







Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Monoamine oxidase-A deficiency is an X-linked disorder affecting the catabolism of serotonin and the catecholamines. The main symptoms are mild intellectual disability and behavioral abnormalities consisting of excessive, sometimes violent aggression. Diagnosis may be inferred from a finding of elevated urinary concentrations of the monoamine oxidase-A substrates, normetanephrine, 3-methoxytyramine, and tyramine, in combination with reduced amounts of the monoamine oxidase products, vanillylmandelic acid, homovanillic acid, 3-methoxy-4-hydroxyphenolglycol, and 5-hydroxyindoleacetic acid. Monoamine oxidase deficiency has been found as an isolated defect affecting monoamine oxidase A and, in combination with a deletion of the Norrie disease gene, as a combined deficiency of monoamine oxidase A and B or an isolated deficiency of monoamine oxidase B. Confirmation of monoamine oxidase-A deficiency is obtained by measurement of the activity of this enzyme in dexamethasone-stimulated fibroblasts. Combined monoamine oxidase-A and -B deficiency has similar biochemical consequences. Presentation is with intermittent hypotonia, stereotypic hand movements, and developmental delay.
|
• Monoamine oxidase deficiency is inherited as an X-linked trait. | |
|
• Monoamine oxidase exists in two distinct forms encoded by separate genes, monoamine oxidase-A and monoamine oxidase-B, and each has preferential affinities for biogenic amine and other amine substrates. | |
|
• Monoamine oxidase deficiency has been described as isolated monoamine oxidase-A deficiency, as a combined monoamine oxidase-A and B deficiency, as a combined monoamine oxidase-A and B deficiency in association with Norrie disease, and as an isolated monoamine oxidase-B deficiency in association with Norrie disease. | |
|
• Individuals with monoamine oxidase deficiency should avoid foods or drugs containing amines. |
|
• Deficiency of monoamine oxidase has been described in association with Norrie disease (an X-linked syndrome characterized by congenital blindness, hearing loss, and variable mental retardation), as an isolated defect affecting only monoamine oxidase-A and as a combined deficiency of both monoamine oxidase-A and B in the absence of Norrie disease. | |
|
• The main symptoms of monoamine oxidase-A (MAOA) deficiency ("Brunner syndrome") are mild mental retardation, behavioral abnormalities, and sleep disorders affecting male members. The most prominent behavioral problem was overt antisocial conduct with excessive, sometimes violent aggression, often triggered by anger. | |
|
• The neurochemical and behavioral abnormalities seen in monoamine oxidase-A deficiency were not found in two brothers who had monoamine oxidase-B deficiency associated with Norrie disease. | |
|
• Combined monoamine oxidase-A and B deficiency with Norrie disease often results in growth failure, severe mental retardation, autistic symptoms, and altered sleep patterns. |
Several disorders of monoamine oxidase (MAO) have been described, involving isolated MAO-A deficiency, combined MAO-A and B deficiencies, MAO-B deficiency with Norrie disease, and combined MAO-A and B deficiencies with Norrie disease. In all but the kindreds with isolated MAO-A deficiency, involvement of deletions of the X chromosome in areas other than the structural genes for MAO-A and MAO-B impedes interpretation of the clinical data in these kindreds.
Isolated MAO-A deficiency (Brunner syndrome; OMIM 300615). In 1993, Dutch geneticist Han G Brunner and colleagues at the University Hospital Radboud Universiteit (Nijmegen, Netherlands) described a large Dutch kindred with a new form of X-linked, nondysmorphic, mild mental retardation that occurred with marked behavioral abnormalities and sleep disorders (11). Detailed information was available from 8 of 14 affected males (this is an X-linked disorder, so only males are affected). Cardiovascular difficulty was also later described (16). The most prominent and distinctive behavioral problem was overt antisocial conduct with excessive, sometimes violent aggression, often triggered by anger. Recorded incidents included stabbing, fighting, attempted rape, and other forced sexual activity. Other abnormal behaviors included arson, exhibitionism, and voyeurism. Except for a unilateral clubfoot in one affected male, no other congenital abnormalities or dysmorphic features were noted. No description was provided of the early childhood behavioral characteristics of affected boys. Unaffected males and obligate female carriers within the family functioned normally.
