

Developmental Malformations
Reducing body myopathy
May. 08, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
In this article, the author reviews and updates information on several anatomical variants of cerebellar hypoplasia in the context of advances in the embryology and genetic programming of cerebellar development. An expanded differential diagnosis based on literature of associated cerebral malformations, neuronal migration disorders, syndromes, genetic diseases, chromosomal abnormalities, and metabolic disorders is discussed, and the clinical, neuroimaging, and pathological findings are reviewed. Features of disorders associated with an enlarged cerebellum and cerebellar dysplasia are briefly mentioned. The difference between primary cerebellar malformations and prenatal disruptions of cerebellar development is discussed. Cognitive and affective disorders associated with cerebellar malformations are highlighted.
|
• Cerebellar hypoplasia is a feature of several neurodevelopmental disorders in which the cerebellum is small in size but normal in shape. | |
|
• The etiology of cerebellar hypoplasia is diverse and includes various syndromes, genetic diseases, chromosomal abnormalities, and metabolic disorders. | |
|
• Prenatal disruptions of cerebellar development may mimic primary cerebellar hypoplasia. | |
|
• Disorganized development of the cerebellum causes cerebellar dysplasia in which the shape and “texture” of the cerebellum appear abnormal due to the presence of abnormal folia pattern or gray matter nodular heterotopia. | |
|
• Macrocerebellum is a rare malformation that may occur as an isolated finding or as part of a few syndromes. | |
|
• Cerebellar hypoplasia, dysplasia, and enlargement are best visualized on MRI. |
Small cerebellums were noted by pathologists of the 19th century. Norman was the first to recognize the "granuloprival" form of selective absence of granule cells, though he attributed the change to a degenerative disease process, as did subsequent authors (110). Numerous genetic defects and associated anomalies, some due to inborn errors of metabolism, are described in association with cerebellar hypoplasia. Cerebellar dysplasia may be associated with cerebellar hypoplasia, hence, their inclusion in this article. Disorders associated with large cerebellar size are rare and are mentioned briefly.
Cerebellar hypoplasia is a feature of several neurodevelopmental disorders and occurs during fetal life. The cerebellar shape (or the affected part) is generally normal or near normal, but its volume is reduced in cerebellar hypoplasia (122). Cerebellar hypoplasia should be distinguished from acquired and progressive cerebellar atrophy in which tissue loss occurs with secondary enlargement of the cerebellar fissures. Differentiating cerebellar atrophy from hypoplasia can be difficult in practice (122; 23; 05); however, newer techniques using diffusion tractography biomarkers are showing an early promise (54). Degeneration of the granular layer may resemble hypoplasia (117). In addition, there is an overlap between cerebellar development and neurodegeneration, which means that they cannot be classified separately at times (101). Atrophic processes beginning in fetal life (eg, in pontocerebellar hypoplasia type 1) are challenging because they are both developmental and degenerative at the same time. Furthermore, many conditions previously regarded as primary malformations are actually acquired disorders in utero and are referred to as cerebellar disruptions (123).
Hypoplasia may affect the vermis selectively with sparing of the lateral hemispheres; the medial borders of the hemispheres may be fused in the midline (as seen in rhombencephalosynapsis), or a space may remain between them. In Chiari I malformation there is cerebellar tonsils herniation; this is not discussed in this article. Chiari II malformation is discussed briefly. Chiari III malformation is associated with cervical encephalocele.
The most common condition is global cerebellar hypoplasia. Selective symmetrical cerebellar hemispheric involvement with sparing of the vermis is more rare than selective vermal involvement and has been described in disruptive cerebellar development secondary to prematurity and in pontocerebellar hypoplasia type 2 (122). Several malformations of the midbrain and hindbrain have been reviewed and are discussed below (01; 05; 104).
The cerebellum plays a prominent and an important role in movement and cognition (149). Therefore, cerebellar dysfunction causes impairment in these functions manifesting in a variety of symptoms and signs. The two most constant features of all cerebellar hypoplasia in infancy are global or gross motor developmental delay and generalized muscular hypotonia (23). Motor skills, such as head control, rolling over, sitting, crawling, standing, and walking, develop late (120). There is no regression or loss of acquired skills. The hypotonia may be mild but more commonly is moderate to severe. If the vermis is selectively involved, axial and proximal limb girdle muscles are affected most.
Rapid alternating movements and fine finger coordination are impaired if the cerebellar hemispheres are involved resulting in dysdiadochokinesia, intention tremor, rebound, and dysmetria on clinical examination (149). Most nonprogressive cases of early-onset ataxia are symptomatic of cerebellar hypoplasia, but a normal-appearing cerebellum is sometimes found in imaging studies of such children (23). Titubation of the trunk when the infant is brought to a sitting posture usually predicts ataxia when the child eventually does walk, usually at 18 to 24 months. Poor balance and wide-based gait may be seen during walking or running. Speech may be dysarthric.
Tendon stretch reflexes are usually preserved, though hypoactive or difficult to elicit, raising a differential diagnosis of primary myopathy or neuropathy. Perinatal denervation of muscle due to ventral horn cells degeneration is a feature of pontocerebellar hypoplasia type 1, a progressive degenerative disease with muscle weakness and diminished or absent reflexes (182). Hypotonia and diminished amplitude of tendon stretch reflexes in cerebellar hypotonia are due to decreased fusimotor muscle spindle activity.
Eye movement abnormalities occur frequently in children with cerebellar hypoplasia. A careful neuroophthalmologic examination is important and helpful (145). Smooth ocular pursuit is usually jerky (ie, saccadic), and saccades may be hyper- or hypometric. The vestibulo-ocular reflex may be impaired. Several types of nystagmus occur, including gaze-evoked, rebound, downbeat, upbeat, and periodic alternating nystagmus. They are usually seen in the most severe cases. Nystagmus may result in decreased visual acuity. Visual acuity may also be diminished if there is an associated optic nerve hypoplasia. Strabismus is often found in children with both cerebral and cerebellar signs. Some children with vermal hypoplasia manifest with infantile-onset saccade initiation delay, previously known as congenital ocular motor apraxia (148).
Because cerebellar hypoplasia may not be an isolated malformation in many instances, but part of dysgenesis that involves the cerebral cortex as well, supratentorial symptoms and signs may be additional features. There is an increased risk of epilepsy, developmental delay of language and cognitive functions, intellectual disability, autism or autistic features, and other psychiatric disorders (120). However, several studies have shown that isolated cerebellar malformations are commonly associated with mild to moderate developmental delay, language deficits, disruptive behavior, autistic features, and disorders of cognitive and affective development, providing supporting evidence for the role of the cerebellum in cognitive and affective functions (149). Investigations in young children with cerebellar malformations have also reported a high prevalence of neurologic, developmental, and functional disabilities (26; 120), including motor, cognitive, language, and social-behavioral dysfunction, which lead to poor quality of life. Vermis abnormalities seem to be associated with expressive language and gross motor delays (26) and negatively impact verbal working memory. Some critics have maintained that it is difficult to prove without detailed neuropathological studies that these malformations are truly isolated to the cerebellum. The presence of epilepsy in cases with apparently isolated cerebellar hypoplasia on neuroimaging indicates that supratentorial structures are also involved (177), perhaps at a microscopic level of the cortex below the limits of MRI resolution.
Cerebellar hypoplasia is a frequent accompanying feature of many major neuroblast migratory disorders of the cerebral cortex, including periventricular nodular heterotopia, polymicrogyria, subcortical band heterotopia, and lissencephaly types 1 and 2 (lissencephaly type 2 is now called “cobblestone malformation”) with a variety of known genetic mutations or deletions (115; 135), as well as in other malformations, such as septo-optic-pituitary dysplasia and holoprosencephaly or lissencephaly/pachygyria (135). These patients usually exhibit supratentorial clinical features, including corticospinal tract signs with exaggerated tendon stretch reflexes; hence, depressed tendon reflexes are not a reliable sign of cerebellar hypoplasia. Congenital muscular dystrophy may be associated with cerebral (eg, cobblestone malformation) and cerebellar dysgenesis, as in Walker-Warburg syndrome, Fukuyama muscular dystrophy, muscle-brain-eye disease, or neural dysgenesis restricted to cerebellar hypoplasia or atrophy (32). Therefore, muscle weakness, contractures, microcephaly, and ocular abnormalities may be evident.
Oligophrenin-1 (OPHN1), with deletions of exons 16 to 17, is one of the first genes identified in X-linked mental retardation and is associated with cerebellar hypoplasia in addition to cerebral cortical dysfunction (199). It causes ataxia, mental retardation, a specific neuropsychological profile, and facial dysmorphism. In general, hydrocephalus and intracranial hypertension are not features of cerebellar hypoplasia, except in some specific types such as the Dandy-Walker malformation.
