







Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Complex II deficiency results from reduction in activity of the mitochondrial enzyme succinate-ubiquinone oxido-reductase. This enzyme complex participates in the mitochondrial electron transport chain to generate ATP and serves in the Krebs cycle. Abnormalities of complex II activity are associated with a wide range of phenotypes. Most affected individuals have an encephalomyelopathy, with various degrees of CNS, skeletal muscle, and cardiac muscle involvement. CNS involvement includes microcephaly, seizures, ataxia, spasticity, developmental delay, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, and radiographic abnormalities compatible with Leigh disease or leukodystrophy. Initial workup includes blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. Final diagnosis depends on muscle biopsy.
|
• Complex II deficiency is caused by a reduction in activity of the mitochondrial enzyme succinate-ubiquinone oxido-reductase. | |
|
• This enzyme complex participates in the mitochondrial electron transport chain to transfer reducing equivalents from succinate to coenzyme Q (as part of the process of generating ATP) and serves a dual role in the Krebs cycle (to convert succinate to fumarate). | |
|
• Abnormalities of complex II activity are associated with a wide range of phenotypes. | |
|
• Most affected individuals have had an encephalomyelopathy, with various degrees of central nervous system, skeletal muscle, and cardiac muscle involvement. | |
|
• Central nervous system involvement has included microcephaly, seizure disorder, ataxia, spasticity, developmental delay, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, and radiographic abnormalities compatible with Leigh disease or leukodystrophy. | |
|
• Initial diagnostic workup of persons with complex II deficiency should include an examination of the blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. Final diagnosis is dependent on muscle biopsy. |
The electron transport chain, located in the inner mitochondrial membrane, is the site of oxidative phosphorylation, the metabolic pathway in which cells use enzymes to oxidize nutrients, thereby releasing energy that is used to reform adenosine triphosphate (ATP).
In this pathway, the NADH and succinate generated in the Krebs citric acid cycle are oxidized, releasing energy to power ATP synthase.
Within mitochondria, complex II links the two key cellular energy-conversion pathways: complex II serves as succinate dehydrogenase (SDH) in the Krebs cycle, and as succinate ubiquinone oxidoreductase, one of five complexes of the mitochondrial oxidative phosphorylation system. In particular, complex II couples the oxidation of succinate to fumarate in the citric acid cycle with the reduction of ubiquinone (coenzyme Q) to ubiquinol in the electron transport chain.
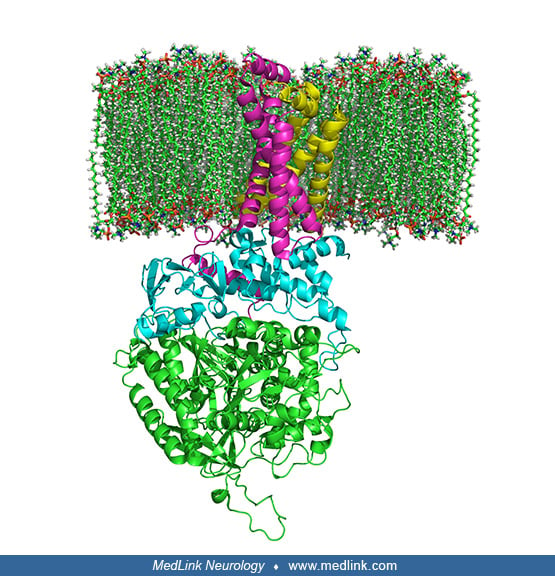
Complex II is a heterotetramer (ie, is protein containing four noncovalently bound subunits, wherein the subunits are not all identical) with four dissimilar subunits: SdhA, SdhB, SdhC, and SdhD.
Two of these subunits are hydrophilic (SdhA and SdhB) and two are hydrophobic membrane anchor subunits (SdhC and SdhD). SdhA contains a covalently attached flavin adenine dinucleotide (FAD) cofactor and the succinate binding site. Human mitochondria contain two distinct isoforms of SdhA (Fp subunits type I and type II). SdhB contains three iron-sulfur clusters: [2Fe-2S], [4Fe-4S], and [3Fe-4S].
The subunits form a membrane-bound cytochrome b complex with six transmembrane helices containing one heme b group and a ubiquinone-binding site.
Two phospholipid molecules, one cardiolipin and one phosphatidylethanolamine, are also found in the SdhC and SdhD subunits in the hydrophobic space below the heme b.
