










Methylmalonic acidemia
Jun. 01, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
The copper-transporting ATPase ATP7A has diverse and important biological functions. Pathologic mutations in ATP7A produce three distinct neurologic syndromes: Menkes disease, occipital horn syndrome, and isolated distal motor neuropathy. Neonatal diagnosis of Menkes disease and early treatment with copper injections enhances survival and can normalize clinical outcomes when mutant ATP7A molecules possess small amounts of residual activity, predictable by a yeast complementation assay. Gene therapy with an adeno-associated viral vector restored ATP7A function and rescued a mouse model of Menkes disease in combination with copper, suggesting a potential therapeutic alternative for individuals with complete loss-of-function ATP7A defects. A newly discovered ATP7A disorder, adult-onset distal motor neuropathy resembling Charcot-Marie-Tooth disease type 2, shares none of the clinical or biochemical abnormalities characteristic of Menkes disease or its milder allelic variant, occipital horn syndrome.
|
• The copper-transporting ATPase ATP7A has diverse and important biological functions. | |
|
• Pathologic mutations in ATP7A produce three distinct neurologic syndromes: Menkes disease, occipital horn syndrome, and isolated distal motor neuropathy. | |
|
• In Menkes disease, severe ATP7A copper transport defects lead to infantile central nervous system neurodegeneration. Plasma catecholamine analysis is valuable for newborn screening, and yeast complementation analysis has predictive value for response to early copper treatment. | |
|
• In occipital horn syndrome, leaky splice junction or hypomorphic missense mutations allow considerable ATP7A-mediated copper transport and largely spare the central nervous system. Symptoms of dysautonomia, related to dopamine-beta-hydroxylase deficiency, and connective tissue problems predominate. Onset is in early childhood. | |
|
• Adult-onset distal motor neuropathy resembling Charcot-Marie-Tooth disease type 2, is caused by unique missense ATP7A mutations and shares none of the clinical or biochemical abnormalities of Menkes disease and occipital horn syndrome. Defective intracellular trafficking underlies this phenotype and implicates an important role of ATP7A in normal motor neuron function. |
In 1962, American pediatric neurologist John H Menkes (1928–2008) and colleagues at Columbia University in New York reported five male infants in a family of English–Irish heritage who were affected with a distinctive syndrome of neurologic degeneration, peculiar hair, and failure to thrive (82; 97; 88). The boys appeared healthy at birth and throughout the first several months of life, but then experienced seizures and developmental regression and ultimately died when aged 7 months to three and half years. The pedigree of the family strongly suggested that the condition was an X-linked genetic disease. Subsequent case reports confirmed that Menkes "kinky hair" disease was a newly recognized syndrome with unique clinicopathologic features, and later work showed that this condition resulted from a disorder of copper metabolism.
In the early 1970s, Australian pediatrician and geneticist David M Danks (1931–2003) and colleagues established that Menkes disease is a human example of abnormal myelination due to copper deficiency (22; 23; 17; 96). Danks recognized that the unusual hair of infants with Menkes disease appeared similar in texture to the brittle wool of sheep raised on copper-deficient soil in Australia. In 1937, Australian veterinary scientists had recognized the critical role of copper in mammalian neurodevelopment through the association of copper deficiency with a peculiar leukodystrophy in ataxic lambs (09). The ewes had grazed in copper-deficient pastures throughout their pregnancies, and their offspring consequently developed symmetric cerebral dysmyelination and gross pathologic changes, such as porencephalic cyst formation and cavitation. After noting the similarity of the hair texture in children with Menkes disease and copper-deficient ataxic lambs, Danks measured serum copper in seven patients with Menkes disease and demonstrated universally low levels. Serum levels of ceruloplasmin, an important copper-carrying ferroxidase enzyme, were also low.
Copper deficiency–associated myelopathy has since been described in various animal species, particularly ruminants, and is generally called swayback or enzootic ataxia. Neuropathological features include Wallerian degeneration and demyelination with microcavitation of the white matter of the spinal cord and brainstem (08). Comparative studies later demonstrated the neuropathological similarity between Menkes syndrome and swayback (109). All of the lesions of Menkes disease were shown to have their counterpart in swayback, with exception of the abnormal dendritic arborization of the Purkinje cells seen in Menkes disease (127).
Recognition of Menkes disease as a disorder of copper metabolism has facilitated diagnosis (ie, by the availability of reliable biochemical markers: low serum copper and ceruloplasmin) and has provided direction to development of an effective treatment for this lethal condition.
In 1987, a female with classic Menkes disease caused by an X-autosome chromosomal translocation was reported (64). This critical observation narrowed the cytogenetic region containing the Menkes locus to Xq13, and cell lines established from this patient ultimately led to cloning of the gene in 1993 (15; 83; 126; 115). This landmark discovery disclosed that the Menkes gene product (ATP7A) is a member of a highly conserved family of cation-transporting ATPases, molecules that function in the transport of ions across cellular and intracellular membranes.
Since the original description of Menkes kinky hair disease, there have been considerable advances in understanding the clinical, biochemical, and molecular aspects of this disorder of copper metabolism and its variants. Early subcutaneous copper injections can normalize neurodevelopmental outcome in some individuals with Menkes disease (approximately 25% in the author’s experience) and mitigate the neurologic effects in others. However, many patients with Menkes disease do not derive substantive benefit from this approach, despite early institution of treatment. Therefore, alternative treatment approaches are needed.
Identification of the Menkes gene (ATP7A) by positional cloning (15; 83; 126) has enabled molecular diagnosis of females who carry pathologic ATP7A mutations and at-risk fetuses in certain families, enhancing prenatal diagnostic preventive efforts. ATP7A is a highly conserved copper-transporting adenosine triphosphatase (ATPase) with homologs in prokaryotic and low eukaryotic systems.
Several excellent model systems for ATP7A copper transporter diseases currently exist, including the ccc2delta strain of the yeast Saccharomyces cerevisiae (which is unable to transport copper into a post-Golgi compartment and consequently requires increased copper concentrations for growth), a copper transport knock-out of the nematode Caenorhabditis elegans, a loss-of-function mutation (DmATP7-/Y) in the fly Drosophila melanogaster, a mutation (calamity) in the zebrafish Danio rerio, several mottled mutants in the mouse, which range in severity and phenotype (particularly the brindled [mo-br] male hemizygote and the blotchy mutant [(mo-blo]), and the macular mutant mouse (48; 60).
|
• In the small intestine, ATP7A regulates copper absorption, and after Cu(I) ions are absorbed into enterocytes, ATP7A is again required to transfer them across the basolateral membrane into the circulation. | |
|
• In other organs and tissues, the ATP7A protein has a dual role and shuttles between the Golgi apparatus and the cell membrane to maintain proper copper concentrations. | |
|
• In the nervous system, ATP7A normally transports copper across the blood-brain and blood-CSF barriers. | |
|
• Menkes disease and related conditions are caused by mutations in ATP7A, which leads to insufficient amounts of copper in the brain and copper accumulation in some other tissues, such as the small intestine and kidneys. | |
|
• As an X-linked disease, Menkes disease typically occurs in males who present at 2 to 3 months of age, with loss of previously obtained developmental milestones and the onset of hypotonia, seizures, and failure to thrive. | |
|
• Findings in Menkes disease have been confused with the effects of child abuse. |
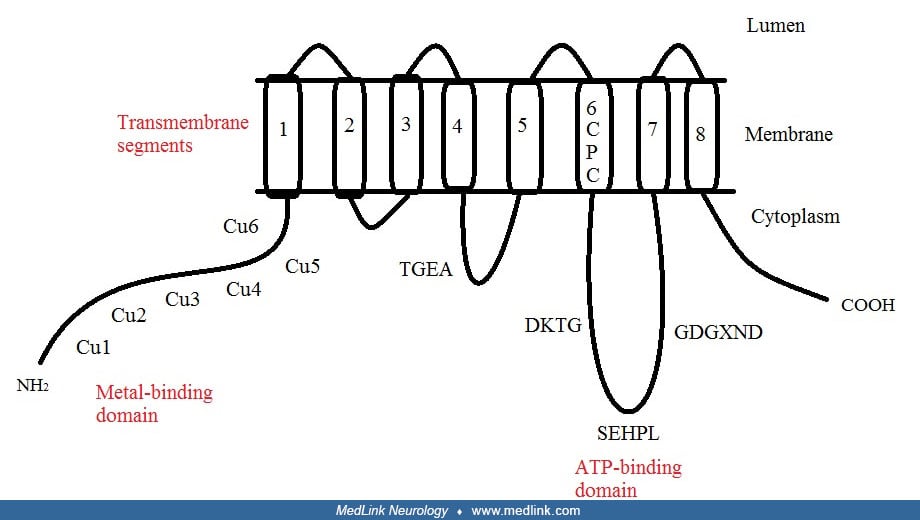
The ATP7A gene is located on the long (q) arm of the X chromosome at Xq21.1. The encoded ATP7A protein, also known as Menkes’ protein (MNK), is a copper-transporting P-type ATPase that uses the energy from ATP hydrolysis to transport copper across cell membranes.