Cerebellar hypoplasia may be classified by anatomical defect in relation to the molecular genetic and embryological development of the cerebellum; this classification is summarized in Table 1. Disorders associated with enlarged cerebellum and cerebellar dysplasia are also included. Similar schemes of classification of cerebellar hypoplasia based on neuroimaging characteristics including that of the fetus have been published as well (18; 134; 05). Classification of abnormal cerebellar foliation and fissuration has also been reported (45).
|
(1) Aplasia of the cerebellum | |
|
(a) Granuloprival type | |
|
(4) Predominant aplasia or hypoplasia of the vermis without fusion of the cerebellar hemispheres | |
|
(a) Joubert syndrome and related disorders | |
|
(5) Aplasia of the vermis with fusion of the cerebellar hemispheres in midline (rhombencephalosynapsis) | |
Truncal titubation in infancy, hypotonia, and ataxia will persist but may improve with further cerebral and cerebellar maturation (23). Associated findings, such as global developmental delay, intellectual disability, autism spectrum disorder, and corticospinal tract signs, are static deficits, but they contribute significantly to morbidity with increasing age (120). In a retrospective study of fetuses with isolated cerebellar hypoplasia caused by presumed hemorrhage on fetal MRI, six of ten cases had normal developmental and cognitive outcomes (154). Mild to moderate deficits were described in three cases, and one case had autism spectrum disorder. Vermis involvement was not a reliable predictor of poor outcome.
A 4-month-old girl presented with poor head control. Pregnancy and delivery were normal. She smiled at 2 months and cooed by 3 months. Parents were unrelated. Family history was unremarkable.
On examination, she was not dysmorphic. Her head circumference was normal for her age. She had head lag. She did not have nystagmus. Funduscopy was normal. Her hands were fisted, and she had cortical thumbs. Her tone was increased, and her reflexes were brisk.
At 8 months, her gross motor development was delayed. She had peripheral hypotonia. Gait ataxia became apparent soon after she started walking at 2 years of age. Upper limbs coordination and speech were normal. Her cognition was normal. Between 4 and 6 years of age, her gait ataxia was mild. She was able to run but was clumsy. At 7 years of age, she developed attention deficit hyperactivity disorder (ADHD) that was responsive to treatment with methylphenidate.
Investigations over the years included normal complete blood count, electrolytes, glucose, calcium, plasma lipid profile, albumin, ALT, AST, TSH, T4, ammonia, lactate, uric acid, amino acids, total and free carnitine, acylcarnitine, alpha fetoprotein, and vitamins E and B12 levels. Very-low-density-lipoprotein (VLDL) receptor and fragile-X DNA testing was unremarkable. Urine organic acid and mucopolysaccharidosis screen were unremarkable. Brain MRIs at the ages of 5 months, 3.5 years, and 5 years showed nonprogressive vermis hypoplasia. Supratentorial abnormalities were not evident.
Introduction. The cerebellum has the longest period of embryological development of any major structure of the brain. Differentiation of neuroblasts in the cerebellar plates along the dorsolateral surface of the developing medulla oblongata begins at about 32 days' gestation; neuronal migration from the external granular layer of the cerebellar cortex is not completed until a year postnatally. Because of this extended period of ontogenesis, the cerebellum is vulnerable to teratogenic insults and metabolic or noxious influences longer than most parts of the brain. However, it is not as vulnerable to prenatal, perinatal, and postnatal hypoxic-ischemic insults (104).
An understanding of cerebellar hypoplasia requires knowledge of both the molecular genetic programming of cerebellar development and classical morphological embryology. Several genes are necessary for not only the differentiation but also the maintenance of specific cell lineages in the cerebellum and are consequently expressed not only in the fetus but also in the adult. Cerebellar granule cells are maintained by the transcription products of Pax-6 and also of the Zinc finger genes, RU49 and Zic (161; 193; 68). A genetic mutation rendering these genes nonfunctional would explain the failure of differentiation or lack of preservation of granule cells in the granuloprival form of global cerebellar hypoplasia.
Nongenetic factors may also cause the selective depletion of granule cells in the fetal cerebellum. Alcohol impairs granule cell migration in the developing cerebellum of a mouse model (77). Experimentally, there are excellent animal models of granule cell depletion during development induced by several teratogenic factors that provide insight into pathogenic mechanisms that probably operate in humans as well. One important model is replicating viral infections, including parvovirus of the embryonic rodent and kitten (103; 156). In human disease, cerebellar hypoplasia may occur as a result of congenital cytomegalovirus infection (17). Varicella encephalitis in older children is a recognized cause of acute cerebellar ataxia but usually does not result in cerebellar atrophy and permanent clinical deficits. Congenital zika virus infection has emerged as an important cause of congenial microcephaly following an outbreak in Brazil in 2015. Early infection during pregnancy is associated with worse outcome including fetal death. Other neurologic features include developmental delay, seizures, spasticity, and congenital contractures (121). A spectrum of brain abnormalities can occur, including neuronal migration abnormalities involving the cerebrum and cerebellum, delayed myelination, ventriculomegaly, cerebral and cerebellar calcification, brainstem hypoplasia, and cerebellar hypoplasia or dysplasia. These abnormalities appear to persist and result from disruption of brain development and destruction of the brain parenchyma. The abnormalities can be detected on fetal ultrasound and MRI (Chimelli and Avvad-Portari 2018; 121; 11; 150).
A second important animal model of selective granule cell depletion in the fetal or neonatal cerebellum is produced with cytotoxic or antimetabolic drugs (191). The external granular layer is capable of mitotic proliferation to repopulate itself following brief or mild exposure to drugs that induces granule cell lysis (157). This reconstitution of the external granule cell layer occurs in the human cerebellum, as well as in experimental animals (132).
After severe neonatal asphyxia, the repopulation of granule cells may be incomplete (196). The external granule cells of the cerebellum and primary olfactory receptor cells are rare examples in the human neonatal brain where neuroblasts retain a capacity for mitotic regeneration. After regeneration, the lamination of the cerebellar cortex is usually irregular and focal hamartomas occur.
Overview. A genetic etiology accounts for many cases of cerebellar hypoplasia. Most hereditary disorders with nonprogressive cerebellar hypoplasia are transmitted as autosomal recessive traits (23). Several may be X-linked (199). Dominant inheritance has been reported but is rare (81; 23). In one subtype of the dominantly inherited spinocerebellar ataxia (SCA) 13, a specific allelic variant (R423H) in the voltage-gated potassium channel KCNC3 (Kv3.3) was found to be associated with a nonprogressive SCA13 phenotype. This specific KCNC3 mutation causes early-onset developmental delay that improves over the years in both motor and cognitive domains, seizures, cerebellar signs, and a nonprogressive cerebellar hypoplasia on MRI (81).
A significant number of patients with isolated cerebellar hypoplasia or dysgenesis of the cerebellar hemispheres remain without a diagnosis (25). The MRI features and genetic causes of several malformations involving the cerebellum with or without brainstem involvement have been reviewed (01). New genetic causes of cerebellar hypoplasia have been reported, with some involving genes that are important in vasculogenesis, thus suggesting abnormal vascular development as a cause of some cerebellar malformations (09).
Cerebellar hypoplasia may be a feature of many disorders of neuroblast migration in the cerebral hemispheres, including periventricular nodular heterotopia caused by a mutation in the filamin-A (FLNA) gene, bilateral fronto-parietal polymicrogyria caused by mutations in the GPR56 gene (115), lissencephaly type 1 due to LIS1 mutations or microdeletion as in Miller-Dieker syndrome, X-linked lissencephaly type 1 with abnormal genitalia caused by ARX gene defect (80), X-linked subcortical band heterotopia due to DCX gene mutations (138), and cobblestone malformations associated with congenital muscular dystrophies (32), such as Walker-Warburg syndrome and Fukuyama disease.
Some of the same genes that are important in mediating radial neuroblast migration in the cerebrum, such as reelin (RELN) and its downstream gene DAB-1, also serve to mediate the inward migration of cerebellar external granule neurons (70). Mutations in the RELN gene are associated with lissencephaly type 1 and cerebellar hypoplasia (115). In addition, mutations in genes involved in microtubule formation and function (eg, TUBA1A, TUBB2B, TUBB3, TUBB5, and TUBG1), have been reported in fetuses and children with malformations of brain development, including cerebellar hypoplasia or dysplasia with or without lissencephaly, pachygyria, and polymicrogyria (63; 135; 96). De novo mutations in MAST1, a microtubule associated protein, were reported to cause mega corpus callosum syndrome with cerebellar hypoplasia and cortical malformations including gyral simplification (170).