Most of the well-described confirmed cases of isolated MAO-A deficiency have derived from the same family, reported in the two seminal papers in 1993 by Brunner and colleagues (11; 10); consequently, this disorder has sometimes been called "Brunner syndrome" after the first author of the original reports, including in reports from Brunner's own research group (06; 81).
In another family, a 5-year-old boy had developmental delay, autistic features, and myoclonic epilepsy, and his mother had mild intellectual disability and recurrent episodes of palpitations, headache, abdominal pain, and abdominal bloating (54). Whole-exome sequencing identified the maternally inherited variant c.410A>G (p.Glu137Gly) in the MAOA gene. Biochemical studies confirmed MAOA deficiency in the child and his mother. Serotonergic symptoms and high serotonin levels were present in the mother. Treatment with a serotonin reuptake inhibitor and dietary modification produced regression of the biochemical abnormalities and partial reduction of symptoms.
A 46-year-old patient with Brunner syndrome had a 15-year history of nightmares (chases, attacks, and fights), sleep-related vocalizations, and motor behaviors (talking, screaming, crying, gesturing, punching, and kicking) (15). Video polysomnography showed REM sleep behavior disorder with excessive tonic and phasic muscle activity in the mentalis and limb muscles as well as dream-enacting behaviors during REM sleep. Clonazepam administration produced a significant reduction in REM sleep behavior disorder symptomatology.
Isolated MAO-B deficiency. No specific defect affecting only MAO-B without involvement of the Norrie gene has been described.
Combined MAO-A and B deficiency (without Norrie disease). In 2010, two males were described with a 240kb deletion of Xp11.3-p11.4 that included both MAO-A and B genes but excluded the Norrie disease gene (88; 87). A similar case was reported in 2012 (60). All had developmental delay, intermittent hypotonia, and stereotypic hand movements. Two brothers presented with severe mental retardation, hand wringing, intermittent hypotonia, lip smacking, and epilepsy (88; 87), and the same phenotype was seen in the son of another family, except epilepsy was not noted (60).
Norrie disease (OMIM 300658). Norrie disease is an X-linked recessive disorder characterized by very early childhood blindness due to degenerative and proliferative changes of the retina, hearing loss, and variable mental retardation. Norrie disease is caused by a mutation in the NDP gene on Xp11, which encodes norrin. The significance of Norrie disease in the present context is that genetic defects of MAO-A and B may co-occur with Norrie disease (27; 73; 74), but Norrie disease may occur without affecting either MAO-A or -B (50).
MAO-B deficiency with Norrie disease. The neurochemical and behavioral abnormalities seen in MAO-A deficiency were not found in two brothers who had MAO-B deficiency associated with Norrie disease (03; 45). Behavior was normal, and both brothers had completed university degrees; the only clinical problems were blindness and deafness, which are features of Norrie disease. Coexistence of cataplexy and abnormal REM sleep organization has also been linked to MAO-B deficiency in boys with Norrie disease (83).
Combined monoamine oxidase-A and B deficiency with Norrie disease. Combined MAO-A and B deficiency with Norrie disease often results in growth failure, severe mental retardation, autistic symptoms, autistic-like behavior, abnormal peripheral autonomic function, atonic seizures, and altered sleep patterns (43; 44; 73; 74; 55; 24; 45),
Other possible cases. In 2001, another family was reported in whom MAO deficiency was suspected based on elevated serum serotonin concentrations. The affected mother presented with a history of flushes, headaches, and diarrhea. Her two sons had moderate intellectual impairment and attention deficit and hyperactivity disorder (20). Discussion relating to this family revealed that similar cases had been previously described (85). In one study, nine individuals exhibited the same features: chronic episodic flushing, diarrhea, headache, psychiatric symptoms, irritability, prostration, increased blood serotonin, and normal or subnormal urinary 5-HIAA concentrations. MAO deficiency was suspected, and metabolic studies supported the MAO deficiency hypothesis (84; 34). Confirmation of the condition by enzyme or mutation analysis was not reported in any of these studies.
Complications of monoamine oxidase deficiency, other than those that occur as a direct manifestation of the disease, have not been described. Potentially lethal hypertensive crises may result from an increased sensitivity to dietary or pharmacological amines (10). In the case of monoamine oxidase-A deficiency, incarceration is likely due to the behavioral abnormalities associated with the disease.