ATP7A is a transmembrane protein with the N- and C-termini both oriented towards the cytosol. Six copper-binding sites at the N-terminal can each bind one copper atom, and eight transmembrane segments form a channel that allows copper to pass through the membrane.
ATP7A is expressed in all tissues except liver. After Cu(I) ions are absorbed into enterocytes by high-affinity copper-uptake protein 1 (CTR1) in the small intestine (43; 16), ATP7A is required to transfer them across the basolateral membrane into the circulation. In other organs and tissues, the ATP7A protein has a dual role and shuttles between the Golgi apparatus and the cell membrane to maintain proper copper concentrations (77). ATP7A normally resides in the Golgi apparatus, where it supplies Cu(I) to various enzymes (eg, peptidyl-α-monooxygenase, tyrosinase, and lysyl oxidase) that are critical for the structures and functions of nervous system, bone, skin, and hair (77; 84). However, if intracellular Cu(I) levels are elevated, ATP7A moves to the cell membrane and eliminates excess Cu(I) from the cell (19; 52; 84).
The major problem in patients with Menkes disease seems to be insufficient amounts of copper in the brain (84). In the nervous system, ATP7A normally transports copper across the blood-brain and blood-CSF barriers. A case of ATP7A somatic mosaicism supported the crucial role of choroid plexus epithelia in delivering copper to the brain (26). Biochemical and molecular investigations of rodent neuroglial cells have confirmed the role of the ATP7A homolog in delivery of copper to the brain (50).
Using the olfactory receptor neuron system as a neurodevelopmental model, Atp7a, the murine homolog, was shown to shift location from the cell bodies of developing neurons to extending axons, with axonal expression of Atp7a peaking during synaptogenesis (29; 30). Axonal outgrowth and synapse integrity are disrupted in the mottled-brindled mouse model of Menkes disease, proving that regulated expression of Atp7a is required for normal neuronal development.
Atp7a trafficks to neuronal processes of hippocampal glutamatergic neurons in response to activation of synaptic N-methyl-D-aspartate (NMDA) receptors (99; 100). This activation, which induces calcium (Ca2+) entry through the receptor after glutamate binding, is also associated with rapid copper efflux from these cells. Atp7a trafficking and copper release are impaired in the mottled-brindled model.
Intracranial vascular tortuosity and bladder diverticula are common among all subtypes of ATP7A-related copper transport disorders, and both have a high risk of complications (24).
Menkes disease and related conditions are caused by mutations in ATP7A. The abnormal ATP7A proteins impair the absorption of dietary copper, fail to supply copper to certain metalloenzymes, or are unable to shuttle back and forth between cell membranes and the Golgi apparatus. Consequently, copper accumulates in some tissues, such as the small intestine and kidneys, whereas the brain and other tissues have unusually low levels.
As an X-linked disease, Menkes disease typically occurs in males who commonly present at 2 to 3 months of age, with loss of previously obtained developmental milestones and the onset of hypotonia, seizures, and failure to thrive. Diagnosis occurs much earlier in affected infants with older affected siblings (36). Characteristic physical changes of the hair and facies, in conjunction with typical neurologic findings, often suggest the diagnosis (06). The appearance of young infants with Menkes disease is less distinctive before the onset of neurodegeneration. Morphological correlates in the brain include atrophy of grey matter, ventriculomegaly, tortuous intracranial vasculature, and white matter signal changes consistent with loss of myelin and axons. In the natural history of classic Menkes disease, death usually occurs by the time the individual with Menkes disease is aged 3 years.
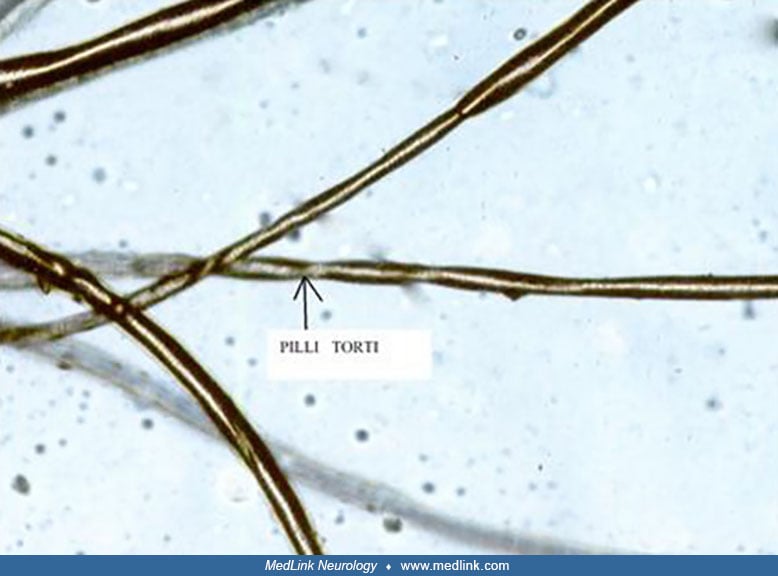
Clinical phenotype. The scalp hair and eyebrows of infants with classic Menkes disease is short, sparse, coarse, and twisted. The hair is often less abundant and even shorter on the sides and the back of the head than on the top. The twisted strands may be reminiscent of those in steel wool cleaning pads. Light microscopy of patient hair illustrates pathognomonic pili torti (ie, 180° twisting of hair shafts) and often other abnormalities including trichoclasis (ie, transverse fracture of hair shafts) and trichoptilosis (ie, longitudinal splitting of hair shafts). Hair tends to be lightly pigmented and may demonstrate unusual colors, such as white, silver, or grey; however, in some individuals with Menkes disease, the hair is pigmented normally.
The face of the individual with Menkes disease is jowly, with sagging cheeks and large ears. The palate tends to be high-arched, and tooth eruption is delayed (13). Noisy sonorous breathing is often evident. Although auscultation of the heart and lungs is usually unremarkable, a pectus-excavatum chest deformity is common. Umbilical or inguinal herniae may be present. The skin often appears loose and redundant, particularly at the nape of the neck and on the trunk.
Neurologically, profound truncal hypotonia with poor head control is invariably present. Appendicular tone may be increased with thumbs held in an adducted, “cortical” posture. Deep tendon reflexes are often hyperactive. The suck and cry are usually strong. Visual fixation and tracking commonly are impaired (37), whereas hearing is normal. Developmental skills are confined to occasional smiling and babbling in most patients with Menkes disease. Growth failure commences shortly after the onset of neurodegeneration and is asymmetric, with linear growth relatively preserved in comparison to weight and head circumference.
