


Peripheral Neuropathies
Carbon disulfide neuropathy
Jun. 11, 2026
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
This article describes the neuromuscular complications of critical illness. The author discusses a large study that links intensive glucose control in critically ill patients to an increased risk of moderate and severe hypoglycemia, both of which are associated with an increased risk of death. Furthermore, he reviews the American Diabetes Association recommendations for a target blood glucose in order to reduce the risk of hypoglycemia in critically ill patients.
|
• After intensive neurorehabilitation, most critical illness neuropathy or myopathy patients recover within 1 to 2 years. | |
|
• The presence of additional central nervous system lesions is associated with poor prognosis for recovery. |
Critical illness polyneuropathy is most often described in patients in the critical care unit with sepsis. Sepsis originally meant putrefaction, a decomposition of organic matter by bacteria and fungi (23). Over time, it has been increasingly associated with a severe systemic response to infection, usually resulting in early death. With improvements in medical and surgical care, survival of many critically ill patients is now prolonged, but the mortality rate may still be greater than 30%, and approximately half a million patients are reported each year with sepsis in the United States (76). Research has been impeded by confusion over terminology, with "Gram-negative sepsis," "sepsis syndrome," and "septic shock" all being applied with uncertainty (18). In attempts to resolve this confusion, the Society of Critical Care Medicine and the American College of Chest Physicians convened a consensus conference in 1992 to standardize definitions (05). Recognizing that a severe systemic response can be evoked in the absence of infection (eg, trauma and burns), the panel coined the term "systemic inflammatory response syndrome" (05).
When systemic inflammatory response syndrome was associated with a documented infection, the term "sepsis" could be applied. Furthermore, the terms "severe sepsis" and "septic shock" were reserved for those patients with organ dysfunction and hypoperfusion or hypotension, despite adequate fluid replacement.
Osler observed muscle wasting as a complication of prolonged sepsis (94). In 1961, Mertens described "coma-polyneuropathies" in patients who had circulatory shock associated with acute intoxication and severe metabolic crises, seemingly due to metabolic and ischemic lesions of the peripheral nervous system (84). Henderson and colleagues described a polyneuropathy in patients with burns (48). Between 1977 and 1981, Bolton and colleagues observed five unusual patients in the critical care unit who showed difficulty in weaning from the ventilator and who had severe weakness in the limbs (16; 17). Comprehensive electrophysiologic and morphologic studies established this condition as a primary distal, axonal degeneration of motor and sensory fibers (17). Because sepsis and multiple organ failure were designated a critical illness by intensivists, the condition was named critical illness polyneuropathy (136). Similar cases of polyneuropathy have been subsequently reported from the United States (103; 42), France (21; 07; 124), Holland (77; 72), Austria (09), Germany (55), and Spain (74).
During the 1990s, researchers began to find, on review of muscle and nerve pathology as well as electrophysiologic studies, that there was a significant component of primary myopathic disease in patients with critical illness. As early as 1972, Karpati and colleagues described in animal models a corticosteroid-induced thick filament myopathy after nerve transection (57). In 1977, MacFarlane reported an acute quadriplegia that developed in a young man after treatment for asthma with high-dose corticosteroids (79). This clinical pattern was recognized in a number of other patients, who were then postulated to have a “functional denervation” due to sepsis and neuromuscular blockade and later developed a thick filament myopathy with exposure to corticosteroids (12). Since these reports, there has been a great deal of interest in elucidating the pathogenesis of what some authors call critical illness myopathy (70). Various other names for this entity have also been used including acute quadriplegic myopathy (52), acute myopathy of intensive care (64), critical care myopathy (33), acute illness myopathy (105), acute myopathy with selective lyses of myosin filaments (108), acute necrotizing myopathy of intensive care (99; 137), and critical illness neuromuscular disease (114).
Critical illness polyneuropathy and myopathy occur as a complication of systemic inflammatory response syndrome or sepsis. The diagnosis of systemic inflammatory response syndrome requires that two or more of these features be fulfilled:
|
• Temperature >38° C or <36° C |
Systemic inflammatory response syndrome has several effects on the nervous system.