Monoamine oxidase-A deficiency is an X-linked disease, and a single large Dutch family has been investigated (11). Evidence for monoamine oxidase deficiency was achieved by a finding of a maximum multipoint LOD score of 3.69 using a CA-repeat polymorphism in the structural gene for monoamine oxidase-A, elevated urinary concentrations of monoamine oxidase-A substrates (normetanephrine, 3-methoxytyramine, and tyramine), and reduced urinary concentrations of the monoamine oxidase-A products (vanillylmandelic acid, homovanillic acid, and 5-hydroxyindolacetic acid) (11). Confirmation monoamine oxidase-A deficiency was obtained by finding zero activity of this enzyme in dexamethasone-stimulated fibroblasts (10) and localization of a nonconservative C-to-T mutation, which caused a change of a glutamine codon (cytosine-adenosine-guanine) to a termination codon (thymidine-adenosine-guanine) at position 296 of the deduced amino acid sequence (10).
Detailed information was available from eight of 14 affected males. Early case details are not available. All had mild mental retardation with IQ scores around 85. No consistent congenital abnormalities or specific dysmorphic signs were noted. Clubfoot was present in one individual. No specific dysmorphic signs were present. All tended toward stereotyped hand movements (eg, hand wringing, plucking, or fiddling), and all had behavioral problems with repeated occurrences of aggression. Aggressive behavior tended to occur during periods of 1 to 3 days and was associated with reduced sleep and night terrors. Abnormal behavior included the following: attempted rape, repeated fighting, stabbing, arson, exhibitionism, voyeurism, and sexual advances to female family members (11).
|
• Monoamine oxidase deficiency is inherited as an X-linked trait. | |
|
• Monoamine oxidase exists in two distinct forms encoded by separate genes, monoamine oxidase-A and monoamine oxidase-B, and each has preferential affinities for biogenic amine and other amine substrates. | |
|
• Monoamine oxidase deficiency has been described as an isolated monoamine oxidase-A deficiency, as a combined monoamine oxidase-A and B deficiency, as a combined monoamine oxidase-A and B deficiency in association with Norrie disease, and as an isolated monoamine oxidase-B deficiency in association with Norrie disease. | |
|
• Individuals with monoamine oxidase deficiency should avoid foods or drugs containing amines. |
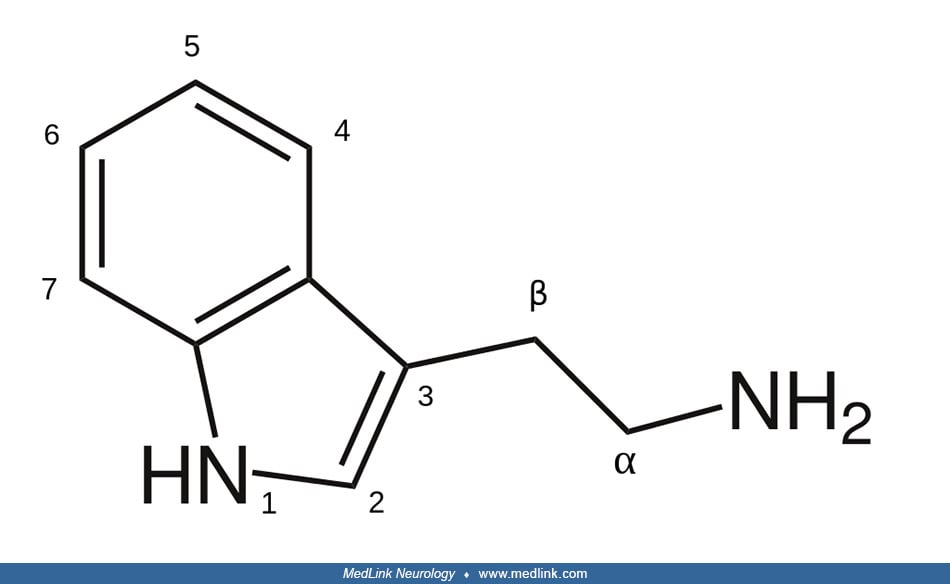
Biochemistry and molecular biology of monoamine oxidases. Monoamine oxidases (EC 1.4.3.4) are a family of mitochondrial-bound flavoproteins that catalyze the oxidative deamination of a variety of biogenic amines to the corresponding aldehydes; the substrates for monoamine oxidases include monoamine neurotransmitters and neuromodulators (eg, histamine; the catecholamines—dopamine, norepinephrine, and epinephrine; and the substituted tryptamines—serotonin and melatonin), and many minor amines (eg, tryptamine and phenylethylamine) (86).