Biochemical phenotype. The biochemical phenotype in Menkes disease involves (1) low levels of copper in plasma, liver, and brain due to impaired intestinal absorption; (2) reduced activities of numerous copper-dependent enzymes; and (3) paradoxical accumulation of copper in certain tissues (ie, duodenum, kidney, spleen, pancreas, skeletal muscle, and placenta). The copper-retention phenotype is also evident in cultured fibroblasts and lymphoblasts, in which reduced egress of radiolabeled copper is demonstrable in pulse-chase experiments. This constellation of biochemical findings denotes a primary defect affecting copper transport that begins with impaired absorption at the intestinal level and continues with failed utilization and handling of whatever copper is conveyed to other cells in the body.
The pathogenic ATP7A gene mutations lead to impairment in copper transport, which, in turn, causes deficiencies of copper-containing enzymes (eg, dopamine β hydroxylase, peptidylglycine alpha-amidating monooxygenase [PAM], cytochrome c oxidase, lysyl hydroxylase, and copper-zinc superoxide dismutase [SOD1]).
Partial deficiency of dopamine beta hydroxylase, a critical copper-dependent enzyme in the catecholamine biosynthetic pathway, is responsible for a distinctively abnormal plasma and cerebrospinal fluid neurochemical pattern in patients with Menkes disease (56; 58; 59).
Deficiency of PAM, a secreted copper enzyme similar to dopamine beta hydroxylase, may contribute to the Menkes disease phenotype (48; 106). PAM is required for removal of the carboxy-terminal glycine residue characteristic of numerous neuroendocrine peptide precursors (eg, gastrin, cholecstokinin, vasoactive intestinal peptide, corticotropin releasing hormone, thyrotropin releasing hormone, calcitonin, vasopressin). Failure to amidate these precursors can result in a 100- to 1000-fold diminution of bioactivity compared to the mature, amidated forms. Although a deficiency of tyrosinase, a copper enzyme needed for melanin biosynthesis, is considered responsible for reduced hair and skin pigmentation in Menkes patients, PAM deficiency may also contribute to this feature through reduced bioactivity of melanocyte stimulating hormone, an amidated compound.
Profound deficiency of copper-dependent cytochrome c oxidase activity is probably a major factor in the neuropathology of Menkes disease. Effects on the brain are similar to those in individuals with Leigh disease (ie, subacute necrotizing encephalomyelopathy), in whom cytochrome c oxidase deficiency is caused by complex IV respiratory chain defects. Lactic acidosis in brain and cerebrospinal fluid suggests widespread disturbance in oxidative metabolism, although, as in Leigh disease, patients with Menkes disease do not have the severe lactic acidemia associated with other complex IV defects (93). Cytochrome c oxidase deficiency probably also contributes to the hypotonia and muscle weakness evident in patients with Menkes disease.
Reduced activity of lysyl hydroxylase, another copper enzyme, normally acts to deaminate lysine and hydroxylysine as the first step in collagen cross-link formation. Decreased lysyl hydroxylase activity significantly reduces the strength of connective tissue investing numerous organs and tissues. In patients with Menkes disease, lysyl lipoxygenase deficiency produces kinky hair, vascular tortuosity, bladder diverticula, and gastric polyps (63; 39; 18).
Deficiency of copper-zinc superoxide dismutase (SOD1) in Menkes disease may lower protection against oxygen free radicals and may have cytotoxic effects, but the relative contribution of partial superoxide deficiency to the neurodegenerative changes in patients with Menkes disease is not clear.
There is also widespread dysregulation of genes involved in cellular responses to oxidative stress, ribosomal translation, signal transduction, mitochondrial function, and immune responses (93).
Frequency. Menkes disease is a relatively rare condition with an estimated incidence of 1 per 100,000 live births to 1 per 250,000 live births. Based on the number of births in the United States (approximately 4.1 million per year), between 16 to 41 infants with Menkes disease are expected to be born in the U.S. each year. One third of these are predicted to be nonfamilial occurrences, representing new mutations.
Similarly, assuming a similar mutation frequency across racial and ethnic groups, and based on the number of births in the world (approximately 130 million per year), between 520 and 1300 infants with Menkes disease are expected to be born each year worldwide.
A study has estimated a much higher birth prevalence of Menkes disease and ATP7A-related conditions than previously appreciated: assuming Harvey-Weinberg equilibrium, the authors found that the allelic frequency of deleterious ATP7A variants predicts a birth prevalence of Menkes disease or ATP7A-related disorders as high as 1 in 8664 live male births (55). Population-based newborn screening studies will be needed to confirm this estimate.
Mortality and morbidity. No reliable approach exists to predict the lifespan of children with Menkes disease, although most of these children die by 3 years of age. Pneumonia, leading to respiratory failure, is a common cause of death, although some patients with Menkes disease die suddenly in the absence of any apparent acute medical process. The major morbidity associated with Menkes disease involves the neurologic, gastrointestinal, and connective tissue (including vasculature) systems.
Typical presentation. Classic Menkes disease often escapes attention in the neonatal period due to its subtle manifestations in neonates (36). However, several nonspecific physical and metabolic findings commonly are cited when birth histories of these babies are reviewed. These findings include premature labor and delivery, large cephalohematomas in individuals born by vaginal delivery, hypothermia that necessitated warming lights or an isolette, hypoglycemia for which early feeding or support with intravenous glucose was instituted, and jaundice that required several days of phototherapy.
Pectus excavatum and inguinal or umbilical hernias are found at birth in some patients with Menkes disease.
Occasionally, unusual hair pigmentation may suggest the diagnosis in newborns. However, the appearance of the hair is often unremarkable at this age. As in healthy babies, newborns with Menkes disease may exhibit no hair or have normally pigmented hair. The pili torti found on microscopic examination of hair from older patients with Menkes disease usually is not evident in the hair of newborns with Menkes disease.
Infants with Menkes disease generally appear to be healthy for the first 4 to 6 weeks of life, although transient neonatal hypothermia and hypoglycemia are not uncommon, and excessive jitteriness has been observed. Often when the infant is approximately two to two and a half months of age, the parents first suspect that "something isn’t right" and voice their concerns to the healthcare provider. By 4 to 5 months, affected individuals develop progressive hypotonia, seizures, failure to thrive, and characteristic coarse, wiry hair. Patients with the classic phenotype may vary in certain respects (eg, presence of functional vision, level of infant personal-social development, and severity of seizures), but they invariably demonstrate profound hypotonia and motor impairment.
Most patients develop seizures from 2 to 3 months of age, accompanied by a neurodevelopmental regression (123), and early seizures are specific for classical Menkes disease among those with ATP7A-related copper transport disorders (24). Seizures occur in more than 90% of symptomatic patients diagnosed at 2 months of age or older, but clinical seizures are seen in only about 13% of patients diagnosed and treated with copper injections in early infancy (6 weeks of age or younger), although about half of these early-treatment cases have abnormal EEG findings (60). The history of epilepsy is usually characterized by three stages: an early stage with focal clonic seizures and sometimes status epilepticus; an intermediate stage with infantile spasms; and a late stage with multifocal clonic seizures, tonic seizures, and myoclonic jerks (93; 10; 124; 123). EEGs in patients with Menkes disease with focal seizures usually show interictal focal epileptiform discharges over the posterior head region without focal slowing; this is followed by modified hypsarrhythmia and anteriorly dominant diffuse slowing with generalized and multifocal epileptiform discharges in myoclonic or generalized tonic seizures (73). Initially, the seizures can be controlled with anticonvulsant therapy, but with disease progression the seizures become extremely resistant to all antiepileptic drugs (10; 124; 123; 73). Once seizures are manifest, treatment with copper histidine is not of clear benefit for controlling seizures (124). Epileptogenesis in Menkes disease is facilitated by impairment of copper-mediated NMDA-receptor function, which enhances neuronal excitability and excitotoxic neuronal injury, leading to a cascade of neuronal dysfunction (93).
Ataxia is an independent indicator of atypical Menkes disease, which has better survival rates than classical Menkes disease (24).
Findings in Menkes disease have been confused with the effects of child abuse (28; 01). Copper deficiency produces connective tissue abnormalities and may result in subdural hematomas, wormian bones, cervical spine defects, rib fractures, other fractures, and spurring of the long bone metaphyses, which are findings that can be confused with nonaccidental trauma, and in this age group particularly with the effects of child abuse.