Cerebellar hypoplasia occurs in “dysequilibrium syndrome” in the Hutterite population in western Canada, an Anabaptist sect with some degree of consanguinity. It is transmitted as an autosomal recessive trait. The pons is also hypoplastic, and cerebral gyration is simplified (61). The defective gene in Hutterite dysequilibrium syndrome has been identified as the very low density lipoprotein receptor (VLDLR) gene, a component of the reelin signaling pathway for neuroblast migration to the cerebral and cerebellar cortices (29). VLDLR-associated cerebellar hypoplasia should be more accurately named VLDLR-associated pontocerebellar hypoplasia because in general, the differential diagnosis of pontocerebellar hypoplasia is different from that of cerebellar hypoplasia. Several mutations in VLDLR have been published thus far in Hutterite, Iranian, Turkish, and Irish/Scottish/German families. The disease phenotype and neuroimaging findings in these families are similar, with the exception of the Turkish families who display quadrupedal locomotion (162). The disorder in the Turkish families is also known as Uner Tan syndrome. It includes a collection of autosomal recessive single gene disorders and manifests with intellectual disability, ataxia, quadrupedalism, and cerebellar hypoplasia. Disease-causing mutations in VLDLR, WDR81, CA8, ATP8A2, and recently recessive tubulin mutations in TUBB2B have been reported in this syndrome (31). Cerebellar vermis and hemispheres hypoplasia or agenesis has also been reported in tuberose sclerosis in addition to cerebellar tubers and cerebellar folia calcification on neuroimaging (159).
Mutations in 35 genes have been described in Joubert syndrome and related disorders. Defective genes include AHI1, NPHP1, CEP41, CEP290, TMEM67, TMEM231, TMEM237, ARL13B, CC2D2A, INPP5E, KIF7, KIAA0586, C5orf42, CSPP1, MKS1, DYNC2H1, CELSR2, and RPGRIP1L, SUFU, and ARL3 (101; 128; 112). The oral-facial-digital syndromes are hereditary conditions that are classified into 13 types and may include cerebellar hypoplasia amongst their multiple anomalies of the face, digits, tongue, and hypothalamic hamartoma (65). Type VI (Varadi-Papp syndrome) is associated with the molar tooth sign on MRI.
Neonatal diabetes mellitus is reported with cerebellar hypoplasia or even aplasia, suggesting an autosomal recessive trait. Mutations in the Ptf1a gene appear responsible for this constellation of findings (172). Other highly inbred families are also described with cerebellar hypoplasia. A mutant zinc-finger protein (ZNF592) was found to cause an autosomal recessive nonprogressive congenital cerebellar ataxia in a large Lebanese family (109). Features of this syndrome, known as CAMOS, include cerebellar ataxia with mental retardation, optic atrophy, and skin abnormalities. Mutations in the ALDH7A1 gene, known to be responsible for autosomal recessive pyridoxine-dependent epilepsy or other neurologic phenotypes, may be associated with brain malformations including cerebellar hypoplasia or dysplasia (167).
Congenital pontine and cerebellar defects are also seen in infants with Möbius syndrome and the oculo-auriculo-vertebral spectrum (Goldenhar syndrome). The cerebellar hypoplasia in Goldenhar syndrome may be asymmetrical and involve only one cerebellar hemisphere (98). Few patients with Aicardi-Goutières syndrome, which causes progressive encephalopathy with basal ganglionic calcifications, have cerebellar hypoplasia (04). Frequent cerebral migration abnormalities (polymicrogyria and heterotopia) and cerebellar abnormalities, including prominence of superior vermis folia, inferior vermis hypoplasia, dysplastic or hypoplastic cerebellar hemispheres, heterotopia, and cysts occur in Aicardi syndrome (71) (this is not the same as Aicardi-Goutières syndrome). Cerebellar hypoplasia is also reported prenatally in the autosomal recessive Juberg-Hayward syndrome associated with callosal agenesis, microcephaly, growth retardation, cleft lip and palate, and radial ray abnormalities (41). Cerebellar hypoplasia may be a feature of Cohen syndrome (181). An association with biliary atresia in the polysplenia syndrome is reported (175). Other associations include ocular anomalies, such as colobomata of the retina or iris and micro-ophthalmia, and other cerebral malformations as well (118). Cerebellar hypoplasia, cerebellar heterotopia, and abnormal cerebellar hemisphere foliation are common features in CHARGE syndrome (coloboma, heart defects, atresia of the choanae, retarded growth and development, and genital and ear abnormalities). Mutations in CHD7 cause CHARGE syndrome (06; 188). Dellman syndrome (oculocerebrocutaneous syndrome) encompasses multiple congenital malformations with brain, eyes, and skin abnormalities. Neuroimaging findings include polymicrogyria, periventricular nodular heterotopia, cerebellar vermis agenesis, and small cerebellar hemispheres (06). Cerebellar hypoplasia may be one of the features of the short telomere syndromes. These disorders can involve almost any organ. Cerebellar hypoplasia occurs uncommonly in dyskeratosis congenita, a hereditary defect in telomere maintenance, but is a characteristic feature of Hoyeraal-Hreidarsson syndrome, a rare severe variant of bone marrow failure and cancer (dyskeratosis congenita) (200). Cerebellar hypoplasia or abnormalities in the shape or size of the cerebellar hemispheres occur only rarely in neurofibromatosis types 1 and 2 (165; 12). Some children with neurocutaneous melanosis have cerebellar hypoplasia in association with hindbrain melanosis (75).
Other etiologies for cerebellar hypoplasia include inborn errors of metabolism such as adenylosuccinate lyase deficiency (79) and mitochondrial disorders. In addition, cerebellar hypoplasia may occur in some of the congenital disorders of autophagy (eg, EPG5-associated Vici syndrome and ATG5-associated autosomal recessive ataxia syndrome) (50), in a few disorders caused by inborn errors of metabolism in the phospholipids biosynthesis pathways (eg, Sengers syndrome, which is characterized by congenital cataract, hypertrophic cardiomyopathy, skeletal myopathy, and rarely cerebellar hypoplasia) (187), and a lethal autosomal recessive disorder with congenital cataract, hearing loss, low serum copper, and ceruloplasmin (73). Bilirubin encephalopathy, or kernicterus, may lead to hyperbilirubinemia-induced cerebellar hypoplasia, particularly in premature infants. Rarely, cerebellar hypoplasia is associated with hematological disorders, particularly pancytopenia and lymphopenia (95).
Alcohol is a frequent toxic and teratogenic factor in the developing fetus that results in cerebellar hypoplasia and causes fetal alcohol syndrome (28). In addition, alcohol interferes with granule cell migration in a mouse model (77). Isotretinoin, a first-generation retinoid, is used to treat severe acne. It is highly teratogenic and can cause fetal retinoid syndrome. Cerebellar hypoplasia is one feature of this syndrome, which can be diagnosed as early as 20 weeks’ gestation (52).
Several external factors can disrupt prenatal and otherwise normal cerebellar development causing hypoplasia, dysplasia, or cleft. These factors include hypoxia, ischemia, hemorrhage, congenital infections, inflammation, trauma, exposure to drugs, drug toxicity, undernutrition, and prematurity (123; 104; 153). Several of these factors and mechanisms have been investigated in perinatal cerebellar injury. Degeneration of the granular layer through any mechanism may resemble hypoplasia (117). Cerebellar hypoplasia of prematurity is multifactorial in origin. Primary risk factors include postnatal glucocorticoid exposure and supratentorial brain injury (59). Other reported factors in premature infants born at less than 28 weeks’ gestational age are severe intraventricular and cerebellar hemorrhage, which in combination, may worsen the cerebellar injury (153). In addition, morphine exposure in preterm neonates was found to be associated with reduced cerebellar volume at term-equivalent age and also poorer motor and cognitive outcomes in early childhood. The clinical features, neuroimaging findings, causes, and outcomes of diseases associated with cerebellar abnormalities encountered in premature neonates have been extensively reviewed (59).
Neuroimaging findings favoring disruption as opposed to primary cerebellar malformation include unilateral cerebellar abnormalities, the presence of hemorrhage/blood products or calcification, and the associated presence of porencephaly (123). Twins, whether monozygotic or dizygotic, have a broad spectrum of brain malformations with patterns of abnormalities that are most consistent with a prenatal vascular pathogenesis, for example, unilateral hypoplasia, asymmetrical cerebellar hemispheres hypoplasia, or cerebellar clefts (113).
Various types of cerebellar malformations, based on the classification presented in Table 1, are discussed below. A summary of the main etiological categories of cerebellar hypoplasia (and dysplasia) are presented in Table 2.
Aplasia of the cerebellum. This is a rare finding on neuroimaging. It may or may not be associated with supratentorial cerebral malformations. The midbrain and upper pons are often hypoplastic or structurally defective. Mutation in the Ptf1a gene leads to complete cerebellar agenesis and neonatal diabetes (172). This gene has a crucial role in cerebellar GABAergic neuronal specification and is known to be involved in pancreatic development (101). Complete cerebellar loss may also be the consequence of a prenatal disruption, eg, from prematurity or prenatal hemorrhage in the setting of neonatal alloimmune thrombocytopenia (123; 153).