Although all the elements of the respiratory chain function in the mitochondria, many of the subunits are encoded genetically in the nucleus and imported into the mitochondria. Complex II is made of four subunits (A, B, C, D), all of which are autosomally encoded (ie, encoded in the nucleus) by the succinate dehydrogenase (SDH) genes (SDHA, SDHB, SDHC, and SDHD). Other factors and cofactors needed for complex II activity are riboflavin, iron, cytochrome b560, and ubiquinone (coenzyme Q). Complex II activity is often measured together with complex III activity as succinate cytochrome C reductase activity.
Isolated complex II deficiency was first inferred in two siblings with mental retardation, seizures, myoclonus, and ataxia (50). Since that report, a variety of other phenotypes have been reported, including Leigh disease (41; 13), leukodystrophy (03; 47), encephalomyopathy (30), Kearns-Sayre syndrome and other mitochondrial myopathies (51; 55), rhabdomyolysis and hemolytic uremic syndrome (43), cardiomyopathy with variable associated neurologic manifestations (psychiatric abnormalities, optic atrophy, oculomotor abnormalities, progressive polyneuropathy) (04; 55; 02; 15), and late-onset progressive neurodegeneration (57). In persons of northern Swedish descent, a myopathic phenotype due to a combination of complex II, complex III, and aconitase deficiencies has been described (25). Complex II (alone or in combination with other elements of the mitochondrial oxidative phosphorylation system) has also been reported to be abnormal in many neurodegenerative disorders, but these biochemical findings are most likely secondary events.
|
• Abnormalities of complex II activity are associated with a wide range of phenotypes. | |
|
• Most affected individuals have had an encephalomyelopathy, with various degrees of involvement of the central nervous system, skeletal muscle, and cardiac muscle. | |
|
• Central nervous system involvement has included microcephaly, seizure disorder, ataxia, spasticity, developmental delay, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, and radiographic abnormalities compatible with Leigh disease or leukodystrophy. | |
|
• Genes for all four of the complex II subunits function as tumor suppressors, with numerous germline and somatic mutations associated with hereditary cancer syndromes, including pheochromocytoma, paraganglioma, renal cell carcinoma, and gastrointestinal stromal tumor. |
Abnormalities of complex II activity are associated with a wide range of phenotypes, perhaps reflecting the underlying biochemical and genetic heterogeneity of the disorder. Factors that may influence the phenotype in any particular case include tissue and organ specificity, other enzyme involvement, and severity of the defect. Most individuals have had an encephalomyelopathy, with various degrees of central nervous system, skeletal muscle, and cardiac muscle involvement. Evidence of central nervous system involvement has included microcephaly, epileptic seizures, ataxia, spasticity, developmental delay, intellectual disability, psychiatric abnormalities, optic atrophy, oculomotor abnormalities, generalized hypotonia, and radiographic abnormalities compatible with Leigh disease or leukodystrophy (12; 03; 47; 31; 30; 15; 37; 17). Muscular involvement has generally taken the form of a slowly progressive proximal weakness, although occasional infants have had severe hypotonia and motor delay (03; 31; 55), or in one case recurrent rhabdomyolysis and hemolytic uremic syndrome (43). Cardiomyopathy is present in some patients and is associated with high mortality and morbidity (03; 31; 02; 37). Although chronic lactic acidosis is frequently present, acidotic crises have rarely been prominent (56; 60), and when present have occurred with severe liver involvement.
The phenotype may depend on which of the subunits or assembly factors of complex II is abnormal: SDHA mutations are associated with Leigh syndrome, leukodystrophy, late-onset optic atrophy, and cardiomyopathy (06; 03); SDHB mutations are associated with leukodystrophy and hypotonia (03); SDHD mutations are associated with early progressive encephalomyopathy, prenatal cardiomyopathy, and fetal or neonatal mortality (30; 02); and SDHAF1 mutations are associated with infantile leukoencephalopathy) (03; 47).
A distinct and remarkably homogenous myopathic phenotype has been described in persons of northern Swedish descent (34; 39; 61; 38; 26; 25), although a similar reported patient was not of Swedish descent (54). Affected individuals are noted to be exercise intolerant from early childhood. Muscle cramps and myoglobinuria are prominent along with hypertrophy of the calves. Acute exacerbations are precipitated by physical activity and are typified by vomiting, malaise, and increased weakness, often to the point where ambulation is precluded. Exacerbations may last up to 6 months. There is no associated encephalopathy, and many individuals have lived into adulthood. The heart is spared.