Urological problems are also frequent in Menkes disease, with bladder diverticula being the most common (69).
Some children with Menkes disease have autonomic impairment with a block in the conversion of dopamine to norepinephrine (mediated by the enzyme dopamine β-hydroxylase) (40); the decreased activity of dopamine β-hydroxylase is not consistently corrected by increasing serum copper levels. P-type ATPase copper transporters ATP7A and ATP7B copper transporters have distinct functions in the regulation of neuronal dopamine-β-hydroxylase (101). Mutations in the gene for ATP7A (ie, ATP7A) underlie both classic and milder phenotypes of Menkes disease.
A neurologically milder allelic variant of Menkes disease is known as occipital horn syndrome (formerly known as Ehlers-Danlos syndrome type IX or X-linked cutis laxa). The occipital horn syndrome is named for the pathognomonic wedge-shaped calcifications that form bilaterally within the trapezius and sternocleidomastoid muscle tendons at their attachment to the occiput in affected individuals (48; 111; 12). This protuberance can be palpated in some patients and is demonstrable radiographically on lateral and Towne’s view skull x-rays or on appropriate sagittal CT scan or MRI images (38). Clinical findings in occipital horn syndrome include lax skin and joints (especially cutis laxa at the nape region), bladder diverticula, inguinal hernias, vascular tortuosity, and normal or subnormal intelligence (87; 12; 38).
Among children with ATP7A-related copper transport disorders, bony exostoses, radial head dislocations, herniations, and dental abnormalities are specific for occipital horn syndrome (24).
One affected boy was 7 months of age when developmental delay was first noted. At 9 months of age, he required surgery for bladder outlet obstruction due to massive diverticula. He was diagnosed with Menkes disease at 22 months, in part because of abnormal hair (pili torti). At 27 months of age, he was able to sit alone, crawl, play with toys, indicate his needs, and voice approximately 20 words with poor articulation. He was ataxic with head bobbing and tremor, and his brain MRI exhibited mild cerebellar hypoplasia. He had a normal EEG and no clinical seizures. His height was between the 25th and 50th percentiles for age, whereas weight and head circumference were both below the 5th percentile. His biochemical parameters (ie, plasma copper, plasma catecholamines, copper accumulation in cultured fibroblasts) did not differ significantly from values in infants with classic Menkes disease.
A severely mentally retarded dysmorphic man with occipital horn syndrome never gained the ability to walk or talk and died at the age of 26 years (87). At autopsy, he had the skeletal anomalies described with occipital horn syndrome. The neuropathologic findings were like those seen in the brains of patients with Menkes syndrome and included neovascularization and extreme reduplication of the cerebral arteries, focal cortical dysplasia, bilateral cerebellar hypoplasia, and cerebellar heterotopias.
Biochemically, plasma copper and ceruloplasmin levels are in the low-normal range, copper egress in cultured fibroblasts is impaired to the same degree as in classic Menkes disease, and activity of fibroblast lysyl oxidase is markedly reduced. Additionally, some patients have signs of autonomic dysfunction (eg, syncope, episodic diarrhea) suggestive of dopamine-beta-hydroxylase deficiency, a relationship likely determined by the role of the ATP7A and ATP7B copper transporters, which have distinct functions in the regulation of neuronal dopamine-β-hydroxylase (101). Neurologic findings are otherwise normal.
Other clinical variants have been reported that involve features of both the classic Menkes and the occipital horn phenotypes.
Individuals with these variants are mildly affected (slight generalized muscle weakness and dysautonomia, including syncope, orthostatic hypotension, and chronic diarrhea) and often escape detection until mid-childhood or later. Patients with occipital horn syndrome have low-normal levels of serum copper and ceruloplasmin and abnormal plasma and CSF catecholamine levels reflecting dopamine-beta-hydroxylase deficiency, as in Menkes disease. The autonomic symptoms that inconvenience many of these patients may also occur in successfully treated patients with Menkes disease and should be amenable to treatment with L-threo-3,4-dihydroxyphenylserine (L-DOPS); L-DOPS is converted to norepinephrine via decarboxylation catalyzed by L-aromatic-amino-acid decarboxylase, which is not copper-dependent. Affected patients have splice-site mutations in the 3' region of the ATP7A gene that impaired but did not eliminate proper RNA slicing; a similar type of splicing mutation was found in an unrelated patient with typical occipital horn syndrome. In 1994, Kaler and colleagues quantitated the amount of proper splicing in cultured cells associated with these mutations at approximately 20% to 30% of normal (48). These defects were the first point mutations reported in the Menkes and occipital horn syndrome gene.
The molecular basis for occipital horn syndrome often involves exon skipping and reduction of correct mRNA processing. More than half of the reported occipital horn syndrome mutations as well as the molecular defect in a mouse model of occipital horn syndrome, mottled-blotchy, involve such aberrant splicing. Other reported cases involve missense mutations in the cytoplasmic portions of ATP7A or just inside the cytoplasmic side of the 7th transmembrane segment (A1362D), deletions in the upstream gene promotor, a 1-base deletion causing a frameshift near the end of ATP7A that removes the di-leucine motif necessary for endocytic recycling of ATP7A, and deep intronic mutations leading to activation of a pseudo-exon (a pseudo-exon is a potential exon in intronic regions of pre‐mRNA that is not normally spliced into mature mRNA) (129). The milder neurologic phenotype in occipital horn syndrome implies that the ability of mutant ATP7A to traffic to the plasma membrane and to pump copper is not completely sabotaged, but that either the quantity of normal ATP7A or the copper-transport ability of the mutant molecule is reduced.
A third clinical phenotype, distal motor neuropathy without overt copper metabolic abnormalities, was identified in association with mutations in the ATP7A copper transporter (65; 130; 53; 41). Distal hereditary motor neuropathies comprise a clinically and genetically heterogeneous group of disorders predominantly affecting motor neurons in the peripheral nervous system. Distal hereditary motor neuropathies have been classified into seven main subtypes based on mode of inheritance, age of onset, distribution of muscle weakness, and clinical progression. Genetic loci for distal hereditary motor neuropathy encode a functionally diverse array of gene products including two cation channels (ATP7A and TRPV4), a transfer RNA synthetase (03), two heat shock proteins (31; 47), and a microtubule motor protein involved in axonal transport (94). The ATP7A-related distal motor neuropathy involves unique missense mutations within or near the luminal surface of the protein, which may be relevant to the abnormal intracellular trafficking shown for these defects and the mechanism of this form of motor neuron disease.
The mechanism for development of the distal motor neuropathy phenotype appears to be distinct from those for Menkes disease and occipital horn syndrome. Subjects with ATP7A-related distal motor neuropathy have no neurologic problems other than motor neuron disease and no clinical or biochemical findings resembling those in subjects with either Menkes or occipital horn syndrome. Specifically, patients with ATP7A-related distal motor neuropathy do not show any of the classic phenotypic hallmarks of mutations at the ATP7A locus, including hair, skin, and joint abnormalities; low serum copper; abnormal plasma catecholamine levels; or renal tubular dysfunction (52). Conversely, individuals with Menkes disease or occipital horn syndrome show no clinical or electrophysiological evidence of motor neuron dysfunction (48; 111).
Clinical findings in this ATP7A allelic variant are limited to progressive distal motor neuropathy with minimal or no sensory symptoms. Signs include distal muscle weakness with curled fingers, pes cavus foot deformities, and diminished or absent deep tendon reflexes (108). Nerve conduction studies demonstrate reduced compound motor amplitudes with generally normal conduction velocities, indicative of a metabolic axonopathy. The late-onset (often adult-onset) character of ATP7A-related distal motor neuropathy implies that the responsible ATP7A mutations (ie, T994I and P1386S) have subtle effects that require years to produce manifest pathological consequences. These mutations induce subtle defects in ATP7A intracellular trafficking that produce preferential accumulation of ATP7A at the plasma membrane compared to wild-type ATP7A (53). In addition, ATP7A (T994I) interacts abnormally with p97/VCP, a protein mutated in two autosomal dominant forms of motor neuron disease (130; 53).
