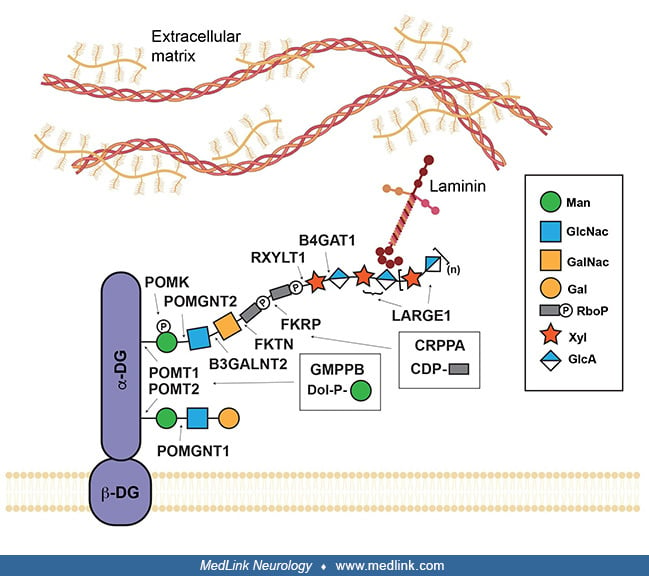
Walker-Warburg syndrome is an autosomal recessive disorder with the characteristic presentation of congenital muscular dystrophy; brain malformations, including hydrocephalus and cobblestone malformation; and eye malformations. It falls at the most severe end of a spectrum of dystroglycanopathies, a group of disorders caused by defects in glycosylation of the transmembrane glycoprotein, dystroglycan. Dystroglycan is part of the dystrophin-glycoprotein complex that links the extracellular matrix to the cytoskeleton in cells in multiple tissues. It is important to note the mutations in most components of this complex lead to different types of muscular dystrophy, highlighting its critical role in muscle function. Mutations in dystrophin result in Duchenne muscular dystrophy, and sarcoglycans are mutated in limb-girdle muscular dystrophy. Research in animal models has shown that the increased severity in dystroglycanopathies lies in the diversity of dystroglycan extracellular binding partners in different tissues during development, leading to distinct phenotypes in the muscle, brain, and eyes. Dystroglycan is synthesized as a precursor molecule that is post-translationally cleaved into alpha and beta subunits. The alpha subunit is outside the cell membrane, where it interacts with proteins containing laminin-G domains in the extracellular matrix through a specialized glycan chain (73; 29). Loss of glycosylation of alpha-dystroglycan in the muscle disrupts interactions with laminins, including the alpha2 subunit, LAMA2, which is mutated in a form of congenital muscular dystrophy with diffuse white matter alterations, and merosin-deficient congenital muscular dystrophy (26). However, cobblestone malformation has also been reported in cases of LAMA2-related muscular dystrophy, indicating that laminin can be involved in neuronal migration deficits (21). Biallelic variants in laminin beta1 (LAMB1) can also cause cobblestone malformations with minimal involvement of eye and muscle (51). Studies in mouse models have shown that interacting proteins in the brain and spinal cord (Slit2, Celsr3) disrupt guidance of axonal processes (71; 40). Other dystroglycan ligands, pikachurin and eye shut homolog, are necessary for the formation and maintenance of photoreceptor synapses transmitting signals from light detection in the retina (56; 41). Agrin, a major regulator of acetylcholine receptor clustering at the neuromuscular junction controlling muscle contraction, is also a well-known alpha-dystroglycan interactor (30). Additional ligands and functions may still need to be identified to understand the multiple players involved in the pathogenesis of these multisystem disorders.