After the development of systemic inflammatory response syndrome, whether it occurs in the critical care unit or on a general medical ward, the earliest nervous system manifestation is septic encephalopathy. Within hours of the appearance of a positive blood culture, careful testing may show impaired attention, concentration, orientation, and writing (131). If the systemic inflammatory response syndrome continues, the patient gradually slips into deep coma, usually without the development of focal signs, seizures, myoclonus, or asterixis. The electroencephalogram is a sensitive indicator of the presence and severity of septic encephalopathy. CT head scans and cerebrospinal fluid examinations are usually unremarkable (130).
If the systemic inflammatory response syndrome can be treated by antibiotics, surgical drainage of an infected focus, inotropic drugs, and fluid replacement, the encephalopathy usually improves rapidly. However, at this time, difficulty in weaning from the ventilator may be noticed. Early studies suggested that the most common neuromuscular cause for this difficulty, after cardiac and pulmonary causes have been excluded, was critical illness polyneuropathy (136; 80; 81). However, clinical signs of neuropathy, including depressed deep tendon reflexes, are present in only half of these patients. Hence, electrophysiologic studies are necessary to establish the diagnosis and to differentiate a myopathic process from neuropathic processes; it is believed that critical illness myopathy is more common than neuropathy. In a Danish case series of ICU weakness, testing via routine electrophysiological examination, quantitative electromyography, and direct muscle stimulation suggested critical illness myopathy in all 35 cases (24). Nonetheless, a more severe neuropathy or myopathy can be suspected when a subacute flaccid quadriparesis or quadriplegia develops in a critically ill patient. The weakness is diffuse and may involve the facial muscles, but ocular muscles are typically spared. Sparing of sensory modalities may provide a clue that it is a myopathic process.
Even though sepsis and multiple organ failure are less common in pediatric as opposed to adult critical care units, a few instances of critical illness polyneuropathy in children have been observed (29; 109; 14; 97).
In a systematic literature review, the prevalence of ICU-acquired weakness ranged from 29% to 56% (34). In the first week of ICU admissions, the rate of muscle mass loss, as measured by serial imaging studies, was 2% per day.
There have been conflicting reports about the effect of critical illness neuromuscular complications on the intensive care unit mortality rate. A systematic literature review found no increase in adjusted mortality of intensive care unit patients with neuromuscular complications but demonstrated prolongation in mechanical ventilation and intensive care unit and hospital stay (113). However, in a prospective cohort of severe sepsis cases who underwent weekly neurologic examinations and nerve conduction studies within 72 hours of developing severe sepsis, an abnormal baseline nerve conduction study was predictive of increased hospital mortality (55%), whereas none of the patients with normal baseline nerve conduction studies died (59). Most of the affected patients had electrophysiologic evidence of both critical illness myopathy and critical illness neuropathy.
The prognosis for a favorable recovery was thought to be as low as 50% (97). One study suggested that children fare better than adults with this condition. Williams and colleagues reviewed the published pediatric cases of neuromuscular complications in the setting of a critical illness (125). A follow-up of 19 cases, 10 patients had full recovery, six had persisting weakness, and three either died or had no noticeable recovery. The follow-up interval in this study was 3 weeks to 2 years. The long-term functional outcome of 42 adult survivors admitted to inpatient neurorehabilitation for critical illness neuropathy (30), myopathy (6), or both (6) has been evaluated (56). After a mean rehabilitation stay of 76 days, there was remarkable recovery on discharge in 57% of cases, which is higher than previously described. On a follow-up mean of 32 months, 74% had a good outcome, with 24 cases achieving it by 6 to 12 months, and the remaining seven subjects between 1 and 2 years. Good outcome correlated with the absence of central nervous system additional lesions.