An overlapping phenotype of combined occipital horn syndrome and distal motor neuropathy has been reported in two brothers resulting from a novel hemizygous ATP7A splice-site variant (NM_000052.7:c.1544-2A> T) (128). The younger brother presented in early childhood with mild global developmental delay, intellectual disability, and chronic diarrhea, whereas the older brother had childhood-onset chronic diarrhea without cognitive impairment. Both developed distal motor neuropathy later in life, and imaging revealed occipital horns. Serum copper and ceruloplasmin levels were mildly reduced. RNA sequencing revealed two aberrant transcript isoforms, one of which may produce a partially functional protein.
Roughly 5% to 10% of patients with ATP7A mutations present with “atypical Menkes disease,” with features that include longer survival, cerebellar ataxia, and developmental delay (114; 07).
One case has been reported with an unusual complex phenotype that resembled Wilson disease (07). The patient was apparently normal until his early teens, when he developed a progressive spastic-ataxic gait, spastic quadriparesis, cerebellar signs (intention tremor, dysmetria, dysdiadochokinesia), uncharacterized nystagmus and dysarthria, and an axonal motor neuropathy with a Romberg sign. Cognition was normal. MRI at age 9 years showed high signal intensity in the globus pallidus bilaterally on T2-weighted images; such abnormalities indicating abnormal copper deposition are characteristic of Wilson disease, a disorder of copper retention (rather than tissue copper deprivation) that is caused by ATP7B mutations. He started using a wheelchair at age 20 years.
Another case has been reported with a patient bearing a missense ATP7A mutation with associated signs of distal motor neuropathy as well as occipital horns, confirming that these disorders are a continuum (32).
A deep intronic ATP7A variant in four males from a single family were variably affected by a predominantly skeletal phenotype characterized by bowing of long bones, elbow joints with restricted mobility that dislocate frequently, coarse curly hair, chronic diarrhea, and motor coordination difficulties (45). Analysis of whole genome sequencing data identified a deep intronic ATP7A variant that results in aberrant splicing. Sequencing of patient cDNA revealed ATP7A transcripts with exon 5 skipping, or inclusion of a novel intron 4 pseudoexon. In both instances, frameshift leading to premature termination are predicted. Quantification of ATP7A mRNA transcripts indicated that most of the transcripts (86%) have non-canonical splicing, with 68% featuring exon 5 skipping and 18% featuring the novel pseudoexon. Intrafamily variability of the phenotypes is thought to result from the stochastic effects of splicing.
|
• Mutations in ATP7A underlie both classic and milder phenotypes of Menkes disease. | |
|
• ATP7A is expressed in all human tissues except the liver. | |
|
• Laboratory findings in Menkes disease include low copper and ceruloplasmin levels, although levels in healthy newborns overlap with those of patients with Menkes disease; copper and ceruloplasmin levels remain relatively low during the first 6 weeks of life of normal infants, so “low” levels are not diagnostic for Menkes disease during this timeframe. | |
|
• An increased placental copper level is another reliable biochemical marker for neonatal diagnosis of Menkes disease. | |
|
• Copper egress in cultured fibroblasts is a definitive diagnostic test and requires propagation of cells obtained from a skin biopsy for at least several weeks before the assay can be performed. |
P-type ATPases function in the regulation of intracellular ion concentrations. Those imbedded in plasma membranes function to extrude their respective ions from the cell. Other P-type ATPases are localized to the membranes of intracellular organelles (eg, sarcoplasmic reticulum, endoplasmic reticulum) and act to sequester ions within their lumens. Interaction with ATP at a specific site induces multiple conformational adjustments that reorient the molecule with respect to the membrane, creating channels for cation translocation from high-affinity binding sites on one membrane side to low-affinity binding domains oriented toward the other side. Alternative splicing that generates multiple isoforms with potential functional differences has been demonstrated for certain ATPases of this class. In particular, intracellular copper transport is regulated by two P-type ATPase copper transporters: ATP7A and ATP7B.
Mutations in ATP7A underlie both classic and milder phenotypes of Menkes disease (48; 59; 74; 116; 117; 119; 85; 21; 65; 103; 35). The vast majority of identified ATP7A mutations are intragenic mutations or partial gene deletions (118). The severity of mutations and amount of residual copper ATPase activity determine the variable clinical and treatment outcomes (54; 49; 59; 49; 66; 67; 27; 110; 84; 103). Even small amounts of functional ATP7A protein permit a mild phenotype (84). Secondary deficiencies of copper-dependent enzymes contribute to some of the clinical manifestations.
ATP7A is expressed in all human tissues except the liver. The messenger RNA transcript is approximately 8.5 kb, with a long 3' noncoding portion and a coding sequence of 4.5 kb. The predicted gene product is a 1500–amino acid molecule that is similar to numerous ion-motive ATPase molecules, including a pair of copper-transporting ATPases (copA and copB) in the bacterium Enterococcus hirae, the ccc2 copper transporter in Saccharomyces cerevisiae, and ATP7B, the Wilson disease gene product. The overall sequence similarity among prokaryotic and eukaryotic cation-transporting ATPases suggests that these proteins have been conserved throughout evolution in response to the need for import and export of various cations across different cellular membranes (92; 76).
In the Menkes gene product, the N-terminal portion has a distinctive recurring amino acid pattern, cysteine-X-serine-cysteine, which is comparable to putative metal binding sites in the bacterial ATPases involved in transport of copper, cadmium, and mercury. Hydrophobicity analysis indicates six to eight transmembrane domains, and the predicted protein demonstrates all of the other functional domains expected of P-type ATPases. The expression of the Menkes gene in nearly all human tissues, the severe consequences of impaired function (ie, Menkes disease), and the high degree of evolutionary conservation all indicate the fundamental importance of the copper transport process that this gene encodes.
Studies in several laboratories localized the ATP7A gene product to the trans-Golgi apparatus, where it is involved in delivering copper to copper-dependent enzymes processed in the secretory pathway of cells (33; 72; 89; 14; 91; 90; 98; 107; 125; 84). In addition, ATP7A relocates in response to increased copper exposure, moving to the plasma membrane of cells where it functions as a pump directly mediating copper exodus from cells (112; 84; 103).
ATP7A exhibits copper-dependent trafficking; ATP7A is found in the trans-Golgi network at low copper concentrations, and in the post-Golgi compartments and the plasma membrane at higher concentrations (103; 104). Phosphorylation is crucial for the exit of ATP7A from the trans-Golgi network, whereas dephosphorylation is crucial for recycling back to the trans-Golgi network. The severity of Menkes disease in individual patients correlates with the cellular localization of ATP7A (103).
WW-PLEKHAs-PDZD11 complexes regulate the localization and function of ATP7A to promote copper extrusion in the elevated copper state (104). PDZD11 interacts with the C-terminus of ATP7A, which contains sequences involved in ATP7A trafficking (104). PLEKHA5, PLEKHA6, and PLEKHA7 bind to the PDZD11 N-terminus through their WW domains. The WW domain, also known as the rsp5-domain or WWP repeating motif, is a modular protein domain that mediates specific interactions with protein ligands. WW-PLEKHAs (PLEKHA5, PLEKHA6, PLEKHA7) recruit PDZD11 to distinct plasma membrane localizations and are required for efficient anterograde targeting of ATP7A to the cell periphery in elevated copper conditions. WW-PLEKHAs promote PDZD11 interaction with the C-terminus of ATP7A.
WW-PLEKHAs and PDZD11 function in the anterograde trafficking of ATP7A either in conditions of basal copper or elevated copper. PLEKHA7 is exclusively localized at the adherens junction/zonula adherens. Under basal copper, few ATP7A-containing membrane vesicles cycle between the plasma membrane and trans-Golgi network, and most ATP7A is in the trans-Golgi network. Elevated copper ATP7A-containing membrane vesicles are trafficked to the cell periphery along microtubule tracks and are tethered to the apical lateral and basal plasma membrane by WW-PLEKHA-PDZD11 complexes.