The identification and study of the genes mutated in Walker-Warburg syndrome have been instrumental in understanding the structure of the functional glycan that binds to extracellular ligands. Mutations in dystroglycan itself (DAG1), leading to primary dystroglycanopathy, have been identified but are rare (09). Most individuals with Walker-Warburg syndrome will have a secondary dystroglycanopathy in which mutations disrupt the function of a glycosyltransferase, an enzyme involved in glycan assembly. Briefly, the ligand-binding glycan consists of a trisaccharide core on the alpha-dystroglycan protein assembled by POMT1, POMT2, POMGNT2, and B3GALNT2, a linker with two ribitol phosphate sugars attached by FKTN and FKRP, and a polysaccharide of variable length started by RXYLT1 and B4GAT1 and extended by LARGE1. Because the function of these proteins has often been defined after the initial gene identification, the gene nomenclature has changed to better reflect their enzymatic activity, and several genes are present in the literature under multiple names: POMGNT2 was also known as GTDC2; RXYLT1 was TMEM5; and B4GAT1 was B3GNT1. Loss-of-function autosomal recessive variants in all these glycosyltransferases are expected to disrupt ligand binding to dystroglycan and have been identified in individuals with Walker-Warburg syndrome (06; 42; 44; 11; 31; 60; 65; 64; 66). However, the genotype-phenotype correlation remains unclear as they can all lead to variable phenotypes on the dystroglycanopathy spectrum, ranging from muscle-eye-brain disease, Fukuyama-type congenital muscular dystrophy, congenital muscular dystrophy with intellectual disability, or limb-girdle muscular dystrophy. Because of the rarity of these disorders, population studies have often focused on the entire dystroglycanopathy spectrum, revealing population-specific mutation frequencies and founder mutations.
Dystroglycan is a transmembrane protein connecting the cell to extracellular ligands, such as laminin. Alpha- and beta-dystroglycan (DG) are shown in purple. The site of action of each glycosyltransferase mutated in dystroglyca...
The first genetic mutation leading to a dystroglycanopathy was identified in Japan in Fukuyama-type congenital muscular dystrophy as retrotransposon insertion in the 3’ untranslated region of the fukutin (FKTN) gene, also known as FCMD (35). FKTN mutations are the second leading cause of muscle disease in children in Japan, and the majority of cases (80% to 87%) are due to this founder mutation, which is also present in China and South Korea (36; 34). Individuals who present with dystroglycanopathy with brain and eye involvement may be given a diagnosis of Fukuyama-type congenital muscular dystrophy if FKTN mutations are found. The presentation of FKTN mutations falls in the spectrum of severe dystroglycanopathies, but brain malformations tend to be less severe; ocular phenotypes are found in fewer cases; and, if present, retinal dysplasia is mild and focal (55). FKTN mutations can cause Walker-Warburg syndrome. A one-base pair insertion leading to protein truncation is a common cause of Walker-Warburg syndrome in the Ashkenazi Jewish population and is included for screening in the gene panel available for carrier testing (28).
The first gene associated with Walker-Warburg syndrome was POMT1, which in a complex with POMT2 attaches the first sugar, an O-linked mannose, to the alpha-dystroglycan protein (06). POMT1 was the most frequently mutated gene in a cohort of 47 fetuses with Walker-Warburg syndrome (32%) (08) and in a cohort of individuals from the Middle East (36%) (43). These studies only included the six genes known at that time: POMT1, POMT2, POMGNT1, FKRP, FKTN, and LARGE1. Subsequent studies including different presentations in the dystroglycanopathy spectrum still showed that POMT1 variants contribute to a large fraction of Walker-Warburg cases. In a British study on congenital muscular dystrophies, POMT1 variants were found in four of 12 Walker-Warburg cases (58). In a Chinese study, there was only one Walker-Warburg case included out of 143 patients, and this individual had a POMT1 mutation (59). Although POMT1 mutations can cause the full spectrum of disease and are usually one of the top three most frequent causes of dystroglycanopathy, these findings suggest that they may be more prevalent in Walker-Warburg syndrome. POMT1 loss of function is expected to completely prevent the assembly of the functional glycan involved in ligand binding.