This reaction requires flavin adenine dinucleotide (FAD) as a covalently bound redox cofactor.
Oxidative deamination by monoamine oxidases involves three main steps: (1) after formation of an FAD-substrate adduct (ie, addition of two or more distinct molecules to form a single reaction product containing all atoms of all of the components) the FAD cofactor is reduced to FADH2 whereas the substrate amine is converted to an imine (ie, a functional group containing a carbon-nitrogen double bond); (2) the imine then dissociates from the enzyme and spontaneously hydrolyzes, producing an aldehyde and ammonium; and (3) FADH2 is reoxidized to FAD, with the formation of hydrogen peroxide from molecular oxygen (08).
|
(1) MAO-FAD + RCH2NH2 ⇒ MAO-FADH2-RCH=NH |
The overall reaction is:
|
• RCH2NH2 + O2 + H3O+ ⇒ RCHO + NH4+ + H2O2 |
Monoamine oxidase is functionally coupled with either an NAD(P)+-dependent aldehyde dehydrogenase (ALDH), which oxidizes the toxic aldehyde products of the monoamine oxidase reaction to the corresponding carboxylic acids, or aldehyde reductase or alcohol dehydrogenase, which reduce the toxic products to alcohols or glycols (a 1,2-diol; ie, an alcohol with 2 hydroxyl groups on adjacent carbon atoms) (59; 08).
Two forms of the enzyme are expressed in most tissues in the outer membrane of the mitochondria; they are designated monoamine oxidase A and monoamine oxidase B based on their differential sensitivity to inhibitors and their preferential affinity for substrates (38; 40). Monoamine oxidase A has a very high affinity for serotonin (120-fold higher than monoamine oxidase B) and, to a lesser degree, norepinephrine, whereas the preferred substrates of monoamine oxidase B are phenylethylamine and benzylamine. The substrate preferences of these isoenzymes are not absolute, and in the absence of one isoenzyme, the other can deaminate some of the available nonpreferred monoamine substrates (17).
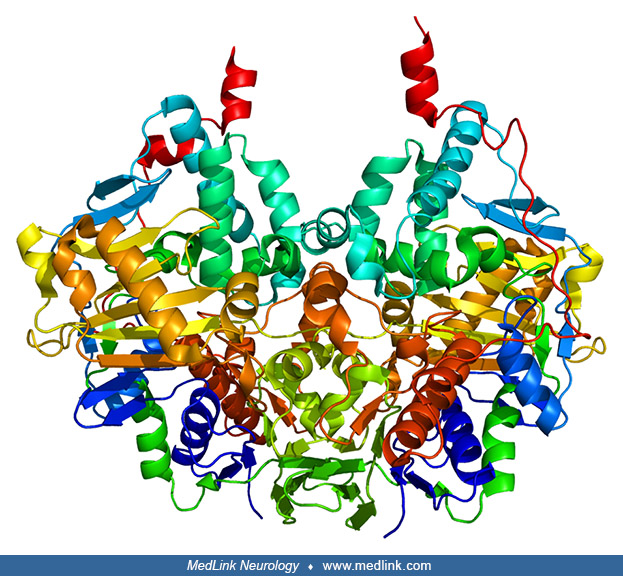
The monoamine oxidase A and monoamine oxidase B are both anchored to the mitochondrial outer membrane through a transmembrane helix within the carboxyl-terminal domain. In their membrane-bound conformations, monoamine oxidase A and monoamine oxidase B are both dimeric (51; 26; 04; 80; 29).
Although both isoenzymes are expressed in most tissues, monoamine oxidase A is the only isoenzyme that is abundant in fibroblasts and placenta, and monoamine oxidase B is the only isoenzyme expressed in platelets and lymphocytes (28). In the brain, monoamine oxidase A is found mainly in dopaminergic and noradrenergic neurons, whereas monoamine oxidase B is the only isoenzyme expressed in the cell bodies of serotonergic and histaminergic neurons and astrocytes (42).