Laboratory findings. Laboratory findings in Menkes disease include low copper and ceruloplasmin levels, although levels in healthy newborns overlap with those of patients with Menkes disease; copper and ceruloplasmin levels remain relatively low during the first 6 weeks of life of normal infants, so “low” levels are not diagnostic for Menkes disease during this timeframe. In general, low ceruloplasmin is a more sensitive and discriminating biomarker for ATP7A-related disorders than serum copper (24).
In any case, lower-than-normal ceruloplasmin levels are not, by themselves, specific for Menkes disease at any age. Low ceruloplasmin levels may be seen in the following conditions: (1) Menkes disease (copper does not adequately cross the intestinal barrier due to ATP7A deficiency); (2) Wilson disease (delivery of copper into the lumen of the ER-Golgi network is absent in hepatocytes due to absent ATP7B); (3) dietary copper deficiency (copper availability doesn't affect the translation of the nascent protein, but the apoenzyme without copper is unstable); and (4) aceruloplasminemia (genetically determined abnormal gene expression of the ceruloplasmin gene).
In contrast to the overlap of copper and ceruloplasmin levels in normal newborns and newborns with Menkes disease, plasma and CSF catechol levels are distinctively abnormal in patients with Menkes disease at all ages, including in the prenatal and newborn periods. High levels of the catechols DOPA, dihydrophenylacetic acid, and dopamine, as well as low levels of dihydroxyphenylacetic acid (ie, the deaminated metabolite of norepinephrine), are hallmarks of the partial dopamine-beta-hydroxylase deficiency invariably associated with Menkes disease and occipital horn syndrome.
An increased placental copper level is another reliable biochemical marker for neonatal diagnosis of Menkes disease.
Copper egress in cultured fibroblasts is a definitive diagnostic test and requires propagation of cells obtained from a skin biopsy for at least several weeks before the assay can be performed. The assay is being replaced by molecular analysis but remains useful for labs with experience in working with radiolabeled copper (half-life of copper-64 is approximately 12 hours) and in the conduct of pulse-and-chase experiments. Rapid molecular diagnostic assays can facilitate early diagnosis as well as carrier testing (75).
Truncating mutations in ATP7A are frequently associated with classical Menkes disease, whereas splice site and intronic mutations are more prevalent in occipital horn syndrome (24).
Expression of Menkes disease in females. Several female patients with Menkes disease have been reported, in whom chromosome rearrangement (eg, translocations), XO/XX mosaicism, or unfavorable lyonization was responsible for expression of the full phenotype (25; 105; 81). In one large family, skewed X-inactivation was associated with neurologic sparing in carrier females (25). In another family, unfavorable switching of skewed X chromosome inactivation led to Menkes disease in a female infant (81).
Samples from individuals I-5, I-6, II-2, II-3, and III-1 were collected. (Source: Matsumoto A, Kano S, Kobayashi N, et al. Unfavorable switching of skewed X chromosome inactivation leads to Menkes disease in a female infant. Sc...
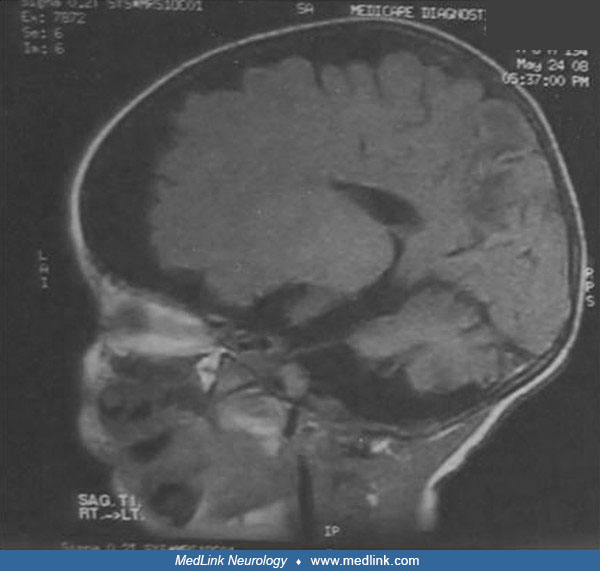
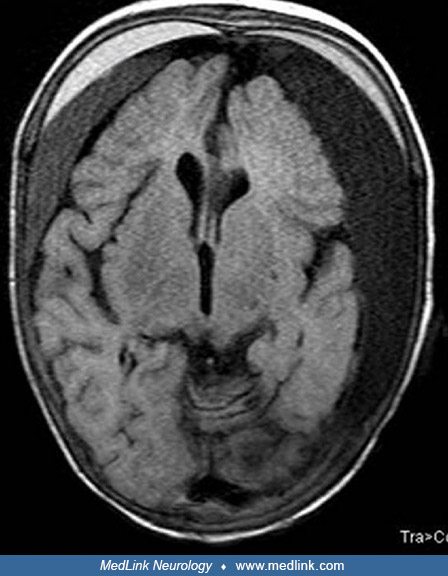
T2-weighted MR imaging shows diffuse atrophy of the cerebrum and delayed myelination in the splenium of the corpus callosum in a female infant with Menkes disease due to unfavorable switching of skewed X chromosome inactivation...
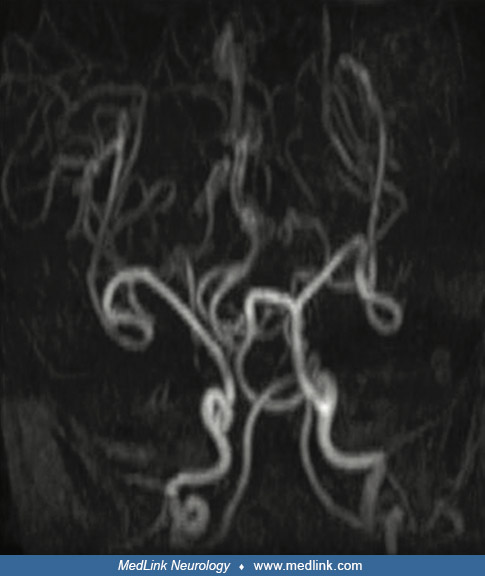
MR angiography showing intracranial tortuous arteries characteristic of Menkes disease in a female infant with Menkes disease due to unfavorable switching of skewed X chromosome inactivation. (Source: Matsumoto A, Kano S, Kobay...
Menkes disease in females manifests with a variable spectrum of clinical findings, but neurodevelopmental disability, hypotonia, and connective tissue abnormalities are uniformly present, whereas other features may or may not be present (eg, seizures, cerebral atrophy, and cerebrovascular tortuosity) (105).
Clinical and biochemical parameters have not been uniformly reliable for detection of female carriers. Serum copper and ceruloplasmin levels are within the reference range in carriers, as are plasma catechol levels. Copper egress in cultured cells of some obligate carriers demonstrate values within the range of males with Menkes disease or intermediate between normal and affected, but many have values that overlap with normal, presumably due to lyonization and apparent selection against the mutant cell type.
Pili torti in the hair of the mother of a patient with Menkes disease is considered definitive proof of her status as a gene carrier. As in prenatal testing, molecular diagnostic approaches aid immensely in carrier detection, once a given family's mutation has been characterized in a male with Menkes disease.
Imaging studies. A variety of imaging studies are often helpful in the evaluation and treatment of patients with Menkes disease or occipital horn syndrome.
Plain radiograph may reveal wormian bones, rib flaring, metaphyseal spurring, and periosteal reactions in the long bones of the limbs (132). In subjects with occipital horn syndrome, skull x-rays may show an occipital exostosis projecting from the line of insertion of the trapezius muscle and wide fontanels on skull x-ray films with no exostoses or deformities elsewhere (38).