Genes for all four of the complex II subunits function as tumor suppressors, with numerous germline and somatic mutations associated with hereditary cancer syndromes, including pheochromocytoma and paraganglioma (07; 22; 46; 05; 06; 03; 18; 49; 27; 16; 10; 14). Renal cell carcinoma may result from SDH mutations (36; 32; 62; 44). Adrenocortical carcinoma may also be a rare manifestation of SDH mutation syndromes (18; 49). Large population studies of apparently nonsyndromic pheochromocytomas have identified a high number of germline mutations in genes of succinate dehydrogenase B and D (45; 21; 49). Other associated tumors include gastrointestinal stromal tumor, pituitary adenoma, and thyroid cancers (49; 23; 64; 28; 10). Persons inheriting an altered copy of one of these genes have a predisposition to developing these otherwise rare benign tumors at a fairly young age. However, no true enzymatic deficiency state has yet been documented in affected individuals or their tumors. The SDHAF2 gene is also a tumor suppressor, and mutations in this gene are associated with paraganglioma.
Succinate dehydrogenase deficiency may also be a contributing mechanism to the progressive β-cell failure of diabetes (35).
The prognosis of complex II deficiency has been highly variable between different affected families. Patients with liver involvement and ketoacidotic crises have generally not survived infancy. Those with myopathy and mild or no cerebral involvement have lived into early adulthood (38). In a kindred, two siblings with age of onset in the mid-40s were alive and well at ages 56 and 62 years (57).
Case 1. A 7-year-old child diagnosed with succinate dehydrogenase deficiency had presented at 1 year of age with encephalopathy and developmental regression following viral illnesses (58). MRI abnormalities were consistent with the clinical diagnosis of Leigh syndrome. Compound heterozygous SDHA variants (c.1328C> Q and c.872A> C) were identified. Treatment with L-carnitine, riboflavin, thiamine, biotin, and ubiquinone produced only mild transient clinical improvement. He is now unable to walk and speak.
Case 2. A 21-year-old woman with succinate dehydrogenase deficiency presented with generalized muscle weakness, easy fatigability, and cardiomyopathy (58). Investigations revealed increased lactate (67.4 mg/dL; reference range 4.5 to 19.8) and plasma alanine (1,272 µmol/L; reference range 200 to 579) levels. Clinical exome sequencing revealed compound heterozygous variants at exon 15 and intron 14 of the SDHA gene.
Case 3. A 25-year-old woman with complete external ophthalmoplegia, short stature, ataxia, cardiac conduction defects, and pigmentary retinopathy compatible with the diagnosis of Kearns-Sayre syndrome was found to have an isolated deficiency of complex II (51).
|
• Complex II deficiency is caused by a reduction in activity of the mitochondrial enzyme succinate-ubiquinone oxido-reductase. | |
|
• In most reported cases of complex II deficiency, a specific biochemical defect has not been documented. | |
|
• This enzyme complex participates in the mitochondrial electron transport chain to transfer reducing equivalents from succinate to coenzyme Q (as part of the process of generating ATP) and serves a dual role in the Krebs cycle (to convert succinate to fumarate). | |
|
• In most cases in which genetic information is available, isolated complex II deficiency appears to be an autosomal recessive disease, with heterozygous carriers being completely asymptomatic. | |
|
• Most patients have had chronic lactic acidosis, but of highly variable degrees. | |
|
• Three of the four subunits of the complex II complex act as tumor suppressors. |
Complex II deficiency is caused by a reduction in activity of the mitochondrial enzyme succinate dehydrogenase (SDH), which is also known as respiratory complex II, succinate-ubiquinone oxido-reductase, or succinate-coenzyme Q reductase (SQR), which is found in the inner mitochondrial membrane. Possible etiologies include direct alteration of one of the four subunits of the complex, or abnormalities of posttranslational processing, complex II assembly (eg, impaired function of assembly factors), mitochondrial protein importation (ie, of proteins formed in the cytosol), or postimportation processing. Loss of function in the succinate dehydrogenase enzyme complex is most often caused by an inherited mutation in one of the four SDHx genes (SDHA, SDHB, SDHC, and SDHD), a mechanism first implicated in familial phaeochromocytoma and paraganglioma (40). In the case of combined complex II and aconitase deficiency, which is most often observed in individuals of Swedish ancestry, an abnormality of proteins containing iron or iron-sulfur is postulated because of the co-involvement of several such proteins (25).