The differences in neurochemical profiles suggest that monoamine oxidase A normally plays a more important role than monoamine oxidase B in the metabolism of the biogenic amines, which is supported by the more severe clinical phenotype seen in individuals with either monoamine oxidase A deficiency or combined monoamine oxidase A and B deficiency.
Monoamine oxidase B was thought to be involved in dopamine degradation, in large part because the therapeutic efficacy of monoamine oxidase B inhibitors in Parkinson disease was attributed to an increase in extracellular dopamine concentration. However, monoamine oxidase A, but not monoamine oxidase B, mainly contributes to striatal dopamine degradation (21). In contrast, monoamine oxidase B, but not monoamine oxidase A, is responsible for astrocytic GABA-mediated tonic inhibitory currents in the striatum (as shown in rats) (21). Therefore, in opposition to the traditional belief, monoamine oxidase A regulates dopamine levels, whereas monoamine oxidase B controls tonic GABA levels (21).
Monoamine oxidase genes. The monoamine oxidase A and monoamine oxidase B genes (MAOA and MAOB, respectively) were cloned in 1988 (02). Both have been mapped to the X chromosome in the p11.3 region in a tail-to-tail arrangement with the 3’-coding sequences separated by about 50 kb (43; 46; 19).
The monoamine oxidase A and monoamine oxidase B genes encode for proteins of 527 and 520 amino acids, with molecular weights of 59.7 and 58.8 kDa, respectively. The 3-dimensional structures of these enzymes have been elucidated (26).
The monoamine oxidase A and monoamine oxidase B genes share approximately 70% overall homology (ie, sequence similarity) and an identical intron-exon organization, each with 15 exons and 14 introns (18). The similarity of the two genes resulted from a common origin: both genes derive from duplication of a common ancestor gene (36).
The coding sequence in the human monoamine oxidase A gene is highly conserved, and observed polymorphisms either affect the third codon position and do not alter the deduced amino acid sequence or should not affect the structure of the protein (77). However, a variable tandem repeat polymorphism exists in the monoamine oxidase A promoter. The polymorphism consists of a repeated sequence present in 3, 3.5, 4, or 5 copies. Alleles with 3.5 or 4 copies are transcribed more efficiently than those with three or five copies of the repeat (71), and the variations have been associated with behavior traits such as impulsivity, hostility, and aggression (52; 14).
Monoamine oxidase disorders. The concentrations of monoamine oxidase A and monoamine oxidase B differ widely in humans, and some variations may predispose individuals to various neurologic and psychiatric diseases, including alcoholism (76), depression and alcoholism (82; 56), bipolar disorder (48; 49; 39; 69; 31; 66), schizophrenia (91), impulsive aggressiveness, antisocial personality (70; 25; 62; 12; 89), and attention-deficit/hyperactivity disorder (47; 68; 57).
Aggressive behavior in animals and humans has been linked to central and peripheral changes in serotonin, dopamine, and norepinephrine metabolism (90; 22; 78; 64; 53; 13). Congenital monoamine oxidase A deficiency and low-activity monoamine oxidase A variants are both associated with a higher risk for antisocial behavior and violence, particularly in males with a history of child maltreatment (41). This is a well-documented example of a gene-environment interaction, ie, a differential effect of a genotype on disease risk in persons with different environmental exposures.
Affected males with X-linked monoamine oxidase A deficiency (Brunner syndrome) had a marked disturbance in their urinary monoamine profile that was compatible with a defect in monoamine oxidase (11). The same group later reported that monoamine oxidase A activity was deficient in dexamethasone-stimulated cultured skin fibroblasts from affected males and that monoamine oxidase B activity was normal (10). The deficient monoamine oxidase A activity was caused by a truncating point mutation (c.886C> T [p.Gln296*]) in exon 8 of the monoamine oxidase A structural gene (MAOA) on chromosome Xp11.3, which changes a glutamine (cytosine-adenosine-guanine) codon to a termination (thymidine-adenosine-guanine) codon (10).
Monoamine oxidase A mutations in Brunner syndrome apparently increase the activity of dopaminergic neurons through upregulation of NMDAR (ie, the N-methyl-D-aspartate receptor) (81).
Two additional families have been identified, if not fully reported; one family was reported with a pathogenic missense mutation (c.797_798GC> TT [p.Cys266Phe]) associated with deficient monoamine oxidase A activity in a family affected by autism, intellectual disability, and abnormal behavior, consistent with the findings of Brunner and colleagues from 1993 (11; 10; 63).