Head CT and brain MRI can assess for gross structural lesions and the degree of myelination. White matter abnormalities reflecting impaired myelination, diffuse atrophy, and ventriculomegaly are typical findings, and these abnormalities can progress rapidly even over a 2-month period (11; 78; 79; 95). Other MRI findings can include increased intracranial vascular tortuosity and basal ganglia abnormalities (132). Although increased intracranial vascular tortuosity may be evident on MRI, it is better visualized with MRA.
The tortuous "corkscrew" appearance of cerebral vessels is well visualized by MRA, and these changes also progress rapidly over a short time (11; 78; 79; 95).
Subdural hematomas are common in infants with Menkes disease, and cerebrovascular accidents can occur in patients with the disease who survive longer (95).
Cystography or pelvic ultrasound reveals diverticula of the urinary bladder in nearly every patient with Menkes disease (69). Routine radiographs often disclose abnormalities of bone formation in the skull (wormian bones), long bones (metaphyseal spurring), and ribs (eg, anterior flaring, multiple fractures), and later, undertubulation (Erlenmeyer flask deformity), which causes wide metaphyses, and metaphyseal flaring, similar to findings in some types of bone dysplasia (02). Also reported are intrapartum skull fractures and cervical spine abnormalities, such as C2 posterior arch defects (121; 46; 34; 70). Some of these bone abnormalities can be mistaken for child abuse (05; 04; 20; 46; 34; 28; 01).
• Four fundamental issues must be addressed in configuring therapeutic strategies for individuals with Menkes disease: (1) the block in intestinal absorption of copper must be bypassed, (2) copper must be made available to the enzymes within cells that require it as a cofactor, (3) copper must be conveyed across the blood-brain and blood-cerebrospinal fluid barriers, and (4) infants with Menkes disease must be identified and treatment commenced early in life before irreparable neurodegeneration occurs. | |
• Unfortunately, at present, a cure for Menkes disease does not exist, and available treatments offer quite limited and typically short-lived benefits. | |
• Although copper replacement does not provide substantial neurologic improvement in all patients with Menkes disease who are treated early or in older individuals with Menkes disease, its use has been associated with modest clinical benefit including decreased seizure frequency and reduced irritability. |
Four fundamental issues must be addressed in configuring therapeutic strategies for individuals with Menkes disease: (1) the block in intestinal absorption of copper must be bypassed, (2) copper must be made available to the enzymes within cells that require it as a cofactor, (3) copper must be conveyed across the blood-brain and blood-cerebrospinal fluid barriers, and (4) infants with Menkes disease must be identified and treatment commenced early in life before irreparable neurodegeneration occurs.
Unfortunately, at present, a cure for Menkes disease does not exist, and available treatments offer quite limited and typically short-lived benefits (118).
Parenteral administration of copper in various forms (eg, copper histidine) restores circulating levels of copper and ceruloplasmin to normal levels, whereas oral copper generally does not (except possibly copper nitriloacetate, in some patients) (48; 50; 51; 42; 71; 120). Subcutaneous copper-histidine supplementation has been standard of therapy in some countries (other than the United States), but long-term administration is not desirable because of nephrotoxicity (86). A systematic review indicated that treatment with copper-histidine is effective to increase survival and reduce neurologic burden of the disease if initiated in the neonatal period (122). However, although low hepatic copper stores are repleted quickly by parenteral therapy, brain copper during treatment increases only gradually, if at all, consistent with trapping of copper within cells comprising the blood-brain barrier.
In patients with Menkes disease who are treated early with copper injections, clinical outcomes have varied. This therapy may only be effective if treatment is initiated within days after birth (122). Approximately 25% of patients with classic Menkes disease who are identified and treated within the first 10 days of life have normal neurodevelopmental outcomes.
Although copper replacement does not provide substantial neurologic improvement in all patients with Menkes disease who are treated early or in older individuals with Menkes disease, its use has been associated with modest clinical benefit including decreased seizure frequency and reduced irritability. There is no evidence that copper treatment influences life span in patients with Menkes disease in a consistent fashion. Given the possibility of small clinical benefits or improved patient comfort in a hopeless disease, decisions concerning copper replacement in symptomatic patients are perhaps best made by the parents, following frank discussion of the limited benefits that can be expected. In instances where the diagnosis is made prior to the onset of neurologic damage, copper replacement is clearly indicated because the prospect of preventing the neurodegenerative features exists, at least for some such individuals.
Proximal renal tubular damage is a known adverse effect of copper overload. These effects presumably relate to exacerbation of the natural tendency of the Menkes kidney to sequester copper. However, the clinical significance of this adverse effect is minor in most treated patients, as renal losses rarely reach the point where replacement (eg, oral bicarbonate) is needed. Concomitant treatment with penicillamine, a copper-chelating agent, has been used in some patients with Menkes disease with the rationale of preventing copper overload.
The carbamic acid derivative, diethyldithiocarbamate, is a chelating agent that forms lipophilic complexes with copper. When fed to rats, diethyldithiocarbamate increases copper levels in the brain. In macular mice that die by age 2 weeks without copper treatment, intraperitoneal administration of diethyldithiocarbamate or dimethyldithiocarbamate resulted in normal survival in the absence of any copper treatment. Furthermore, survival correlated with increases in macular brain copper levels. In mice that received no exogenous copper and 200 mg/kg of dimethyldithiocarbamate, the brain copper level was the same as in normal controls.
These experiments suggest that the lipophilic complex was able to bypass the block in macular brain copper uptake. Although toxicity considerations may prohibit long-term use of such agents in humans, the principle that lipid-soluble complexes can enhance copper transport across cellular membranes deserves further attention with respect to treatment of individuals with Menkes disease.
Gene therapy for patients with Menkes disease is a theoretical possibility although there are a number of potential problems with this approach. Because gene therapy requires targeting of specific organs, a disease like Menkes disease, which affects nearly every cell in the body, may not be amenable to full correction. Assuming the brain is the target organ, concern regarding the potential toxicity of gene therapy viral vectors would be heightened. The gene needs to be delivered to many cells, and expression needs to be sustained. The gene product, when produced, has to be targeted to the appropriate membrane. Coproduction of native mutant forms of the copper ATPase could inhibit proper function of the normal molecule expressed by the introduced gene. Parenteral copper replacement would likely remain necessary in order to circumvent the defect in intestinal absorption. Despite these significant caveats, functional characterization of the Menkes copper ATPase, progress in developing systems of gene delivery to the brain in the Menkes mouse model, and the severe nature of the untreated condition may render Menkes disease a candidate for gene therapy in the future.
Whatever mode of treatment for Menkes disease is employed, intervention at the earliest possible moment is of paramount importance, as the window of opportunity before neurologic injury is no longer than several months.
L-threo-dihydroxyphenylserine (L-DOPS; Droxidopa®) is an agent for possible treatment of the partial dopamine-beta-hydroxylase deficiency in Menkes disease and occipital horn syndrome. L-DOPS is a synthetic amino acid converted to norepinephrine by the enzyme aromatic-L-amino acid decarboxylase. Administering this compound to patients with Menkes disease increases levels of norepinephrine and DHPG (the deaminated metabolite of norepinephrine) because the block in dopamine-beta-hydroxylase activity is bypassed. L-DOPS should correct the typical neurochemical abnormalities in plasma, and possibly in CSF if L-DOPS crosses the blood-brain barrier (which is not established).
However, the precise contribution of dopamine-beta-hydroxylase deficiency to the Menkes disease phenotype is unclear. Congenital dopamine-beta-hydroxylase deficiency, a rare autosomal dominant disorder, is characterized by noradrenergic denervation and adrenomedullary failure (80; 113). Norepinephrine and epinephrine are undetectable in plasma, urine, and cerebrospinal fluid, whereas plasma dopamine levels are elevated. Severe orthostatic hypotension is the most important clinical feature; other clinical features include blepharoptosis, hyperflexible joints, high palate, sluggish deep tendon reflexes, and mild normocytic anemia. In addition, elevated dopamine levels cause hypoprolactinemia, reduced REM sleep, increased slow-wave sleep, and sodium loss, despite low blood pressure. Treatment of patients with dopamine-beta-hydroxylase deficiency using L-DOPS, which is converted directly into norepinephrine, results in a sustained relief of orthostatic symptoms. Similarly, in patients with occipital horn syndrome, treatment with L-DOPS may correct neurochemical abnormalities and resolve dysautonomic symptoms, which include orthostatic hypotension and chronic diarrhea.