Differentiation of a cerebellar ridge at the margin of the fourth ventricle. Rarely, the cerebellar primordium along the rhombic lip begins to form, but medial growth is arrested early so that the cerebellum consists only of an elevated ridge along the anterior and lateral margins of the fourth ventricle. The cerebellar neuroepithelial cells do mature, however, and Purkinje cells, granule cells, and Bergmann glia are all represented and form an imperfect but architecturally recognizable laminated cerebellar cortex within that ridge. This represents a minimal development and is unlike the global cerebellar hypoplasia, in which the cerebellum is small but completely formed. Some cases reported as "cerebellar agenesis" are not total because some residual cerebellar cortex is retained (62). The almost complete lack of cerebellar tissue is compatible with normal life span and possible employment in simple jobs. However, such patients have motor deficits and impaired learning/intellectual ability (30).
Global cerebellar hypoplasia. Global cerebellar hypoplasia, or diffuse hypoplasia of the cerebellum, is associated with diverse causes (122). Several syndromes, disorders caused by genetic mutations, and chromosomal diseases such as trisomies 13 and 18 are well-documented causes (01). Monosomy 1p36, a terminal deletion syndrome (34), and ring chromosome 6, a rare condition (14), also result in cerebellar hypoplasia as well as other cerebral anomalies, such as partial callosal agenesis.
Hypoplasia of the cerebellar vermis and hemispheres are a feature of a syndrome caused by mutations in NEUROD1 gene. The syndrome is also associated with neonatal diabetes, sensorineural hearing loss, learning difficulties, and visual impairment (139). Familial cerebellar hypoplasia and pancytopenia may be associated with chromosomal instability, but this is not demonstrated in all cases (95). Acrocallosal syndrome is associated with pons, vermis, and cerebellar hemisphere hypoplasia (53).
Brainstem disconnection is a rare syndrome associated with several features, including absent vermis, cerebellar hemispheric hypoplasia, and a disconnection between the midbrain and pons or between the pons and the medulla (49). It is unknown whether the etiology is genetic or acquired in origin, but prognosis is mostly unfavorable (124).
Global cerebellar hypoplasia may be only one component of much more extensive malformations of the brain that include small cerebral hemispheres with pachygyria or lissencephaly and agenesis of the corpus callosum (115; 135). Similar extensive brain malformations with microcephaly, widespread band-like intracranial calcification, and cerebellar hypoplasia have been reported in a distinctive form of congenital infection-like syndromes (03).
In hereditary degenerative diseases, the distinction between "hypoplasia" and "atrophy" may pose a semantic problem (122; 23). Examples of progressive metabolic and degenerative diseases, active both prenatally and postnatally, that may cause a congenitally small cerebellum include:
(1) Some spinocerebellar degenerations
(2) Several diseases associated with pontocerebellar hypoplasia
The number of disorders associated with small pons and cerebellum has steadily increased over the last 2 decades. There are now at least 11 types of pontocerebellar hypoplasia labelled as pontocerebellar hypoplasia type 1 to type 11. Their genotype and phenotype overlap (06; 05). In pontocerebellar hypoplasia type 1, there is pontocerebellar hypoplasia and progressive motor neuron degeneration. Mutations in the RNA exosome component gene EXOSC3 were reported to cause pontocerebellar hypoplasia and spinal motor neuron degeneration in several families (182). Respiratory chain defects may cause a similar disease phenotype (44).
Mutations in EXOSC8 were reported in 22 infants from three independent families. The disease is severe and progressive and is associated with hypomyelination, spinal motor neuron disease, and cerebellar and corpus callosum hypoplasia (24). Global cerebellar hypoplasia was reported in one form of spinal muscular atrophy with lower extremity dominance. A mutation in BICD2 was found (55).
Mutations of the CASK gene were found to cause a brain malformation, manifesting as an X-linked disease with progressive microcephaly, severe intellectual disability, dystonia, and severe pontocerebellar hypoplasia in females with a similar but more variable male phenotype, which may include severe epileptic encephalopathy or nystagmus (199). Global cerebellar hypoplasia (or atrophy) is a feature in some patients with Galloway-Mowat syndrome, a rare autosomal recessive disorder caused by mutations in WDR73 gene. Other features may include microcephaly, epilepsy, dysmorphic features, intellectual disability, spasticity, ataxia, movement disorder, and nephrotic syndrome (76).
Cerebellar hypoplasia is also part of the cerebral dysgenesis in congenital muscular dystrophy (32). Congenital disorders of glycosylation (CDG) cause nearly 70 genetic diseases. They affect multiple organs, and most involve the nervous system. Type 1a is the most frequent N-linked CDG (85). It is an autosomal recessive multisystemic disorder of glycosylation that involves the cerebellum and is associated with changes in transferrin. It is consistently associated with pontine and cerebellar hypoplasia. Other clinical features include dysmorphic features, inverted nipples, short stature, spinal and other bony deformities, osteopenia, failure to thrive, hypothyroidism, hepatomegaly, esotropia, slowly progressive retinopathy, developmental delay, stroke-like episodes, seizures, peripheral neuropathy, hypotonia, and ataxia as expression of the cerebellar hypoplasia. Some cases are associated with coagulopathies. The diagnosis may be made from a blood sample with transferrin isoelectric focusing or, more quantitatively, by transferrin chromatography or Western blot analysis. The test, however, may not show any abnormality (85). If the diagnosis is still suspected in the presence of normal transferrin, it should be confirmed on a skin biopsy or molecular genetic testing including single-gene testing, multigene panel, or exome sequencing depending on the phenotype (85). A novel syndrome with abnormal glycosylation in the N-glycan biosynthesis pathway has been delineated (105). It is caused by steroid 5α-reductase type 3 defect and is associated with cerebellar ataxia, vermis or global cerebellar hypoplasia, early visual impairment, and variable eye malformations, such as optic nerve hypoplasia, retinal coloboma, and congenital cataract.
Other metabolic disorders with global cerebellar hypoplasia include Zellweger syndrome, molybdenum cofactor deficiency, sulfite oxidase deficiency, mucopolysaccharidoses, and nonketotic hyperglycinemia (122).
In the agranular (or granuloprival) form of global cerebellar hypoplasia, the cerebellum is small and of low weight. Histologically, neuronal elements of the cerebellum may be lost and replaced by extensive gliosis. The cerebellar cortex of both vermis and lateral hemispheres exhibits a selective loss of granule cells (110). Granuloprival cerebellar hypoplasia may be sporadic or follow a familial, probably autosomal recessive pattern (110).
Ethanol adversely affects cerebellar granule cells and their migration in mice, suggesting a cause for a small cerebellum and impaired coordination in children with fetal alcohol syndrome (77).
Global cerebellar hypoplasia may also be secondary to disruptive processes prenatally, eg, congenital infections including cytomegalovirus neurologic disease (123).
Predominant aplasia or hypoplasia of the vermis without fusion of the cerebellar hemispheres. Predominant or selective hypoplasia of the cerebellar vermis may be inherited in a variety of genetic transmission patterns, including the autosomal recessive Joubert syndrome and related disorders, where it occurs commonly (128). Vermal hypoplasia also occurs as a component of the Dandy-Walker malformation and as a part of tectocerebellar dysraphism with or without occipital encephalocele. Tectocerebellar dysraphism with occipital encephalocele is not a unique disease and may be seen in several disorders such as Joubert syndrome and related disorders (127). Deletions or duplications involving FOXC1 on 6p25.3 are associated with Dandy-Walker malformation or vermis hypoplasia (07). FOXC1 is expressed in the mesenchyme covering the developing cerebellum and is required for cerebellar development (07). Heterozygous ZIC1 and ZIC4 deletion are also associated with Dandy-Walker malformation (166). Posthemorrhagic cerebellar disruption was reported to mimic Dandy-Walker malformation in a fetus (91).
Most cases of cerebellar vermal aplasia leave a space filled with subarachnoid fluid between the medial sides of the two cerebellar hemispheres, as is typical in Joubert syndrome and related disorders (112). Joubert syndrome and related disorders are the subject of another MedLink article.
Cerebellar vermis hypoplasia or other posterior fossa anomalies occur in Ritscher-Schinzel syndrome, which is also known as the 3C syndrome because it manifests with craniofacial, cardiac, and cerebellar defects. Vermis hypoplasia may be a feature in Micro syndrome (02). Congenital fibrosis of the extraocular muscles syndrome may be associated with vermis hypoplasia, with cerebellar atrophy and progressive ataxia developing later (195).
A novel brain malformation with defect in axonal guidance was described and named pontine tegmental cap dysplasia (19). It is associated with vermis hypoplasia and malformation. The cerebellar hemispheres shape and size are normal, although mild hypoplasia of the cerebellar hemispheres may occur. A protruding abnormal structure projects from the pons into the fourth ventricle. Patients with this malformation present with various degrees of learning disabilities, signs of cerebellar dysfunction, and cranial neuropathies. In contrast, medullary tegmental cap dysplasia, another rare brainstem malformation in which an abnormal mass protrudes from the posterior medullary surface, can be seen in a few diseases, eg, Joubert syndrome and fibrodysplasia ossificans progressiva syndrome. Patients with MRI displaying this anomaly show heterogenous clinical, neuroimaging, and postmortem findings consistent with the presence of multiple etiologies and pathophysiological findings. The cerebellar vermis and, to a lesser extent, the cerebellar hemispheres may be hypoplastic (58).