Deficiency of monoamine oxidase A leads to increased urinary concentrations of its substrates, normetanephrine, 3-methoxytyramine, serotonin, and tyramine, and reduced urinary concentrations of the amine metabolites, vanillylmandelic acid, homovanillic acid, 3-methoxy-4-hydroxyphenolglycol, and 5-hydroxyindoleacetic acid (11; 01; 45).
Molecular simulations of the rate-limiting step of MAO-A-catalyzed serotonin degradation for known Brunner syndrome MAOA variants showed that the R45W mutation causes a 6000-fold slowdown of enzymatic function, whereas the E446K mutation causes a 450-fold reduction of the serotonin degradation rate, both of which effectively function as a gene knockout (65). In another study, the C266F mutation causes an approximately 18,000-fold slowdown of enzymatic function, which is equivalent to a MAOA gene knockout, whereas the V244I mutant causes a somewhat smaller, yet still significant, 300-fold slowdown (67).
Monoamine oxidase enzyme with serotonin (SRO) docked in the active site (shown in inset on the left) and reacting with the flavin adenine dinucleotide (FAD) cofactor. The locations of the mutated residues of two mutation varian...
Scheme of the rate-limiting step of MAO-catalyzed serotonin degradation, involving a hydride transfer between the reacting carbon atom of serotonin (SRO) vicinal to the amino group, to the nitrogen atom of the flavin adenine di...
Norrie disease. Norrie disease is an X-linked recessive disease caused by loss-of-function mutations in the Norrie disease pseudoglioma gene on chromosome Xp11.4, which encodes the protein norrin. Males with Norrie disease have childhood blindness due to degenerative and proliferative changes of the retina, and many develop sensorineural deafness and a progressive mental disorder, often with psychotic features.
MAO-B deficiency with Norrie disease. Two brothers (ages 29 and 31 years) were reported with a complete selective loss of monoamine oxidase B activity due to a microdeletion of the Norrie disease and monoamine oxidase B genes (45). The distal deletion breakpoint lay in intron 5 of the monoamine oxidase B gene, with the deletion extending proximally into the Norrie gene (45). The two individuals with monoamine oxidase B deficiency combined with Norrie disease did not show the same neurochemical abnormalities that are seen in monoamine oxidase A deficiency but instead showed increased urinary phenylethylamine levels (45). Confirmation of monoamine oxidase B deficiency was obtained by measurement of the activity of this enzyme in platelets (45).
Combined monoamine oxidase A and B deficiency without Norrie disease. Combined monoamine oxidase A and B deficiency (in the absence of Norrie disease) in two families was caused by microdeletions in chromosome Xp11.3 exclusively encompassing the MAOA and MAOB genes (88; 87; 60).
Central monoamine neurotransmitter metabolism has not been investigated in individuals with monoamine oxidase A deficiency, but in an individual with combined monoamine oxidase A and B deficiency, the concentration of serotonin in cerebrospinal fluid was elevated, and concentrations of homovanillic acid (a dopamine metabolite) and 5-hydroxyindoleacetic acid (a serotonin metabolite) were undetectable (60). Therefore, altered central monoamine metabolism is likely responsible for the aggressive behavior seen in individuals with monoamine oxidase A deficiency; however, because monoamine oxidase A deficiency also raises serotonin concentrations, it provides an interesting exception to the low-serotonin paradigm of impulsive aggression (09). REM sleep deprivation may also be involved because monoamine oxidase A inhibitors suppress REM sleep in humans (23), and REM sleep deprivation produces shock-induced fighting in animals (79).
Animal models. Mouse models are available for deficiencies of monoamine oxidase A (13; 72; 05; 08), monoamine oxidase B (37), and combined monoamine oxidase A and B deficiency (17; 08; 60; 75; 58).
Most significantly, transgenic mice lacking monoamine oxidase A show violent behavior, providing confirmatory evidence for the link between abnormal behavior and monoamine oxidase A deficiency (13; 07; 41). Combined antagonism of 5-HT2 and NMDA receptors reduces the aggression of monoamine oxidase A knockout mice (30).
In MAO-B knockout mice, phenethylamine has been shown to be a substrate of monoamine oxidase B in the paraventricular thalamic nucleus, where the enzyme is highly expressed (58); phenylethylamine is likely a substrate of monoamine oxidase B in the paraventricular thalamic nucleus as well as other tissues.
