


Sleep Disorders
Sleep-related urologic dysfunction
Jul. 06, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Narrowing of the cerebral aqueduct of Sylvius is termed aqueductal stenosis. Cerebrospinal fluid flow is restricted but still occurs. Aqueductal atresia, by contrast, is a total obliteration of the cerebral aqueduct, leaving only a few ependymal clusters and rosettes in its place that enable no CSF flow. The aqueduct is the conduit between the third and fourth cerebral ventricles. When narrowed, CSF accumulation dilates the upstream lateral and third ventricles and causes ventriculomegaly that often can be detected in fetal ultrasound and MRI images as early as the second trimester. The consequences and treatment of this condition are discussed in this article.
Stenosis of the cerebral aqueduct of Sylvius:
|
• May be congenital (genetic origin) or acquired. | |
|
• May be isolated; associated with other brain malformations, vascular anomalies, or genetic syndromes; or secondary to acquired lesions that infiltrate, compress, or cause inflammation and gliosis in periaqueductal tissue. | |
|
• Posthemorrhagic hydrocephalus may be due to intra-aqueductal blockage by blood descending from the lateral and third ventricles in intraventricular hemorrhage in the perinatal period. | |
|
• Restricts the circulation of CSF, resulting in dilatation of the lateral and third ventricles increased intraventricular pressure. | |
|
• May result in potentially reversible thinning of the cerebral mantle and corpus callosum. | |
|
• May be progressive, debilitating, or even fatal. | |
|
• Does not cause epilepsy. | |
|
• Treatment of resulting hydrocephalus may be traditional ventriculoperitoneal shunt or, preferentially, by endoscopic third ventriculostomy. |
Hydrocephalus has been depicted in the descriptive literature of the seventeenth and eighteenth centuries and has presumably occurred throughout human history. The most frequent cause of hydrocephalus in the perinatal period is isolated aqueductal stenosis.
The three leading causes of congenital hydrocephalus are neural tube defects (meningomyelocele), aqueductal stenosis, and posterior fossa malformations (35). This article is confined to the discussion of isolated aqueductal stenosis and secondary changes. In this condition, causes and timing are often unclear. However, in some cases the cause is known, wherein malformations compress the aqueduct, resulting in primary stenosis. These include encephalocele, Dandy-Walker malformation, and lumbosacral meningomyelocele (often with Chiari malformation). Though isolated aqueductal stenosis is the most frequent form, a large percentage of patients manifest additional anomalies (42).
One study (N=12) showed the variability in progression of fetal ventriculomegaly due to aqueductal stenosis (44). One-third of cases were terminated as further progression could not be observed because of severe ventriculomegaly; others progressed rapidly in the third trimester, some evolved slowly throughout the second and third trimesters, and others remained stable. Premature delivery or cystic parenchymal change occurred in those who experienced rapid development; except for the stable cases, all required surgical intervention.
Clinical findings in the newborn include bulging anterior fontanelle, splayed cranial sutures, decreased activity, feeding difficulty, vomiting, apnea, bradycardia, setting-sun sign, developmental delay, and failure to thrive; findings in older patients include abnormal hypothalamic function, upward gaze, spasticity, blindness, and intellectual decline (12). Acute bilateral ptosis has been reported, with full eye movement; resolution was achieved with third ventriculostomy (66).
Imaging studies, both fetal ultrasound and MRI and postnatal MRI, reveal generalized ventriculomegaly and nonvisualization of the cerebral aqueduct as seen in midsagittal and axial MRI views (05). An additional feature is obliteration of the subarachnoid space over the cerebral convexities so that the brain appears closely pushed against the inner table of the calvarium. By contrast, in adult normal-pressure hydrocephalus, this subarachnoid space is generally preserved (67).
An important negative feature is that even severe ventriculomegaly with increased pressure in hydrocephalus is not epileptogenic. However, if the hydrocephalus due to aqueductal atresia is associated with other cerebral dysgeneses, particularly involving the cerebral cortex, seizures may be a manifestation of the cortical lesions.
In one study of isolated aqueductal stenosis, mortality averaged 40%, with over one-half of deaths occurring in the first three postnatal months (33). In that study, 95% of patients underwent ventricular shunting, but only 10% were developmentally normal at long-term follow-up. This latter finding has been borne out in another study showing the poor prognosis of shunting in pediatric patients with hydrocephalus (23). In that study (follow-up period at least 20 years), one third of patients were deceased, one half were employed, and nearly one third had a severe mental handicap.