In most reported cases of complex II deficiency, a specific biochemical defect has not been documented. This enzyme complex participates in the mitochondrial electron transport chain to transfer reducing equivalents from succinate to coenzyme Q (as part of the process of generating ATP) and serves a dual role in the Krebs cycle to convert succinate to fumarate. It is also responsible for regulating mitochondrial respiratory function by responding to the cellular iron level, thereby influencing cellular growth (63). In addition, complex II is essential for Ca2+ signaling, which is necessary for mitochondrial and cellular function at multiple levels: impaired Ca2+ signaling caused by deficiency or chronic inhibition of complex II results in membrane potential loss, reduced production of ATP, and increased production of reactive oxygen species (42).
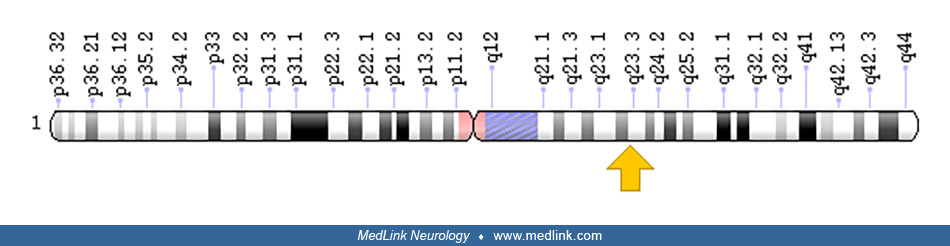
Complex II is composed of four subunits with molecular weights of 70 kd (succinate dehydrogenase A) encoded by the SDHA gene on chromosome 5p15, 27 kd (succinate dehydrogenase B) encoded by the SDHB gene on chromosome 5p15, 15 kd (succinate dehydrogenase C) encoded by the SDHC gene on chromosome 5p15, and 13 kd (succinate dehydrogenase D) encoded by the SDHD gene on chromosome 5p15.
Succinate dehydrogenase contains three iron-sulfur centers and binds a single flavin adenine dinucleotide (FAD) moiety covalently. Succinate dehydrogenase is unique in having a dual role in the essential energy-producing processes of a cell: in addition to its function in reduction of ubiquinone (coenzyme Q) as part of complex II in the aerobic electron transfer chain, it also catalyzes the oxidation of succinate to fumarate in the citric acid cycle (49).
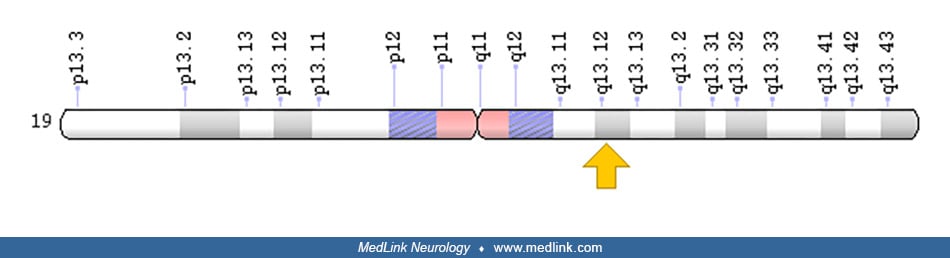
Two complex II assembly factors have been identified and characterized. Succinate dehydrogenase complex assembly factor 1 (SDHAF1), encoded by the SDHAF1 gene on chromosome 19q13.12, is a protein that resides in the mitochondria and is essential for SDH assembly, but does not physically associate with the complex in vivo.
SDHAF1 promotes maturation of the iron-sulfur protein subunit SdhB of the complex II catalytic dimer (ie, SdbA plus SdhB) and protects it from the deleterious effects of oxidants. Succinate dehydrogenase complex assembly factor 2 (SDHAF2), encoded by the SDHAF2 gene on chromosome 11q12.2, is a mitochondrial protein needed for the flavination (ie, attachment of the cofactor FAD) of a succinate dehydrogenase complex subunit required for activity of the complex.
In addition, the SDHAF2 gene is a tumor suppressor, and mutations in this gene are associated with paraganglioma. Cytochrome b560 is also associated with mature complex II.