In addition to the glycosyltransferases described above, gene mutations in other proteins involved in glycan assembly and function can be found in cases with Walker-Warburg syndrome. CDP-L-ribitol pyrophosphorylase A (CRPPA), also known as ISPD, was identified as another cause of this severe dystroglycanopathy (53; 70). CRPPA is involved in the synthesis of the ribitol donor, CDP-ribitol, that FKRP and FKTN use to insert ribitol into the functional glycan (22). The pathogenesis in this case involves the metabolism of the sugar, and CDP-ribitol supplementation is currently being studied in preclinical models as a therapeutic approach for CRPPA mutations and for hypofunctional variants in FKRP and FKTN in which increased concentrations of the sugar donor may be beneficial. GDP-mannose pyrophosphorylase B (GMPPB) variants are another cause of metabolic or tertiary dystroglycanopathy disrupting the synthesis of the mannose donor instead (13). GMPPB mutations usually cause presentations on the less severe end of the spectrum, but isolated cases of muscle-eye-brain disease have been reported (13; 03).
POMK variants have also been reported in Walker-Warburg cases (31). POMK (previously SGK196) is an O-mannose kinase that phosphorylates the initial O-linked mannose and enables the elongation of the polysaccharide chain, matriglycan, synthesized by LARGE1 (67).
Another common cause of dystroglycanopathy that can occasionally lead to Walker-Warburg syndrome is POMGNT1. Mutations in POMGNT1 were initially identified in muscle-eye-brain disease (72). POMGNT1 encodes an O-mannose beta-1,2-N-acetylglucosaminyltransferase, adding an acetylglucosamine to mannose, and has the same function as POMGNT2. POMGNT1 was originally thought to contribute to the core trisaccharide in the alpha-dystroglycan laminin-binding glycan. However, it appears to act in a different glycosylation cascade on O-linked mannose, and it facilitates the formation of the functional glycan and cell-cell adhesion (37; 47). Thus, although POMGNT1 mutations can be found in cases with Walker-Warburg syndrome (61), they more frequently lead to muscle-eye-brain disease, which presents with less severe brain malformations and longer lifespan (25).
A few genes were identified to phenocopy the brain malformations in cases suspected with Walker-Warburg syndrome. LAMB1 mutations lead to cobblestone malformation with minimal involvement of eye and muscle (51). A TUBB3 de novo missense variant was reported in a deceased infant with a suspected Walker-Warburg phenotype (hydrocephalus, brainstem kinking, optic atrophy, elevated CK) (50). Cobblestone-like changes can also be found in conjunction with polymicrogyria in severe cases with mutations of the transmembrane receptor GPR56 (05). GPR56 loss of function affects the integrity of the pial basement membrane and can lead to similar neuronal migration defects. Severe hydrocephalus with cerebellar hypoplasia and brainstem kinking was also observed prenatally in cases with biallelic loss-of-function variants in FLVCR1 whose homolog FLVCR2 is linked to a form of proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (54; 12). However, this is most likely a phenocopy leading to congenital hydrocephalus due to a disruption in mitochondrial function in neuronal progenitors (07).
In closing, in the past two decades, remarkable advances have been made in the understanding of Walker-Warburg syndrome and the closely related Fukuyama-type congenital muscular dystrophy and muscle-eye-brain disease. As of April 2025, the following genes have been associated with Walker-Warburg syndrome: POMT1; POMT2; POMGNT2 (GTDC2); POMGNT1; B3GALNT2; FKPR; FKTN; CRPPA (ISPD); RXYLT1 (TMEM5); GMPPB; B4GAT1 (B3GNT1); POMK (SGK196); GMPPB; LARGE1; DAG1 (06; 42; 65; 64; 66; 44; 53; 70; 11; 31; 60). Their function converges on a specific glycan of alpha-dystroglycan that interacts with different proteins in various tissues, including muscle, brain, and eye, leading to a complex phenotype.