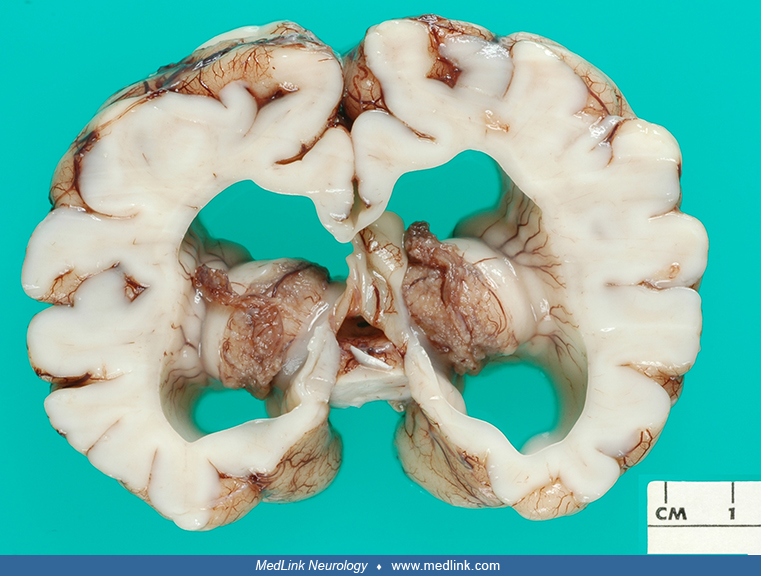
The following photographs depict CNS tissue from four individuals with aqueductal stenosis. In the first three, ventriculomegaly is evident in coronal sections of cerebrum; in the fourth image, an anomalous aqueduct is shown microscopically. Three postnatal patients had additional, extracranial anomalies that led to death--in two cases before shunting was instituted. For privacy concerns, additional clinical details are not provided.
The cerebral aqueduct of Sylvius develops from the central canal within the central embryonic neural tube at the level of the mesencephalic vesicle, which matures to become like the midbrain.
Pathogenesis. The neural tube is characterized by bilateral symmetry and cephalization and is achieved by the establishment of three axes with genetic gradients of expression in each opposing directions along each axis. The cerebral aqueduct is more affected by genes acting in the dorsoventral gradient of the vertical axis, ie, “dorsalizing” gene families such as PAX (48; 47; 48; 51). In some malformations, such as holoprosencephaly, aqueductal stenosis occurs with mutation in genes of each of the three axes; if the rostrocaudal gradient of the longitudinal axis reaches as far caudally as the midbrain, aqueductal atresia usually occurs. The atresia is enhanced by genes of the dorsoventral gradient of the vertical axis and genes of the mediolateral gradient of the horizontal axis, such that the colliculi are noncleaved or fused as a midline dome of the tectal roof of the midbrain and the oculomotor nuclei of each side are fused in the midline, but the ventrally situated cerebral peduncles remain paired and separate (50). The facial dysmorphisms midfacial hypoplasia frequent in lobar and semilobar holoprosencephaly are due to the mesencephalic neural crest not forming or migrating properly and does not occur in cases of alobar holoprosencephaly in which the rostrocaudal gradient in the longitudinal axis does not extend as far caudally as the mesencephalon (50). Stenosis or atresia of the cerebral aqueduct usually also involves poor development of the roof plate and its ependymal processes that form the dorsal medial septum extending to the intercollicular fissure. In isolated aqueductal stenosis, the genetic/neuropathological pathogenesis probably is similar but has been less studied and is not yet well documented.
When fully developed, the aqueduct courses through the midbrain; it is bordered by periaqueductal (or central) gray matter, the tegmentum ventrally, and tectum dorsally. In the newborn, the aqueduct is about 0.5 mm in diameter by 13 mm in length; it is narrower at each end and somewhat wider at its center (18). In the adult, this variability continues. The aqueduct averages 1 to 1.4 mm in diameter and about 14 mm in length. Dimensions for adults, obtained by MRI, differ from postmortem measures and vary with age, with diameter increasing and length decreasing as patients get older and the cerebrum shrinks (37). The cerebral aqueduct initially is lined by primitive neuroepithelial cells, which differentiate as ependyma and a transient form a pseudostratified epithelium, which soon becomes a simple (one cell thick) cuboidal ependymal epithelium, as with the rest of the ventricular system. Loss of p73 in ependymal cells during the perinatal period can lead to aqueductal stenosis by obliteration of the ependymal-lined lumen (20).