Various chromosomal abnormalities have been demonstrated in a few patients with vermal aplasia, but their significance is unclear. About 75% of affected children have a borderline low IQ or low IQ. Trisomies 13 and 18 are the chromosomal disorders most consistently associated with cerebellar hypoplasia (93). Other less constant associations include partial trisomy 12q and monosomy 21q (36), trisomy 17 mosaicism (163), an autosomal recessive trait mapped to 17p (171), combined 16q22-qter and 9pter-q22 duplication (119), 22q11.2 deletion with polymicrogyria and cerebellar hypoplasia (133), distal 5p deletions (cri-du-chat syndrome) (37), 2q22-23 deletion that includes the ZFHX1B gene (160), the fragile X syndrome (106), Prader-Willi syndrome with an interstitial deletion of 15q11-q13 that may cause an asymmetrical cerebellar hypoplasia (164), and Rett syndrome (107).
A microdeletion in exons 16 to 17 of the oligophrenin-1 (OPHN1) gene at Xq12 is associated with cerebellar hypoplasia. It is inherited as an X-linked recessive trait (199). A mutation in the ABCB7 gene was identified using whole-genome sequencing in several males from a Mongolian family with X-linked pattern of early-onset nonprogressive cerebellar ataxia, developmental delay, and cerebellar hypoplasia on MRI. The gene encodes a mitochondrial protein transporter (129). Other X-linked disorders with cerebellar dysgenesis have been reviewed (199).
Hypoplasia or aplasia of the vermis is found in some infants with Smith-Lemli-Opitz syndrome, a disorder of cholesterol synthesis associated with defective expression of the gene "Sonic hedgehog" (87). Beta-ureidopropionase deficiency is a rare metabolic disease associated with cerebellar hypoplasia (16). Some cases of mitochondrial cytopathy with dysfunction of respiratory chain enzymes are associated with cerebellar hypoplasia, either as an isolated or predominant abnormality (94). In Leber congenital amaurosis, vermal hypoplasia is described in some patients; however, they also have multiple systemic anomalies, not just severe visual impairment (194).
Selective posterior vermal agenesis or hypoplasia is sometimes referred to as the “Dandy-Walker variant,” but this designation often is misleading because some have nothing in common with the Dandy-Walker syndrome; a meticulous neuropathological study of four such fetuses at 21 to 24 weeks’ gestation showed abnormal migration of external granule cell precursors from the rhombic lip of His and a paucity of Bergmann glial cells (140).
A selective hypoplasia or aplasia of the posterior vermis, not due to Dandy-Walker malformation or Joubert syndrome and related disorders, has been recognized. Some cases are associated with Beckwith-Wiedemann syndrome and congenital anomalies of multiple organ systems (140). Some children with leukemia also present with hypoplasia of the vermis (40). Isolated inferior vermis hypoplasia may be associated with motor and language delays (92).
Planimetric studies of MRI by several investigators have shown hypoplasia of the cerebellar vermis and, to a lesser extent, the cerebellar hemispheres in autistic children (141), but other studies using similar techniques failed to demonstrate significant alterations from normal size of the cerebellum or fourth ventricle in infantile autism (60).
Aplasia of the vermis with fusion of the cerebellar hemispheres in midline. The condition in which the vermis is absent and the two cerebellar hemispheres and the dentate nuclei of the two sides are fused in the midline is called rhombencephalosynapsis (104). It is the subject of another MedLink article.
Unilateral or asymmetrical cerebellar hypoplasia. These are rare abnormalities ranging from unilateral absence of one cerebellar hemisphere to mild hypoplasia or asymmetry of the cerebellar hemispheres with or without an intact vermis. Such abnormalities have been reported in twins (113). The condition can be detected prenatally by careful ultrasound examination. A favorable developmental and cognitive outcome is likely when the vermis is spared (125).
The lateral hemispheres of the cerebellum also undergo selective degeneration in certain hereditary diseases, such as olivopontocerebellar atrophy and other spinocerebellar degenerations. This should not be confused with hypoplasia.
Rarely, cerebellar hemisphere aplasia is unilateral and likely caused by an ischemic infarction in fetal life (125; 123). In some cases, the fetal cerebellar infarct is hemorrhagic (198). Genetic abnormalities may predispose and indirectly cause unilateral or asymmetrical cerebellar hypoplasia. For example, mutations in COL4A1 may cause cerebellar hemorrhage with subsequent disruption in cerebellar development (100). Unilateral cerebellar aplasia/hypoplasia is reported in patients with Aicardi syndrome (71); in patients with PHACES syndrome (posterior fossa anomalies, hemangioma, arterial anomalies, cardiac abnormalities/ aortic coarctation, eye abnormalities, sternal or ventral defects), which can be diagnosed prenatally, though agenesis/dysgenesis of the vermis or global cerebellar hypoplasia also occur (88); and in patients with osteogenesis imperfecta caused by recessive WNT1 mutations (08). Fetal cerebellar infarction may result in apparent cerebellar hypoplasia due to congenital cytomegalovirus (46) or congenital varicella infections (178), though ischemic lesions might be occurring in addition to direct viral involvement of developing neurons.
In general, hemicerebellar hypoplasia is considered a prenatal disruption of an otherwise normal cerebellar development caused by various external factors in-utero (eg, ischemia, hemorrhage, infection, toxicity, or trauma) rather than perinatal in origin (125; 123), with rare exceptions as stated above.
Asymmetric cerebellar hemispheric hypoplasia is reported in Prader-Willi syndrome (164), Goldenhar syndrome (98), and osteogenesis imperfecta caused by recessive WNT1 mutations (08), but this is considered to be a rare feature in genetic disorders.
The clinical spectrum varies from normal or near normal development to ataxia (limbs or truncal) and marked learning disability (125).
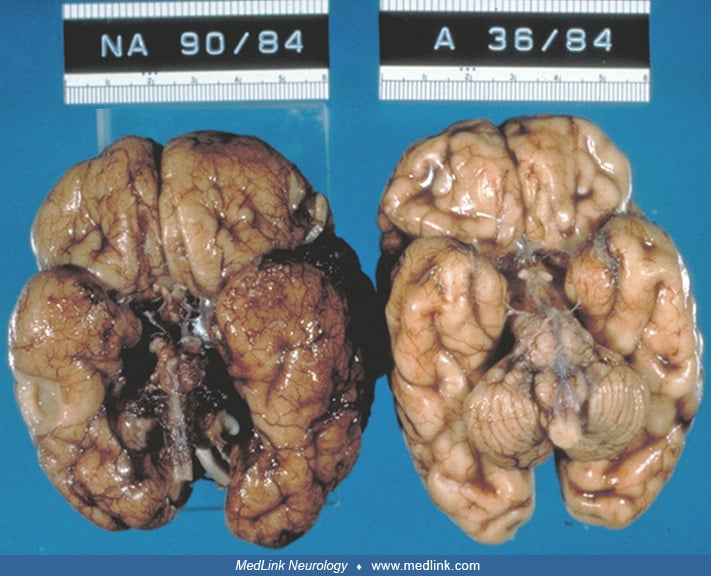
Focal dysplasia of the cerebellar cortex. Dysplasia limited to the cerebellar cortex may be demonstrated by MRI and appears as an irregular bumpy gray-white matter interface. It usually involves the lateral hemispheres more than the vermis. The folial pattern may appear abnormal, or there may be gray matter nodular heterotopia. Cerebellar cortical dysplasia is commonly associated with cerebral malformations (45), such as holoprosencephaly, some types of congenital muscular dystrophy (32), GPR56-related polymicrogyria (136), or chromosomal disorders, but they can also occur in isolation (45; 25). In Joubert syndrome and related disorders, the superior cerebellar vermis is usually dysplastic, and folial disorganization involving the cerebellar hemispheres may also occur (122; 128). Cerebellar dysplasia may occur secondary to disturbances in cerebellar cortical development caused by small infarcts, hemorrhages, or other acquired lesions. The reported bottom-of-the fissure dysplasia is likely caused by cerebellar watershed injury (189). Cerebellar cortical dysplasia is demonstrated sometimes at autopsy as an incidental finding that was clinically asymptomatic in life and that has no evident etiology. Prenatal diagnosis is challenging (89). A study reported that fetal ultrasonography was better at detecting cerebellar cortical dysplasia caused by various etiologies than fetal MRI (47). Fetal ultrasonography performed in 12 fetuses, between 22 and 34 gestational weeks with cerebellar cortical dysplasia, showed that the cerebellar fissures were disorganized and arranged in an oblique or vertical fashion rather than the regular horizontal fashion around an arc as is the case in fetuses with a normally developed cerebellar cortex. Small focal zones of abnormal lamination or disorganized folia might be difficult to identify on imaging. In some patients with a congenitally small cerebellum, the Purkinje neurons are not reduced in total number, but many are displaced heterotopically into the molecular zone in both the vermis and lateral cerebellar hemispheres.