The prevalence and incidence of Walker-Warburg syndrome are not well established. The prevalence of congenital muscular dystrophies, including the dystroglycanopathies, are reported to be 0.5 to 0.9 X 100,000 in different European populations (58). Multiple reports have analyzed mutation frequencies in dystroglycanopathies, including Walker-Warburg syndrome, muscle-eye-brain disease, and limb-girdle muscular dystrophy, in different populations in Europe (24; 58) and in China (59). However, cases have been reported all over the world, often with different founder mutations in each population (43). For Ashkenazi Jews, the carrier frequency for FKTN mutations was reported to be 1:79 (43; 15). Fukuyama-type congenital muscular dystrophy is the second most common cause of childhood muscular dystrophy in Japan, where a retrotransposon insertion in the 3’ of FKTN is the most prevalent haplotype in as many as 80% individuals with Fukuyama-type congenital muscular dystrophy (36).
Autosomal recessive inheritance entails a 25% risk of subsequent siblings to affected individuals. Hydrocephalus and encephalocele are candidates for early detection via fetal ultrasound. Ultrasound diagnosis is possible as early as 14 weeks (38). In an at-risk pregnancy (earlier sibling affected), a very early manifestation of a Walker-Warburg phenotype was achieved at 11 weeks of gestation using a high-resolution transvaginal ultrasound probe (01).
Identification of genes associated with Walker-Warburg syndrome now allows precise molecular diagnosis in about 60% to 70% of affected families. An FKTN founder variant is included in the Ashkenazi Jewish carrier testing panel, and clinical genetic testing for the FKTN retrotransposon insertion is available for individuals of East Asian descent. Dystroglycanopathy genes are included in both neuromuscular disease panels and brain malformation panels.
The differential diagnosis of Walker-Warburg syndrome includes Fukuyama-type congenital muscular dystrophy and muscle-eye-brain disease. Fukuyama-type congenital muscular dystrophy is the most prevalent form of congenital muscular dystrophy in Japan and has essential features in common with Walker-Warburg syndrome, though the brain and eye phenotypes are less severe (55). Similarly, there is an overlap of findings in the three critical organs with muscle-eye-brain disease: pachygyria and cortical dysplasia, hypoplasia of the pons, and hypoplasia/dysplasia of the cerebellum (25).
Since the discovery of the common role of alpha-dystroglycan in these disorders, the discovery of dystroglycan deficiency and genetic analyses have taken prominence in diagnosis, and these disorders have become closely linked as dystroglycanopathies affecting the muscle, brain, and eye. Mutations in the same genes may cause any of these three disorders. Clasped (hyperabducted) thumbs may occasionally be seen in Walker-Warburg syndrome. These, in combination with hydrocephalus in males, may suggest X-linked hydrocephalus. Meckel-Gruber syndrome has to be considered for differential diagnosis in the case of occipital encephalocele.
A combination of cerebral abnormalities and congenital muscular dystrophy is found in merosin-deficient congenital muscular dystrophy, which features a severe cerebral myelin deficiency without gross functional deficiencies. Partial lissencephaly may be found in merosin-deficient congenital muscular dystrophy, especially in the occipital regions, leading to an initial diagnosis of dystroglycanopathy (63).
The following table lists the main similarities and differences between Walker-Warburg syndrome, Fukuyama-type congenital muscular dystrophy, and muscle-eye-brain disease.