Copper. Parenteral copper (administered intravenously or subcutaneously) in a variety of formulations has been used to treat individuals with Menkes disease and occipital horn syndrome.
Whether any particular preparation is superior to another in terms of neurologic outcomes is not clear; the biology of the Menkes transporter suggests that uptake of copper into cells is not dependent on the chemical form in which copper is introduced by subcutaneous injection. Copper chloride, copper histidine, and copper sulfate have been used in humans, although all of the experiences with very early treatment of Menkes disease have been with copper histidine. Copper chloride and copper sulfate are available commercially in the United States, whereas copper histidine is not (although copper histidine is available for use at the National Institutes of Health) (59). Both copper chloride (cupric chloride) and copper sulfate (cupric sulfate) can produce proximal renal tubular damage, presumably by exacerbating copper sequestration in the kidneys of patients with Menkes disease, but the clinical significance is generally minor because renal losses rarely reach the point where replacement (eg, oral bicarbonate) is needed. Cupric sulfate can produce significant injection-site inflammation.
Neonatal diagnosis and early treatment with copper injections enhance survival in patients with this disease and can improve clinical outcomes, particularly if mutant ATP7A molecules retain small amounts of residual activity and if treatment is initiated early, before patients become symptomatic (52; 53). In a clinical trial of copper histidine for Menkes disease, improvements were demonstrated in gross motor, fine motor/adaptive, personal-social, and language development, and also in growth in head circumference, among subjects who received early treatment prior to onset of symptoms (53). Mortality at the age of 3 was higher (50%) among subjects who were older and symptomatic when treatment began than among patients who were asymptomatic when therapy was initiated (29%). Despite early treatment, most patients die before 5 years of age, and the remaining patients are severely retarded in neurodevelopment (68).
Unfortunately, even prenatally initiated copper replacement is inadequate to correct Menkes disease caused by severe loss-of-function mutations (44). In one reported case, copper histidine (900 μg per dose) was administered directly to the fetus by intramuscular injection (into the fetal quadriceps or gluteus) under ultrasound guidance (44). Fetal copper and ceruloplasmin levels rose to approximately three times baseline levels with treatment, as assessed by percutaneous umbilical cord sampling. After pulmonary maturity was confirmed biochemically, the baby was delivered at 35.5 weeks and daily copper histidine therapy (250 μg subcutaneously, twice daily) was initiated. Nevertheless, despite early intervention with copper replacement and marked improvement of biochemical parameters in blood, the infant was hypotonic, had developmental delay and electroencephalographic abnormalities, and died of respiratory failure at five and a half months of age. The patient's ATP7A mutation (Q724H) severely disrupted mRNA splicing and produced an essentially complete absence of ATP7A protein.
The ethical considerations surrounding copper replacement treatment in patients with Menkes disease who manifest significant neurologic symptoms were reviewed by Sheela and colleagues (102).
L-DOPS. L-DOPS is a synthetic amino acid that is converted in vivo to norepinephrine by the enzyme aromatic-L-amino acid decarboxylase. Administering L-DOPS to patients with Menkes disease can increase levels of norepinephrine and DHPG (the deaminated metabolite of norepinephrine) because the block in dopamine beta hydroxylase is bypassed. L-DOPS should correct typical neurochemical abnormalities in plasma of patients with Menkes disease (and possibly also in CSF, if L-DOPS crosses the blood-brain barrier). The starting adult dose of L-DOPS is 250 mg orally, twice daily; this can be titrated as tolerated based on levels of plasma and CSF catecholamine levels, and DOPA/DHPG and DOPAC/DHPG ratios. Incremental dosage increases should not exceed 50% of the prior dose. The pediatric dose of L-DOPS is 20 mg L-DOPS (estimated 5 mg/kg body weight) orally, twice daily. Adverse effects (eg, nausea, vomiting, headache, dry mouth, elevated transaminase levels) have been observed in less than 1% of adults in whom this compound has been used for a number of dysautonomia syndromes. Because many of these symptoms are impossible or difficult to detect in infants, this agent is generally withheld in this age group, especially if any adverse effects (eg, excessive irritability) are temporally associated with administration.
Genetic counseling is an important component. As an X-linked recessive trait, the ATP7A is transmitted by asymptomatic females who are carriers to 50% of their male offspring (who are affected) and to 50% of their female offspring (who are typically asymptomatic carriers). Conversely, 50% of both male and female offspring are healthy. Thus, the overall risk of having a child with Menkes disease for a woman who is a documented female carrier is one in four (25%) for each pregnancy (ie, one in two chance that the sex is male, multiplied times the one in two chance that the male inherits the Menkes gene). Counseling, carrier testing, and, if indicated, prenatal diagnosis to female relatives of a documented gene carrier can be offered.
No specific contraindication exists for immunizations of infants with Menkes disease because the pertussis component is acellular. Seasonal vaccination against influenza is recommended.
Prophylaxis against urinary tract infections (eg, trimethoprim/sulfamethoxazole) is warranted in patients with bladder diverticula.
Physical or occupational therapy can maximize developmental attainment in patients with Menkes disease. Aspects of these therapies can also be taught to parents for application in the home.
Menkes disease has a substantial emotional impact on the family. Parents of patients with Menkes disease often have the pain of watching the transition from apparent good health to profound impairment within the first several months of life. Anger, disbelief, guilt, and anxiety regarding an uncertain future are common reactions. Unfortunately, no reliable way to predict the life span of children with Menkes disease exists; however, most of these children die by the time they are 3 years of age. Pneumonia leading to respiratory failure is a common cause of death, although some patients with Menkes disease die suddenly in the absence of any acute medical process.
|
• Genetic counseling and prenatal diagnosis (when available and desired) can facilitate prevention and early identification of Menkes disease. | |
|
• Approximately one-third of Menkes disease cases result from new mutations. | |
|
• For pregnancies in known or suspected female carriers, prenatal diagnosis of Menkes disease on biochemical grounds is available. |
Genetic counseling and prenatal diagnosis (when available and desired) can facilitate prevention and early identification of Menkes disease. However, approximately one-third of all incidents of Menkes disease cases result from new mutations.
For pregnancies in known or suspected female carriers, prenatal diagnosis of Menkes disease on biochemical grounds is available from the John F Kennedy Institute in Glostrup, Denmark. Carrier status must be suspected in a woman and her female relatives (mother, sisters, daughters) following the diagnosis of Menkes disease in a son. Recurrence risk in future pregnancies of such women may be as high as 25%.
Abnormal egress of radiolabeled copper in cultured amniocytes (reduced compared to normal, ie, a higher percentage of copper retained by cells) was the basis of the original prenatal testing. When techniques for obtaining fetal tissue earlier in gestation (ie, chorionic villus sampling) became available, diagnostic criteria derived from analysis of those tissues were developed (elevated copper content and abnormal copper egress in cultured chorionic cells). In using chorionic villus copper content as the marker, it is necessary to avoid contamination from either the instrument used to obtain the sample or by incomplete separation of the maternal deciduum. The most reliable biochemical marker using chorionic villus specimens has been retention of radiolabeled copper in cultured chorionic cells after a 20-hour pulse and 24-hour chase. Knowledge of the abnormal gene in Menkes disease enables prenatal testing by molecular means for families in which the proband's mutation has been characterized (50).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Douglas J Lanska MD MS MSPH
Dr. Lanska of the University of Wisconsin School of Medicine and Public Health has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neurogenetic Disorders
Jun. 01, 2026
Neurogenetic Disorders
May. 08, 2026
Neurogenetic Disorders
Apr. 30, 2026
Neuropharmacology & Neurotherapeutics
Apr. 23, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026
Neurogenetic Disorders
Apr. 14, 2026