Third ventricular diameter in congenital aqueductal stenosis is inversely related to the size of a massa intermedia in the third ventricle (54). The massa intermedia is a residual incomplete thalamic embryonic cleavage at the time of formation of the interhemispheric fissure and is a continuum of the medial thalamic nuclei of the two sides, not a commissure or axonal fascicle as once thought and not a “thalamic adhesion” as it had been erroneously labelled by some authors. The massa intermedia is a common incidental finding by MRI or neuropathologically and is of no clinical consequence and asymptomatic, not causing third ventricular obstruction even if large. In some species of mammals, such as horses, the massa intermedia is usually enormous, nearly filling the third ventricle but not causing obstructive hydrocephalus (Sarnat HB, personal observation, unpublished).
Chronic aqueductal compression by a mass lesion may lead to periaqueductal gliosis and at least partial obliteration of the lumen. Localized swelling in the periventricular and periaqueductal tissue, in particular, also contributes to narrowing and eventual obstruction of the lumen of the aqueduct (07). Dysgenesis of the mesencephalic tectum associated with aqueductal stenosis or atresia is more likely related to genetic gradients than to compression (61). Rarely, an aqueductal web may obstruct CSF flow (25; 68).
Classification. Aqueductal stenosis was previously classified as malformative or acquired (45). In the former, three types were recognized: true stenosis or due to an anomalous septum (both rare) or stenosis due to forking of the lumen. When forking is identified by careful microscopy, two channels are separated by normal CNS tissue.
The absence of gliosis in the young patient may reflect the early age of tissue damage at a time when the fetus is unable to mount a reaction (02). At present aqueductal stenosis may be reclassified into three forms: (1) isolated; (2) associated with other brain malformations, genetic syndromes, or specific identified genetic mutations; (3) acquired, induced by chronic pressure on the midbrain tectum, infiltration by midbrain tumors or inflammation as might be associated with congenital, or early postnatal viral encephalitis.
Without history or corroborating imaging, the cause of congenital stenosis is sometimes unknown, although a perturbation in embryonic development may be postulated in some cases. Because the aqueduct is so narrow, it can be obstructed by debris arising in the ventricular system (for example, sloughed cells or purulent material from an infection or blood products stemming from a hemorrhage). Intraventricular hemorrhages in late fetal life or the neonatal period may cause blockage of the cerebral aqueduct by intra-aqueductal thrombosis (posthemorrhagic hydrocephalus). Chiari II malformation may result in narrowing of the aqueduct and often accompanies lumbo-sacral meningomyelocele. Some cases also include bilateral congenital agenesis of the foramen of Monro for CSF flow from the lateral ventricles into the third ventricle (30). Approximately 5% of cases of isolated aqueductal stenosis are X-linked.
Other midline defects in the brain may accompany aqueductal stenosis in complex brain malformations. Rhombencephalosynapsis (fusion of the medial surfaces of the cerebellar hemispheres and dentate nuclei with hypoplasia or absence of the vermis) is associated with aqueductal stenosis in a significant number of cases. In one review of 20 affected children, 13 (65%) had aqueductal stenosis and hydrocephalus; the finding was uniform in those with complete rhombencephalosynapsis (65). In another study of 30 patients with aqueductal stenosis, one-half manifested rhombencephalosynapsis (29). Rhombencephalosynapsis is associated with aqueductal stenosis in some cases of alobar holoprosencephaly, but most often rhombencephalosynapsis occurs in septo-optic-pituitary dysplasia without aqueductal stenosis (see the MedLink topic “Rhombencephalosynapsis”). Mesencephalosynapsis is also associated with aqueductal stenosis and can be isolated or syndromic (19).
Apart from syndromic aqueductal stenosis as part of a complex brain malformation, a genetic basis for isolated aqueductal stenosis has not been well established, but chromosomal and copy number variants and several single gene mutations have now been demonstrated (17). An example is a pathogenic variant of CCDC88C gene in fetuses with prenatal hydrocephalus and aqueductal forking and atresia, confirmed neuropathologically; this gene encodes the protein DAPLE, which contributes to ependymal cell planar polarity by inhibiting the WNT signaling pathway (36).