Migration of the external granular layer is not complete until 1 year of age; therefore, postnatal, as well as prenatal insults, may result in focal dysplasia of the cerebellar cortex. The radial processes of the specialized Bergmann astrocytes serve as guide fibers for migrating granule cells within the folia. Genetic or molecular defects could impair cerebellar foliation and fissures formation if they adversely affect the formation or function of Bergmann glial fibers (35; 42). The Huwe1 gene, which codes for ubiquitin ligase, regulates Bergmann glia differentiation. Loss of Huwe1 in Bergmann glia led to extensive cerebellar cellular disorganization and layering abnormalities in a knockout mouse strain (42). There were severe granule neuron migration defects as well as persistence of ectopic clusters of granule neurons in the external granular layer. Β1-integrin expression in Bergmann glia is crucial to the proper development of the cerebellar cortex, and its deletion in knockout mice led to cerebellar hypoplasia, fusion of the cerebellar folia, impaired migration of the granule cells along glial fibers, and abnormal positioning of Bergmann glia and development of its processes (57).
Focal or extensive hamartomas of the human cerebellar cortex may occur in genetic or acquired diseases that affect the radial glial fibers of the Bergmann cells. Perinatal asphyxia in the premature or term infant may result in loss of both Purkinje cells and granule cells and incomplete regeneration of the external granular layer, with resulting hamartomas of the cerebellar cortex (196). Cytotoxic destruction of the external granular layer and subsequent regeneration may also cause hamartomas, with abnormal orientation of Purkinje cells, abnormal arborization of Purkinje cell dendrites, and failure of dendritic spines to make synaptic contact with parallel fibers (191).
Cerebellar dysplasia is a feature in Chudley-McCullough syndrome, an autosomal recessive disease with sensorineural hearing loss and partial agenesis of the corpus callosum (48). Cerebellar cysts, vermis hypoplasia and dysplasia, and abnormal shape of the fourth ventricle (enlarged, elongated, and square-like) have been reported in Poretti-Boltshauser syndrome (51). The disease manifests with ataxia, variable intellectual disability, language impairment, defective saccade initiation, and frequent occurrence of myopia or retinopathy. Several mutations in LAMA1, a subtype of laminin, have been identified as the cause of this disease and are summarized in a report (51). The phenotype has expanded with three further individuals who also had obsessive compulsive traits, tics, and anxiety (179). The authors also demonstrated that LAMA1 deficiency can change cytoskeletal dynamics, leading to altered cell adhesion and migration, which likely have consequences on dendrite growth and axonal formation. Cerebellar dysplasia occurs commonly in several types of tubulinopathies (96). Patients with cerebellar and basal ganglia dysplasia, who also had a subtle irregular pattern of gyri and sulci on brain MRI, have been reported. They were found to have mutations in TUBA1A, TUBB2B, TUBB3, TUBB5, and TUBG1 genes (136; 63). The dysplasia occurred more commonly in the cerebellar cortex, usually involved the cerebellar hemispheres, especially the right, and occurred most frequently in the postero-superior region. The cerebellar folia were oriented abnormally, but there were no cysts or cerebellar signal abnormalities (136). Dysplasia of the developing cerebellum may occur in association with medulloblastoma of the vermis early in life, probably as a true congenital tumor (192).
Macrocerebellum and macrohemicerebellum. Macrocerebellum is an enlarged cerebellum that is detected on neuroimaging, which is not due to neoplasia or dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos syndrome). Global developmental delay, hypotonia, eye movement abnormalities, and other cerebellar signs are the main clinical findings in children. Imaging reveals an enlarged cerebellum, which can be verified on quantitative analysis. The enlarged cerebellum may cause mass effects on the brainstem (104). Signal abnormalities in the cerebral white matter may also be seen (43). Thickening of the cortical gray matter of the cerebellar hemispheres and ventriculomegaly/hydrocephalus has been described in some patients (126). In addition, abnormalities in the cerebellar white matter were reported in one child using diffusion tensor imaging (74). Macrocerebellum can be a feature of few syndromes, including Sotos, Williams, megalencephaly-capillary malformation, and Rubinstein-Taybi syndromes (126; 06, 43). No cases have been examined histopathologically. The cerebellar volume is also significantly increased in children with nevoid basal cell carcinoma syndrome and achondroplasia (158; 116). Disproportionate cerebellar volume increase has been documented at 6 and 21 months following ventriculoperitoneal shunting. The infant was born at 35 weeks’ gestation and was shunted at 4 months of age for suspected post hemorrhagic hydrocephalus. Six months later, he developed symptomatic Chiari type 1 malformation that required posterior fossa decompression (69).
Postnatal cerebellar enlargement has been reported in Costello syndrome (64) and in mucopolysaccharidoses type I (10). A child with macrocerebellum, epilepsy, mild developmental delay, dysautonomia, gut malrotation, and impaired gut motility was found to have a microdeletion in chromosome 16q24.1–q24.2 on genomic microarray analysis (155). In Joubert syndrome and related disorders, the cerebellar hemispheres may be enlarged, thus mimicking macrocerebellum (128).
Macrohemicerebellum may accompany a minority of cases of hemimegalencephaly (“total hemimegalencephaly”), either isolated or syndromic, usually in association with epidermal nevus syndrome or Proteus syndrome (56).
Midsagittal cleft of the vermis (rhombencephaloschisis) and unilateral cerebellar cleft. These are very rare anomalies. Unilateral cerebellar clefts may follow fetal cerebellar hemorrhage and usually involve the cerebellar cortical gray matter, which may extend all the way to the fourth ventricle in some cases. They may be associated with supratentorial abnormalities. The clinical features are highly variable (123).
Chiari II malformation. This malformation commonly accompanies spina bifida meningomyelocele. It is associated with dysplasia of the vermis and hindbrain herniation through foramen magnum (147). The distorted cerebellum is small, which is thought to be secondary to tissue loss caused by cerebellar crowding and compression within a small posterior fossa. In that sense, Chiari II malformation is not a primary malformation of the cerebellum but is secondary to mechanical forces. Cerebellar hypoplasia followed by progressive atrophy of the cerebellum has been called the “vanishing cerebellum” in serial MRI studies (27). Anomalous development of cerebral and cerebellar structures in spina bifida meningomyelocele is discussed in detail in a review (78).
Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease). This is a rare slow growing hamartomatous disorder that causes disruption of the cerebellar cortical cell layers. It usually starts in the cerebellar hemispheres and causes folia enlargement. It may be found incidentally or from symptoms due to its progressive expansion (159). It may be associated with Cowden syndrome. It is not a neoplasia as was previously thought.
|
(1) Congenital infections (eg, cytomegalovirus; zika virus) |
|
|
Embryology of cerebellum. The formation of rhombomeres of hindbrain compartments, which restrict cellular movement and concentrate clones of cells of the same type to form structures of the brain, is accomplished by homeobox-containing genes of several families; the most important being Wnt, En, Hox, and embryonic growth-related (Egr in humans or Krox in mice). Detailed reviews and a consensus paper on cerebellar development are available (90; 21; 176). The mesencephalic neuromere, just rostral to rhombomere 1, may also contribute. The origin of the deep cerebellar nuclei is rhombomere 2. Genetic knockout mice and chicks with inactivation of the Wnt-1 or En-1 genes fail to develop a cerebellum, and En-2 defective mice have global cerebellar hypoplasia (83; 183). Another factor essential to cerebellar development is fibroblast growth factor-8 (21; 176). Other regulator genes, particularly of the Pax (paired) and Wnt families, are expressed later for specific cellular differentiation and conservation of identity at maturity. More details on the anatomical, cellular, and genetic programs underlying cerebellar development are found in a book on the topic (99).
The classical morphological development of the cerebellum explains the timing and nature of vermal hypoplasia in particular. The cerebellum begins development in the fifth week (stage 14). The alar plate of the most anterior segment of the rhombencephalon (rhombomere 1) undergoes a rapid proliferation of neuroepithelial cells that grow laterally and then medially to form a ridge along the anterior and lateral walls of the future fourth ventricle. The anterior ridge continues to grow medially and the two sides meet and fuse in the midline by the seventh week (stage 19), coinciding with the early appearance of the vestibular commissure (111). This lateral to medial growth of the cerebellar primordium within the Cartesian grid developed for caudal regions of the brain is actually more of a posterior to anterior growth because the creation of the rhombic lip displaces cells distributed axially along the dorsal midline of the anterior/posterior axis to a lateral/medial orientation (68). An alternative view suggests unpaired primodium (tuberculum cerebelli) forming an inverted V-shaped structure straddling the midline.