In most cases in which genetic information is available, isolated complex II deficiency appears to be an autosomal recessive disease, with heterozygous carriers being completely asymptomatic (50; 13). Several kindreds have been reported with a Leigh disease phenotype (09; 48; 33), and a conditional mouse model of complex II deficiency has been developed that develops a Leigh-like syndrome (01). Affected patients of northern Swedish ancestry show autosomal recessive inheritance (34). In a family with the phenotype of late-onset optic atrophy, ataxia and myopathy, a missense mutation was detected on a single allele of the 70 kd subunit (SdhA) in two affected siblings, raising the possibility of autosomal-dominant inheritance (08). A recessive homozygous succinate dehydrogenase D (SDHD) mutation (ie, p.Asp92Gly) causes prenatal cardiomyopathy, a severe mitochondrial complex II deficiency, and fetal or neonatal mortality (02).
Most patients have had chronic lactic acidosis but of highly variable degrees. Two patients have had intermittent lactic acidotic crisis. All patients with elevated lactate have also had elevation in the ratio of lactate to pyruvate, as would be expected from disruption of the electron transport chain. Elevation of alanine levels is frequently seen in both blood and urine. Serum carnitine levels are uniformly low.
No detailed postmortem data are available on patients with complex II deficiency. Muscle biopsy specimens frequently show abnormal variation in fiber size and accumulation of lipid droplets; modified Gomori trichrome staining often reveals ragged-red fibers. Electron microscopic analysis of muscle mitochondria shows disruption of the normal architecture. Patients of the northern Swedish phenotype do not show ragged-red fibers but do have glycogen and lipid droplets in skeletal muscle in addition to iron-rich granular inclusions (26).
Up to two-thirds of all carotid body paragangliomas are associated with SDH deficiency (53). All four subunits of succinate dehydrogenase are tumor suppressor genes predisposing to paraganglioma, but only mutations in the SDHB subunit are associated with an increased risk of metastasis (24). Persons with mutations in the succinate dehydrogenase B (SDHB), succinate dehydrogenase C (SDHC), and succinate dehydrogenase D (SDHD) genes generally do not have a complex II deficiency in normal tissue, although complete and selective loss of complex II activity has been documented in an associated pheochromocytoma (20). Succinate dehydrogenase D (SDHD) mutation shows the most complex genetics, with autosomal dominant transmission and incomplete penetrance when maternally transmitted (07). The spectrum of mutation in succinate dehydrogenase D (SDHD) has included both truncating and nontruncating changes, and as would be expected, the trans allele is inactivated in associated tumor material. A wide spectrum of mutations is also seen in succinate dehydrogenase B (SDHB) (05), but only one kindred with a mutation at the ATG start site of succinate dehydrogenase C (SDHC) has been reported (46).
Secondary mitochondrial abnormalities have been noted in multiple neurodegenerative diseases, but a mechanism has been elucidated only in Friedreich ataxia. Frataxin, the mitochondrial protein responsible for Friedreich ataxia, functions successively as an iron chaperon and iron store by coupling iron oxidation with stepwise assembly (52; 29). The aberrant gene is thought to reduce iron availability and solubility and increase oxidative damage.
|
• Complex II deficiency is a rare condition. |
Complex II deficiency is a rare condition. A relatively mild phenotype limited to exercise intolerance and myopathy is seen in persons of northern Swedish descent.
Paragangliomas are rare tumors occurring at a rate of approximately 1 in 30,000 with 25% of tumors being familial.
|
• Prenatal detection of at-risk pregnancies has not been reported for complex II deficiency. | |
|
• The succinate dehydrogenase B gene should be systematically tested in patients with nonsyndromic pheochromocytoma. |
Prenatal detection of at-risk pregnancies has not been reported for complex II deficiency. In a single reported case, decreased succinate cytochrome C reductase activity was measured in a patient's fibroblasts, suggesting that measurement in amniotic cells is possible (13). This result must be interpreted with caution, however, as these enzymes seem to have a great deal of tissue specificity and in another case (51) no abnormality in succinate dehydrogenase was seen in fibroblasts despite a clear decrease in muscle activity. Mutational analysis of the succinate dehydrogenase A gene may provide a feasible alternative to biochemical analysis. The succinate dehydrogenase B gene should be systematically tested in patients with nonsyndromic pheochromocytoma (19).