Acquired stenosis, even with late onset, may be due to compression from a mass. Such masses may be tectal tumors that infiltrate or compress such as gliomas and pinealomas, pineal cysts, supracollicular lipomas, or vascular malformations (59; 11; 27; 06; 64). Compression of the aqueduct also may result from a persistent and enlarging suprapineal recess of the third ventricle, which usually is a transitory fetal structure or adjacent scarring or gliosis (49). Some of these mass lesions may be congenital. In this sense, a “congenital” narrowing, which is one present at birth, may have an acquired basis.
By older estimates, isolated aqueductal stenosis occurs in two in every 1000 births (42). With the increase in diagnostic abilities, this figure would be expected to change. High quality registry-based data worldwide may clarify the reasons for the higher incidence of hydrocephalus and aqueductal stenosis in lower income parts of the world (60).
Congenital isolated aqueductal stenosis cannot be prevented.
Isolated aqueductal stenosis must be differentiated from cases associated with other CNS or extracranial anomalies. This requires careful imaging. Venous anomalies, including arteriovenous fistulas involving the great vein of Galen, can compress or otherwise obstruct the cerebral aqueduct and imitate the presentation of congenital aqueductal stenosis (27; 06). Perinatal intraventricular hemorrhages in preterm neonates can obstruct the aqueduct (41). Spontaneous third ventriculostomy may occur in some cases of congenital aqueductal stenosis (43). Aqueductal webs in adults occasionally alter CSF flow and can be detected by MRI flow dynamics (68).
Prenatal imaging employs ultrasound in the second trimester and, if fetal hydrocephalus is suspected, it is followed by additional high-resolution ultrasound and fetal MRI in the late second or third trimester (12; 16; 57). The chief diagnostic criterion for ventriculomegaly is an atrial diameter greater than 10 mm measured ultrasonographically. Additional craniometric data based on a large international study have been published (10). Ventriculomegaly due to aqueductal stenosis may be differentiated from other malformative or disruptive causes: brain parenchyma may be compressed, but not otherwise reduced in mass; lateral ventricles are enlarged anteriorly and posteriorly; the third ventricle is dilated, with transverse diameter greater than 2 mm; the choroid plexus is dangling; the fourth ventricle is normal; and, cranial size and configuration are normal with normal-appearing posterior fossa (16). Additional findings may implicate aqueductal stenosis as the cause of enlarging ventricles, chiefly funnel-shaped aqueduct and even direct visualization of an obstructing web or blood clot in the aqueduct (26). Venous malformations require MR angiography in addition to the standard imaging sequences (27; 06). Agenesis of the internal carotid artery may be associated with aqueductal stenosis (64). Perinatal intraventricular hemorrhages often obstruct the aqueduct causing hydrocephalus with dilatation of the third and lateral ventricles (41).
In addition to CNS imaging, a total anatomic survey is indicated to rule out additional anomalies. Infectious causes (eg, rubella, cytomegalovirus, toxoplasmosis) are possible and need to be explored by appropriate testing, including maternal serology. A hematologic workup is indicated in cases of fetal hemorrhage. Detailed anatomic and genetic workup is necessary to confirm or rule out syndromes or specific etiologies (for example, L1CAM mutation or X-linked hydrocephalus). Expectant management should be arranged, in an effort to offset the risk of premature birth with that of progressing hydrocephalus (16). In cases of pregnancy termination, fetal autopsy is important to further delineate findings.
Ventricular shunting has been used for decades, whereby CSF drains from a ventricle to the peritoneal cavity, or less often, the systemic venous return or right atrium. Shunts may become blocked over time and require revision. Milder forms of aqueductal obstruction, those developing from the formation of thin membranes for example, can be treated with aqueductoplasty, sometimes combined with stent placement. Surgical approaches for this procedure may be pre-coronal, suboccipital, or trans-fourth ventricle via the foramen magnum and are used in both children and adults (46; 09). Lumboperitoneal shunting has proven successful, particularly in patients with slit ventricles or ventriculoperitoneal shunt failure (38).
A more recent approach than ventriculoperitoneal shunting is the use of endoscopic third ventriculostomy, which provides equally good results with fewer complications both intraoperatively and later (53; 08; 58; 10; 43; 03; 21; 55; 31). It can be safely used in infants less than a year of age and in patients with an aqueductal web as the cause of obstruction (25; 31). Many neurosurgeons now consider endoscopic third ventriculostomy to be the treatment of choice in hydrocephalus secondary to aqueductal stenosis.