Cerebellar development is dependent on signaling from the isthmic organizer region (90). Wnt and Fgf signals from the isthmic organizer region are important for positioning the cerebellar primodia during early embryogenesis along the anterior/posterior axis of the neural tube (21; 176). The lateral part of the alar neuroepithelium is continuous with the thin membranous roof plate covering the fourth ventricle. This portion was formerly designated the "rhombic lip of His" or the "germinal trigone," but it is now recognized that the rhombic lip is only a part of the cerebellar primordium. By 16 weeks, the cerebellum is a formed structure completely covering the fourth ventricle. Lmx1a is expressed in the roof plate and is required to segregate roof plate lineage from neuronal rhombic lip derivatives. In addition, it maintains the cerebellar rhombic lip and is critical for posterior vermis morphogenesis (39). The rhombic lip progenitors that express Lmx1a give rise to granule cells that end up in the posterior vermis. The absence of Lmx1a is associated with premature regression of the rhombic lip and posterior vermis hypoplasia in mice (39).
The cerebellum originates from two different germinal plates (90). The plate at the junction of the roof of the fourth ventricle (the ventricular zone) gives origin to all cerebellar GABAergic cells, including Purkinje cells, Golgi cells, basket cells, stellate cells, GABAergic cells of the deep cerebellar nuclei, and some glial elements. The deep stratum of the cerebellar plate generates the large neurons of the dentate and other deep cerebellar nuclei. The deep cerebellar nuclei, including the dentate, emboliform, fastigial, and globose nuclei, develop from the deepest layer of the cerebellar plate at 60 to 80 days' gestation (131). All the cerebellar nuclei are recognized at 16 weeks, but mature neurons with Nissl substance are not identified by light microscopy until 23 weeks; degenerative changes in some neurons as programmed cell death are found at 21 to 23 weeks' gestation (190).
A second germinal plate forms immediately beneath the pia that covers the cerebellar plate posterolaterally and spreads over the entire surface of the cerebellar cortex as the external granular layer. This primordium in the subventricular zone in the rhombic lip also gives rise to postmitotic neuroblasts that migrate ventrally along the external surface of the brainstem to form a transitory embryonic structure, the corpus pontobulbare. Neurons of the corpus pontobulbare eventually form the pontine, inferior olivary, and arcuate nuclei. The brainstem nuclei are intrinsic parts of the cerebellar system in terms of their fiber connections and functions. The timing of events in cellular differentiation and synaptic interactions of neurons in the cerebellar system is precise and well synchronized (169).
The cerebellar progenitor ventricular zone expresses Ptf1a, which gives rise to all cerebellar GABAergic neurons, whereas the dorsally located rhombic lip expresses Math-1 (also called ATOH1), which gives rise to all cerebellar glutamatergic neurons including granule cells and glutamatergic cerebellar nuclei. The RP58 (ZNF238) gene encodes a zinc-finger transcription factor. It is essential for the growth, foliation, lamination, and organization of the cerebellum. When deleted, it causes severe cerebellar hypoplasia in mice knockout models. It is also required for the development of both glutamatergic and GABAergic neurons (20). Bmp-derived signal from the roof plate is important for rhombic lip induction and Math-1 expression. Notch1 activity from the ventricular zone serves as an antagonist to Bmp. Therefore, interactions between Bmp and Notch1 signal pathways are thought to regulate cerebellar progenitor development (101).
Mouse models have localized reelin expression to granule cells progenitors. RELN signaling is considered to be essential in the proliferation of granule cells progenitors and also in Purkinje cell migration and maturation. CHD7, a chromatin remodeling factor, is also critical for the proliferation of granule cells progenitors and their survival.
The external granular layer of the cerebellar cortex is first recognized in the eighth week (stage 23), arising from the rhombic lip. Purkinje cells are generated when the number of granule cells is still small, at 9 to 10 weeks (111). The external granular layer begins to form as an extension of the rhombic lip in the posterolateral aspect of the cerebellar primordium, from where it spreads laterally and rostrally over the surface of the developing cerebellar cortex (169). The rostral part of the cerebellum originates from the caudal portion of the embryonic mesencephalic vesicle, corresponding well to the domain of the homeobox genes En-1 and Wnt-1.
Certain genes are associated not only with differentiation of specific cell types in the cerebellum, but also are important for their maintenance after maturity. Bergmann glial cells express the protein gene products of Pax-3, whereas granule cells of the cerebellum express the transcripts of Pax-6, Zic-1, and Math-1 (161; 22). Purkinje cells also express Pax-3. The Wnt-3 gene is essential for cellular development and maintenance, and its continued expression in Purkinje cells also depends on interactions with granule cells (143).
A precise ratio is maintained between the number of Purkinje cells in relation to the number of granule cells. Humans have a higher ratio than simpler mammals. The ratios in studies of humans were 1 of 2991 and 1 of 3300, whereas the mouse has a ratio of 1 of 778, and the rat has a ratio of 1 of 449 (86; 82). The regulation of granule cell proliferation is mediated by the gene "sonic hedgehog" (Shh) and its receptor "patched" and is closely associated with yet another gene "smoothened." "Patched" protein is localized to granule cells and sonic hedgehog to Purkinje cells, thus, providing a molecular substrate for signaling between these synaptically-related neurons to establish the needed amount of granule cell production (168). "Sonic hedgehog" in Purkinje cells acts as a mitogen on external granule cells (184). In humans, mutation of the "patched" gene is associated with basal cell carcinoma of the skin and with medulloblastoma of the cerebellum and, thus, “patched” gene is the etiology of at least a subset of human primitive neuroectodermal tumors (186).
Another known function of "sonic hedgehog" is for the synthesis of cholesterol. If cholesterol synthesis is impaired and the precursor molecule 7-dehydrocholesterol accumulates, as occurs in the Smith-Lemli-Opitz syndrome, a high proportion of the children have holoprosencephaly, as well as defects of the cerebellar vermis in particular (87).
Afferent fibers growing into the cerebellum, parallel fibers of granule cells, and complex dendritic trees of Purkinje cells all contribute to the formation of a molecular zone that separates the Purkinje cell layer from the external granular layer. The Purkinje cell layer is initially several cells thick, but with further organization of the cerebellar cortex the Purkinje cells spread out to form a single cell layer.
Granule cells continue to migrate from the external granular layer, through the molecular and Purkinje cell layers, to their mature positions within the interior of the folium. This process of granule cell migration continues throughout fetal life and beyond the immediate postnatal period until almost 1 year of age in humans. The potential for cerebellar dysgenesis, thus, extends well beyond fetal life. Further details on granule cell migration are available (77).
Unlike the sequence in the cerebral cortex in which synaptogenesis always follows neuronal migration, granule cell axons synapse with Purkinje cell dendrites before moving into the interior of the folia (185). However, granule cells do not form "glomeruli" in the external granular layer for receiving afferent input from axons originating outside the cerebellum as they do in their permanent site in the internal granular layer (102).
Basket cells, whose axonal terminals surround the cell bodies of Purkinje cells, begin to form shortly after the external granular layer first appears. These neurons remain in the molecular zone in the adult. The lamina dissecans is a transitory fetal zone between Purkinje cells and the internal granular layer. It consists of thick preterminal and terminal axonal segments. It forms at 20 to 21 weeks' gestation and persists for only about 10 weeks (131).
Climbing fibers from the inferior olivary nuclei, spinocerebellar tracts, and other sources outside the cerebellum arrive and form synapses with Purkinje cell dendrites in the cerebellar hemispheres from the 28th week through the early postnatal period.
Granule and Purkinje cells progenitors are known to have cilia. Cilia proteins that are critical for cilia genesis and maintenance are required for expansion of the granule progenitor population (38). Several mutations in ciliary genes that code for proteins required for cilia function or formation were found to cause Joubert syndrome and related disorders (101; 112).
Thyroid hormone is needed for cerebellar development. It has multiple effects on cerebellar development (90). Thyroid hormone regulates the development of axodendritic connections between granule and Purkinje cells. Hypothyroidism occurring during cerebellar development impairs neuronal migration, differentiation, and proliferation (13).
In summary, the cerebellum develops in a manner opposite that of the cerebrum: rather than lateral growth, it undergoes medial growth; rather than paired hemispheres forming by cleavage of a primordial midline cerebral vesicle or prosencephalon, the cerebellar primordia are the paired alar plates that grow toward the midline and fuse. If the fusion is incomplete or defective, the vermis is imperfectly formed or absent altogether. Midline defects in the cerebellum, thus, are due to lack of medial growth and limited fusion. Granule cells arise from a different primordium than all other cellular elements of the cerebellar cortex. As with other parts of the brain, genetic programming is the basis for cellular differentiation and interactions; some malformations are attributed to genetic mutations.
The U.S. national birth defects prevention study reported a birth prevalence of nonsyndromic cerebellar hypoplasia of 1.3 per 100,000 persons during 1997 to 2011. In their population-based, case-control study, cerebellar hypoplasia cases were more likely than controls to be from multiple pregnancy, born prematurely, and have low birth weight (72).