The presence of progressive encephalomyelopathy and lactic acidosis usually suggests a mitochondrial process early in the presentation of these patients. Other mitochondrial disorders with similar presentations include MERRF, MELAS, and other disorders of the respiratory chain (especially complex IV, cytochrome oxidase). Elevations in blood lactate are seen in several conditions, including disorders of pyruvate metabolism (such as isolated pyruvate dehydrogenase deficiency and pyruvate carboxylase deficiency), disorders of oxidative phosphorylation (such as cytochrome oxidase deficiency), alterations of the Krebs cycle (primarily fumarase deficiency), organic acidopathies (such as propionic acidemia and methylmalonic acidemia), and mitochondrial encephalomyopathies (such as MERRF and MELAS). Abnormal carnitine levels can be seen in many amino and organic acidopathies and are a nonspecific finding.
Hereditary tendency to pheochromocytoma is seen in von Hippel-Lindau disease, multiple-endocrine neoplasia type 2 and neurofibromatosis I. It is not yet clear if some cases of hereditary paraganglioma will be due to other genetic loci.
|
• Initial diagnostic workup of persons with complex II deficiency should include an examination of the blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. | |
|
• Cranial imaging and EEG may be helpful in documenting the extent of cerebral involvement. | |
|
• Initial differentiation between the various diagnostic possibilities may be made on the basis of serum lactate, pyruvate, and amino acid levels and urine organic acids. | |
|
• Final diagnosis is dependent on muscle biopsy. |
Initial diagnostic workup of persons with complex II deficiency should include an examination of the blood pH, lactate, pyruvate, creatine phosphokinase, and liver enzymes. Routine spinal fluid analysis is not indicated. EMG studies may document a subclinical myopathy. Investigation of the heart with ECG and echocardiography is imperative, as many of these individuals have had cardiac involvement, which is associated with significant mortality and morbidity.
Cranial imaging and EEG may be helpful in documenting the extent of cerebral involvement (12; 03; 47; 31; 55).
In vivo proton magnetic resonance spectroscopy has identified a characteristic pattern in complex II deficiency: a singlet at 2.40 ppm originating from the two equivalent methylene groups of succinate (11). Accumulation of succinate in disordered cerebral white matter was also detected in a patient with SDHAF1-related complex II deficiency by in vivo proton MR spectroscopy (47).
Initial differentiation between the various diagnostic possibilities may be made on the basis of serum lactate, pyruvate, and amino acid levels and urine organic acids. Final diagnosis is dependent on muscle biopsy. Muscle biopsy specimens should first be examined for the presence of ragged-red fibers and structural abnormalities of the mitochondria (55). Enzyme histochemistry and immunohistochemistry are helpful in identifying and categorizing types of complex II deficiency (55). Enzyme activities may be measured in whole muscle preparation or isolated mitochondrial fractions. Commonly determined values are succinate dehydrogenase activity (which measures two of the four subunits of complex II), succinate cytochrome C reductase activity (which measures complex II and complex III activity), reduced nicotinamide-adenine dinucleotide cytochrome C reductase (which measures combined activities of complex I and complex III), and complex IV (cytochrome C oxidase). Although the enzymes of the electron transport chain are ubiquitously expressed, enzyme levels determined in other tissues (eg, leukocytes or fibroblasts) must be interpreted with caution because of the tissue and organ specificity of these enzymes.
|
• Little information is available about the treatment of complex II deficiency. | |
|
• Based on anecdotal evidence, consideration should be given to treatment with coenzyme Q and riboflavin. |
Little information is available about the treatment of complex II deficiency.
A promising observation was made regarding coenzyme Q10 in CSF, muscle, lymphoblasts and fibroblasts of two sisters with Leigh syndrome. Muscle biochemistry showed succinate cytochrome c oxidoreductase (complex II to III) deficiency and both clinical and biochemical abnormalities improved remarkably with coenzyme Q10 supplementation (59).
Three other children with complex II deficiency were treated with riboflavin with apparent benefit (12): two of the children with early-onset leukoencephalopathy remained stable or improved neurologically with riboflavin treatment, whereas another child with growth retardation and severe hyperlactacidemia remained neurologically normal as lactate decreased to near-normal levels. Supplementing riboflavin in the growth medium of cultured fibroblasts from these children produced a 2-fold increase of complex II activity, an effect that did not occur in controls.
Several persons with the Swedish phenotype due to combined complex II, complex III, and aconitase deficiency have had uneventful pregnancies (34), as did another set of siblings (57).
Several affected children with complex II deficiency have undergone anesthesia without incident.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026