Despite early discouraging results with fetal shunting decades ago, safe prenatal ventriculo-amniotic shunting seems increasingly possible with developments in diagnosis, shunt and valve implements, and surgical technique (13; 40). This will require a carefully controlled prospective study (16). Families of infants requiring shunting may suffer stress and may benefit from intervention, especially in cases of increased morbidity and socioeconomic challenges (01). A multicenter North American study demonstrated that accurate prenatal diagnosis of aqueductal stenosis not only can be made by precision fetal neuroimaging but at gestational ages amenable to in utero intervention (15).
Neuroendoscopic surgical techniques are now employed with increasing frequency in cases of intraventricular infection to lyse and remove purulent material or other debris and irrigate the ventricular cavities with antibiotic saline; secondary hydrocephalus in such cases may require additional shunting (24). Endoscopic septum pellucidotomy may be required for unilateral hydrocephalus. The neural and vascular anatomy of the septum have been described, and ideal areas for surgery have been identified (04). Aqueductal stenosis due to pineal cyst has been treated by endoscopy, with resolution in about 80% of cases (11). However, endoscopic aqueductoplasty has a high risk of long-term failure and is not recommended by some authors who recommend endoscopic third ventriculoscopy as a preferred surgical approach (39). Third ventriculostomy can relieve hydrocephalus secondary to aqueductal stenosis.
Functional outcome is highly variable but appears to be based largely on the severity of ventriculomegaly and presence and effect of additional malformations (34; 42). Patients who are born prematurely and develop hydrocephalus secondary to CNS hemorrhage have reduced motor function and difficulties in behavioral and intellectual development, hearing, and vision (22). In one long-term (mean 5.9 years) follow-up study of 41 patients, nearly one-half had epilepsy and well over one-half were developmentally delayed; three-quarters were shunt-dependent; one-third were normal neurologically (62). The short- and long-term outcomes of fetuses diagnosed prenatally were poorer in those with complex aqueductal stenosis, but ventricular rupture itself did not impact the prognosis (57).
Rare transitory visual impairment was described in a 16-year-old East Indian girl after third ventriculostomy, but she sustained no permanent visual loss (55). An adolescent boy had a tremor associated with hydrocephalus from an aqueductal web; his tremor resolved after third ventriculostomy (25).
A prospective comparison of 81 patients treated with either third ventriculostomy or primary shunting revealed no difference in outcomes between the two groups (800 days median follow-up) or between those who sustained ventriculostomy failure and required later shunting (32). Stenosis may recur following aqueductoplasty alone and may require re-operation with stent placement; transient vertical diplopia or upgaze weakness have been observed in a small subset of patients (46).
A multicenter North American Fetal Therapy Network study demonstrated that women carrying a fetus with aqueductal stenosis were at risk for preterm delivery, Caesarean section if the head is too large for safe vaginal delivery, and maternal morbidity; therefore, prenatal treatment of fetal hydrocephalus may at times improve not only fetal by also maternal outcomes (14).
The search for predictors of outcome continues, one finding being the effacement of sulci seen on imaging, which is taken as evidence for cortical maldevelopment and reduced prognosis (34).
The recurrence risk for isolated aqueductal stenosis is estimated at 4% (63). This estimate is based on older studies and could change in the future as diagnosis improves. The hereditability of X-linked hydrocephalus is different of course and is indication for genetic counseling.
Delivery of an infant with severe ventriculomegaly and expansion of the calvaria is complex and in all likelihood requires caesarian section. The value of prenatal diagnosis and preparation for the delivery cannot be overstated.
An obvious but important point is that the pharmacokinetics of infants and children differ from those of adults and must be considered in patients requiring anesthesia (28). Anesthetic approaches must take into account immature neural pathways and the possibility of developing propofol infusion syndrome but otherwise tend to vary among practitioners and institutions (56).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Sleep Disorders
Jul. 06, 2026
Sleep Disorders
Jul. 05, 2026
General Child Neurology
Jun. 24, 2026
General Child Neurology
Jun. 10, 2026
Epilepsy & Seizures
Jun. 02, 2026
General Neurology
May. 13, 2026
General Child Neurology
May. 12, 2026
Developmental Malformations
May. 08, 2026