Mice lacking monoamine oxidase A and monoamine oxidase B exhibit (1) decreased proliferation of neural stem cells beginning in late gestation and persisting into adulthood; (2) significantly increased monoamine levels, particularly serotonin; and (3) anxiety-like behaviors as adults (60).
|
• Monoamine oxidase deficiency is a rare disease. |
Monoamine oxidase deficiency is a rare disease that has been incorporated into a worldwide research project focused on primary and secondary neurotransmitter disorders (61). A single large, Dutch kindred has been described with monoamine oxidase A deficiency (11). Three related boys and two brothers in a separate family have been described with monoamine oxidase B deficiency in association with Norrie disease (03; 45; 83). Three children from two families have been identified with combined monoamine oxidase-A and B deficiency in the absence of Norrie disease (88; 87; 60).
The major symptoms of monoamine oxidase-A deficiency are mild mental retardation and behavioral abnormalities, mainly violent aggression (11). These clinical characteristics are nonspecific, but measurement of monoamine oxidase substrates and products in urine should differentiate individuals with monoamine oxidase-A deficiency from those with monoamine oxidase-B deficiency or other conditions. Monoamine oxidase-A deficiency leads to a characteristic elevation of urinary concentrations of the monoamine oxidase-A substrates, normetanephrine, 3-methoxytyramine, and tyramine, in combination with reduced amounts of the monoamine oxidase products, vanillylmandelic acid, homovanillic acid, 3-methoxy-4-hydroxyphenolglycol, and 5-hydroxyindoleacetic acid (11; 01).
A similar urinary pattern of biogenic amines and metabolites has been found in individuals with X chromosome deletions involving the Norrie disease gene and both the monoamine oxidase-A and monoamine oxidase-B structural genes (73; 74; 55; 24), and in individuals with combined monoamine oxidase-A and B deficiency in the absence of Norrie disease (88; 60). Isolated monoamine oxidase-A deficiency and combined monoamine oxidase-A and B deficiency can be distinguished from those with Norrie disease because Norrie syndrome is characterized by congenital blindness and progressive deafness (27; 73; 74). The concentrations of the above amines and metabolites are normal in individuals with monoamine oxidase-B deficiency who have no behavioral or mental problems. The only biochemical abnormality in monoamine oxidase B deficiency is an increase in the concentration of urinary phenylethylamine (45).
Urinary concentrations of all monoamine oxidase products are also reduced in conditions affecting the overall biosynthesis and turnover of serotonin and the catecholamines. These include the inborn errors that affect tetrahydrobiopterin metabolism and aromatic L-amino acid decarboxylase (33). Individuals with these conditions usually present in early infancy, and often with severe neurologic impairment; however, cases with only mild neurologic symptoms have been reported. A decrease in the urinary concentrations of the monoamine oxidase substrates, as well as the metabolites, distinguishes these conditions from monoamine oxidase deficiency.
Other conditions associated with altered behavior and changes in biogenic amine metabolism include pheochromocytoma, neuroblastoma, and diseases such as Parkinson disease, where a specific defect affects the nigrostriatal dopamine system. In these, as well as most other conditions affecting biogenic amine metabolism, either a combined decrease or increase of monoamine oxidase substrates and products takes place.
|
• Monoamine oxidase-A deficiency should be considered in any X-linked condition that shows linkage to proximal Xp as well as in families where male members show prominent behavioral disturbance. | |
|
• Measurement of neurotransmitters and metabolites in urine can identify the disease and distinguish between affected males, carrier females, and normal controls. | |
|
• Monoamine oxidase-A deficiency causes an increase in the urinary concentrations of the monoamine oxidase substrates, serotonin, normetanephrine, 3-methoxytyramine, and tyramine, and a reduction in the concentrations of the monoamine oxidase products, vanillylmandelic acid, homovanillic acid, 3-methoxy-4-hydroxyphenolglycol, and 5-hydroxyindoleacetic acid. | |
|
• Monoamine oxidase A deficiency can be confirmed by measurement of the enzyme activity in dexamethasone-stimulated fibroblasts. | |
|
• Monoamine oxidase-B deficiency has only been described in association with Norrie disease. |
Positive diagnosis of monoamine oxidase-A deficiency has only been reported in detail in one kindred in which males affected by the disease presented with mild mental retardation and violent aggressive behavior (11).