|
Features |
WWS |
F-CMD |
MEB |
|
Progressive hydrocephalus |
All cases |
None, ventricular system may be enlarged |
Minority |
|
Frontal lobe fusion |
Variable |
Occasional |
No |
|
Neocortical dysplasia |
Cobblestone lissencephaly |
Polymicrogyria mainly frontal, cobblestone lissencephaly |
Frontal and temporal, cobblestone lissencephaly |
|
Cerebral white matter |
Abnormal due to white matter edema |
Delay in myelination |
Multifocal abnormalities or normal |
|
Cerebellar vermis |
Hypoplastic |
Normal size |
Hypoplastic |
|
Pancerebellar cortical dysplasia |
Yes |
Yes |
Yes |
|
Flat ventral pons |
Yes |
Yes |
Yes |
|
Posterior encephalocele |
Occasional |
No |
No |
|
Anterior and posterior eye chamber abnormalities |
Common findings include corneal opacities, Peters anomaly, cataracts, or persistent primary vitreous |
Mild to severe myopia in minority |
High myopia, glaucoma (buphthalmos), cataracts |
|
Retinal dysplasia or detachment |
Both commonly present |
Focal retinal “round” lesions occasionally found |
Retinal dysplasia common, no detachment |
|
Microphthalmia |
Majority, can be asymmetric |
No |
No |
|
Neuromuscular Weakness |
Yes |
Yes |
Yes |
|
Muscular dystrophy with negative alpha-dystroglycan staining |
Yes |
Yes |
Yes |
|
Creatine kinase Elevated |
Yes |
Yes |
Yes |
|
Severe cognitive and sensory delay |
No motor milestones, no cognitive development |
Some head control achieved, unsupported sitting and walking exceptional |
Some head control achieved, unsupported sitting and walking exceptional |
|
Survival |
Most die before 1 year; rare survival beyond 2 years |
Variable survival; puberty may be reached |
Variable survival; puberty may be reached |
|
| |||
Due to severe brain malformations, prenatal detection is possible during routine ultrasounds, and a fetal MRI is recommended in sites where available. Genetic analyses can be performed through chorionic villus sampling and a brain malformation panel, including dystroglycanopathy genes and other causes of congenital hydrocephalus.
In the newborn, examination of the eyes should include an active search for anterior chamber abnormalities in combination with symptoms of retinal dysplasia. MRI is the method of choice to delineate all aspects of the cerebral abnormalities (32; 57). Besides the detection of cobblestone malformation, midbrain-hindbrain involvement helps to differentiate cobblestone complex from classic lissencephaly. These findings include hypoplasia or dysplasia of the vermis and cerebellar hemispheres, small cerebellar cysts, midbrain hypoplasia or dysplasia, pontomedullary kink, and pontine midline cleft (32).
Hypoglycosylation of alpha-dystroglycan in biopsied muscle offers good evidence for a dystroglycanopathy diagnosis. However, genetic testing is now recommended for a combination of hypotonia and brain and eye findings to search for variants in the known genes using a brain malformation or neuromuscular disorder panel or exome sequencing. Recessive mutations in LAMB1 may cause cobblestone lissencephaly with minimal involvement of eye and muscle.
Two genes not related to alpha-dystroglycan glycosylation, GPR56 and TUBB3, are also included in the malformation panels. In case of negative results with severe hydrocephalus, FLVCR1 could also be tested.
No specific therapy is available. Seizures may be treated with standard antiepileptic drugs. Hydrocephalus requires surgical treatment by ventriculoperitoneal shunt, and encephaloceles may require excision of extracranial neural and meningeal tissue and closure of the defect, at least the cutaneous surface.
Cases with prenatal diagnosis should be referred for genetic counseling.
Prenatal detection has been accomplished before 20 weeks’ gestation and through routine ultrasound in the third trimester. Given the variability of expression, it is not certain that detection is possible in all cases.
Prenatal diagnosis by gene analysis is possible.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Maria Chiara Manzini PhD
Dr. Manzini of Rutgers University has no relevant financial relationships to disclose.
See Profile
Ganeshwaran H Mochida MD PhD
Dr. Mochida of Boston Children's Hospital and Harvard Medical School has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuromuscular Disorders
Jun. 16, 2026
Neuromuscular Disorders
May. 27, 2026
Neuromuscular Disorders
May. 21, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026