In a study from the central region of Saudi Arabia, where 8140 newborns were admitted to the neonatal intensive care unit (NICU) of 94,210 live births over a 10-year period starting in 2001, CNS malformations were found in 248 of 627 neonates in the NICU who had neuroimaging after excluding neonates with hypoxic-ischemic encephalopathy and neonates with CNS complications secondary to prematurity (66). Posterior fossa abnormalities were found in 44 of the 248 neonates. More specifically, “cerebellar hypoplasia” and “vermis hypoplasia” were described in 10 instances as primary malformations and in 13 instances as associated with other CNS malformations. The authors reported several factors that could influence the rates of CNS malformations reported, including the high consanguinity rate in Saudi Arabia (ranging between 50% and 90%), the rarity of termination of pregnancy and performing a postmortem examination, and the exclusion of still births and neonates who did not have any clinical problems in the immediate neonatal period (66).
Avoiding teratogens (eg, isotretinoin) and alcohol during pregnancy is essential. Otherwise, no means of preventing cerebellar hypoplasia are available for the majority of cases.
Because of severe hypotonia and delayed motor development, neuromuscular diseases, such as the congenital myopathies, congenital muscular dystrophies, and spinal muscular atrophy (Werdnig-Hoffmann disease), should be considered.
Tumors of the cerebellum or fourth ventricle may be congenital or appear early in infancy. An example is medulloblastoma. Hypotonia may be a clinical expression of vermal involvement by the tumor at an early age, but generally signs of increased intracranial pressure, such as rapid growth of the head size, bulging fontanelle, setting sun sign of the eyes, and clonus, help distinguish mass lesions from hypoplasia of the cerebellum. Nonneoplastic lesions of the cerebellum that may appear early include cerebellar hemorrhages in preterm infants, infarcts and cysts of the cerebellum (even from fetal life), and arachnoid cysts of the posterior fossa.
Both architectural disorganization and gliosis of the cerebellar cortex may follow radiation injury in the human infant as with the experimental animal (174). In addition, cerebellar hypoplasia should be distinguished from cerebellar sclerosis and atrophy after perinatal asphyxia, encephalitis, or other acquired lesions (137). Histological findings of severe neuronal loss and gliosis may be similar. The distinction between cerebellar atrophy and hypoplasia may be as difficult with imaging, (23), and neuropathological examination is sometimes inconclusive in chronic states (122). The principal progressive degenerative diseases that may be confused with cerebellar hypoplasia include several diseases associated with pontocerebellar hypoplasia, olivopontocerebellar atrophy (114), and the spinocerebellar degenerations.
Many major cerebral malformations, of which cerebellar hypoplasia is only one component, and the metabolic and degenerative diseases with which cerebellar hypoplasia is associated are discussed in the "Etiology" section.
Absence of granule cells selectively should be distinguished from postmortem autolysis. Granule cells are more resistant to hypoxia than Purkinje cells, but lyse more readily as a postmortem change. Loss of granule cells may also occur as a hypoxic or ischemic injury, however, sometimes called "état glacé," but this loss is patchy between and within folia rather than uniform.
If the diagnosis is suspected from the clinical features, or even if the diagnosis is not apparent but a cerebellar lesion is evident, then a brain MRI is the diagnostic test of choice (104). MRI enables quantification of the size and volume loss of cerebellar tissue (122; 15). It also provides important details on the shape of the cerebellum and the integrity of other structures within and outside the posterior fossa (146; 104). MRI of the fetus in utero also facilitates the prenatal diagnosis of cerebellar hypoplasia or other posterior fossa anomalies (67; 197) and may also identify additional supratentorial malformations and systemic malformations that may have been missed on prenatal ultrasonography (134; 89; 197). Advanced MRI techniques using diffusion tractography biomarkers, and in particular fractional anisotropy, have shown an early promise in a pilot study in differentiating between cerebellar hypoplasia and atrophy (54). Their clinical use needs to be investigated further. CT may provide the diagnosis as well, but the resolution is not as good, and only axial views, not the important sagittal views, are technically feasible. Cranial ultrasound was formerly a poor technique to resolve posterior fossa structures well enough to be diagnostic, even in preterm neonates; newer technology, though, can now provide fetal transcerebellar diameter (180). Prenatal imaging, particularly sonography, is sometimes predictive of chromosomal defects (180). Chromosomal microarray analysis provides additional value in the prenatal diagnosis of posterior fossa anomalies including fetuses with normal karyotype. Higher detection rate was noted in fetuses with cerebellar hypoplasia (151).
Although stage-specific normal measurements are available for the fetal cerebellar vermis on ultrasonography and MRI (134), important pitfalls in the radiological diagnosis and management of fetal cerebellar disorders may still occur and require postnatal MRI confirmation (92; 97).
If a muscle biopsy is performed to rule out neuromuscular disease as a cause of hypotonia and motor delay that was not diagnosed through genetic testing, the finding of congenital muscle fiber-type disproportion or of fiber-type predominance may suggest the need for MRI of the posterior fossa to look for cerebellar hypoplasia or more extensive cerebral dysgenesis (152; 108). The findings of "congenital myopathies" on biopsy are probably due to suprasegmental (upper motor neuron) influences on spinal motor neurons during the histochemical stage of human muscle development at 20 to 28 weeks' gestation involving bulbospinal pathways, but probably not due to the corticospinal tract, which matures later (152). These histochemical aberrations are not demonstrated in all cases of cerebellar hypoplasia, however, and some are associated with a normal muscle biopsy or with a nonspecific maturational delay of histochemical differentiation of myofibers. A muscle biopsy diagnosis of congenital muscular dystrophy may be associated with cobblestone malformation/pachygyria and cerebellar hypoplasia (32).
Electrophysiological studies are not helpful unless cerebral disease also is evident, and seizures are present, in which case an EEG is indicated.
High blood glucose may be indicative of PTF1A-related cerebellar agenesis. Serum muscle enzymes (eg, creatine kinase) are normal in cerebellar hypoplasia but may be helpful in patients with suspected congenital muscular dystrophy. Isoelectric focusing of serum transferrin or more quantitative chromatographic carbohydrate-deficient transferrin assay or Western blot analysis should be performed if congenital disorders of glycosylation are suspected (06). If congenital infections are suspected, serology should be done. In the early disease course of ataxia telangiectasia, when telangiectasias have not appeared yet, alpha fetoprotein measurement may be helpful, especially when evidence for a progressive disease course has not become apparent. Rarely, some inborn errors of metabolism present with nonprogressive early-onset ataxia. Therefore, blood amino acids and urine amino and organic acids may be indicated (23).
Genetic studies, including karyotype, FISH, array-CGH (comparative genome hybridization) analyses, whole exome sequencing, whole genome sequencing, and metabolic testing, are indicated, especially if dysmorphism or other associated clinical or imaging features suggest a particular chromosomal abnormality, genetic defect, or an inborn error of metabolism (173; 142). Studies have reported that the diagnostic yield of exome sequencing is relatively high in patients with cerebellar hypoplasia (09) as well as in patients with cerebellar atrophy or hypoplasia as the two conditions can sometimes be difficult to distinguish from each other, especially earlier on (142). Of the 176 families investigated, genetic variants implicated in several diseases were identified in 54.5% of the cases (142). Additional testing following whole exome sequencing using a bioinformatic tool proved helpful in identifying mutations in novel genes not previously known to cause early-onset ataxia with cerebellar hypoplasia (130).
No specific medical or surgical treatment is available. Multidisciplinary approach to management is needed to address the various medical, behavioral, educational, and social issues (144). Referral to rehabilitation and other supportive services (eg, social services) is advised. Physiotherapy, occupational therapy, and speech and language therapy are helpful. Patients with cerebellar dysfunction benefit from intensive rehabilitation therapy (33). Coordinative training is reported to improve foot placements. In addition, static and movable robotic training provide a promising new approach in patients with cerebellar ataxia. When robotic training is added to conventional training or neuromodulation of the cerebellum, additional benefits may be realized. Wearable dynamic orthoses represent a potential aid to assist gait. They can reduce body sway and stabilize the trunk and lower limbs. Encouraging the family to contact support groups (disease-specific or National Ataxia Foundation) is usually worthwhile. Prenatal counselling should be offered where relevant. Obstructive hydrocephalus may be a treatable complication in selected types of cerebellar hypoplasia, but these are the exception. For example, in Dandy-Walker malformation, a ventriculoperitoneal shunt may be indicated, but ventriculocystoperitoneal shunting may be a more specific and effective treatment (84).
Teratogens such as isotretinoin, phenytoin, and alcohol must be avoided during pregnancy.
There are no specific contraindications. In the case of Joubert syndrome and related disorders, the anesthesiologist should be aware of the tendency to apnea and hyperpnea, which may be exaggerated during postoperative recovery.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Michael S Salman MBBS BSc MSc MRCP PhD
Dr. Salman of the University of Manitoba has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MD FRCPC MS
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
May. 08, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 24, 2026
Developmental Malformations
Apr. 16, 2026
Developmental Malformations
Apr. 16, 2026