Nevertheless, monoamine oxidase-A deficiency should be considered in any X-linked condition that shows linkage to proximal Xp (11) as well as in families where male members show prominent behavioral disturbance. Measurement of neurotransmitters and metabolites in urine can identify the disease and distinguish between affected males, carrier females, and normal controls (11; 01; 45). It is important that subjects not eat amine-rich foods (ie, bananas or dates) prior to testing. Monoamine oxidase-A deficiency causes an increase in the urinary concentrations of the monoamine oxidase substrates, serotonin, normetanephrine, 3-methoxytyramine, and tyramine, and a reduction in the concentrations of the monoamine oxidase products, vanillylmandelic acid, homovanillic acid, 3-methoxy-4-hydroxyphenolglycol, and 5-hydroxyindoleacetic acid (11; 01). Monoamine oxidase A deficiency can be confirmed by measurement of the enzyme activity in dexamethasone-stimulated fibroblasts (10). Monoamine oxidase A activity cannot be used to detect carrier females, as their activities fall in the control range (10).
Monoamine oxidase-B deficiency has only been described in association with Norrie disease (03; 45; 83). The major clinical symptoms are those associated with Norrie disease. Clinical features that might be found in an isolated case of monoamine oxidase B deficiency are unknown but may include cataplexy and abnormal REM sleep (83). Increased urinary phenylethylamine should raise suspicion of monoamine oxidase B deficiency, which could be confirmed by measuring enzyme activity in platelets (45).
Combined monoamine oxidase-A and B deficiency (in the absence of Norrie disease) has been described in two families. Three affected male children showed intermittent hypotonia, stereotypic hand movements, and developmental delay. Array comparative genomic hybridization (88; 87) and-oligonucleotide-SNP chromosomal microarray (60) were used to identify two separate microdeletions in the region chromosome Xp11.3-p11.4 that encompassed both monoamine oxidase-A and B. Urine and cerebrospinal fluid were examined from one child. Urinary neurotransmitters and metabolites resembled those seen in individuals with monoamine oxidase-A deficiency, and cerebrospinal fluid showed elevation of serotonin and undetectable concentrations of the serotonin metabolite, 5-hydroxyindoleacetic acid, and the dopamine metabolite, homovanillic acid.
|
• No specific treatment for monoamine oxidase deficiency has been described. | |
|
• In patients lacking normal monoamine oxidase A and monoamine oxidase B function, potentially lethal hypertensive crises may result through an increased sensitivity to dietary or pharmacological amines. | |
|
• Complications of these amine-induced hypertensive crises can include intracerebral hemorrhage, cardiac arrhythmias, and cardiac failure. | |
|
• Individuals with monoamine oxidase deficiency should avoid foods or drugs containing amines. |
No specific treatment for monoamine oxidase deficiency has been described.
Dietary intervention by avoiding foods rich in amines may be beneficial because monoamine oxidase A and monoamine oxidase B are essential for inactivating dietary and pharmacologic amines, including m-tyramine, p-tyramine, and phenylethylamine (45). In patients lacking normal monoamine oxidase A and monoamine oxidase B function, potentially lethal hypertensive crises may result through an increased sensitivity to dietary or pharmacological amines (10). Tyramine and phenylethylamine can cause hypertensive crises by acting as indirect sympathomimetics at sympathetic nerve terminals and in the adrenal glands (35). Complications of these amine-induced hypertensive crises can include intracerebral hemorrhage, cardiac arrhythmias, and cardiac failure (35). Affected individuals should not eat aged and fermented foods that contain high amounts of tyramine (60). In particular, aged cheeses (eg, blue, cheddar, Swiss, Gorgonzola, Gouda, Parmesan, Romano, feta, and Brie) and aged, dried, fermented, pickled meats (eg, bacon, sausage, liverwurst, pepperoni, salami, ham, hot dogs, and corned beef) must be avoided.
Because of the behavioral abnormalities, many procedures, even dental procedures, require special management. Furthermore, anesthesia presents serious risks of fatal arrhythmias in monoamine oxidase A deficiency. Successful anecdotal management with general anesthesia for dental treatment in a patient with MAO-A and MAO-B deficiency has been reported (32).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026