Knowledge of the presence of critical illness myopathy or neuropathy aids management on the ventilator and, in particular, indicates that the patient has a neuromuscular problem that may prolong care in the critical care unit. If the polyneuropathy is mild, recovery is expected to occur within a matter of weeks. However, if the polyneuropathy is severe, recovery may take months. In especially severe cases, recovery may not occur (126). Recovery from myopathy should occur within weeks to months as well, unless muscle necrosis is severe. A 1-year follow-up study of 15 critically ill patients (six with critical illness polyneuropathy, six with critical illness myopathy, and three with a mixed picture) was performed. Eight of the 15 patients survived, and those who did not survive succumbed to their underlying illnesses, not as a complication of their neuromuscular disorders. Of the eight survivors, only two were not ambulatory at 1 year. Those two patients were found to have absent compound muscle action potentials on electrophysiological studies at 1 year. Thus, neurophysiological studies performed at intervals can aid in determining the prognosis of recovery (58).
In a prospective study by Leijten and colleagues in the Netherlands, critical illness polyneuropathy was associated with increased mortality rate and rehabilitation problems (73). However, rehabilitation therapy is likely to be beneficial in speeding the recovery of these patients (88). Literature confirms the author’s experience that the prognosis for recovery from critical illness myopathy is more favorable than that of critical illness neuropathy alone (44), or combined myopathy and neuropathy of critical illness (60). Dysphagia is associated with complicated and prolonged stay in the intensive care unit, impaired weaning from mechanical ventilation, impeded rehabilitation, and increases morbidity and mortality (106).
In comparison to 30 controls, 115 patients previously admitted to the intensive care unit were identified to have, on RNA sequencing, mitochondrial dysfunction, disruption of lipid metabolism, and increased fibrosis (117). On multivariate analysis, these patterns of RNA expression were associated with more prominent weakness at 5 years. Of interest, early parenteral nutrition in the ICU and glucocorticoid use were associated with this abnormal RNA pattern of expression
A 65-year-old woman was admitted with an acute onset of pneumonia, confusion, and hypotension. She had a previous history of hypertension and chronic obstructive pulmonary disease. She quickly suffered a respiratory arrest, requiring intubation and transfer to the critical care unit. Sputum and blood cultures grew Streptococcus pneumoniae. She was treated with penicillin G, tobramycin, and cloxacillin, but developed the adult respiratory distress syndrome. Neurologic examination disclosed drowsiness and extensor plantar responses, but normal motor power and tendon reflexes. Both CT head scan and CSF were normal. An EEG revealed mild generalized suppression. Over the next several weeks, she developed elevated levels of serum amylase and hepatic enzymes, a pneumothorax, a bronchopleural fistula, and phlebitis. She required several courses of aminoglycosides for recurrent infections and had mildly elevated trough levels of serum tobramycin. The diffuse encephalopathy became severe, consistent with septic encephalopathy. Two months following admission, she was greatly improved but still unable to breathe independently. She was more alert, but had areflexia and severe weakness in all limbs.
Median and common peroneal nerve conduction studies revealed normal conduction velocities and distal latencies, but reduced amplitudes of compound muscle and sensory nerve action potentials. The sural compound action potential was absent. The diaphragm potential was absent on bilateral phrenic nerve stimulation. Needle EMG showed numerous trains of fibrillation potentials and positive sharp waves (+4) in quadriceps, medial gastrocnemius, first dorsal interosseus, and right deltoid and right chest wall muscles.
At 3 months, muscle strength had improved, but she remained areflexic and ventilator dependent. Compound muscle action potential amplitudes had improved. Sural nerve potentials had returned, and needle EMG revealed less abnormal spontaneous activity in various muscles.
At 4 months, she was weaned from the ventilator. Tendon reflexes had returned in the arms. Compound muscle and sensory nerve action potentials had improved further, and spontaneous activity had subsided with the appearance of polyphasic voluntary motor units. Six months following admission, she was discharged ambulatory, with a mild right foot drop and absent ankle jerks.
At a 2-year follow-up examination, she was mildly weak on dorsiflexion of the right foot and had absent ankle jerks. Nerve conduction studies were normal. Thus, she had made an almost full recovery from severe critical illness polyneuropathy.
Many authors have speculated that sepsis is the cause of critical illness polyneuropathy (136; 126). The neuropathy tends to be more severe with increasing time in the critical care unit, hyperglycemia, hyperosmolality, and decreased serum albumin concentrations (126). Humoral and cellular factors have also been implicated in this process (95; 22; 27; 13; 55). Increased E-selectin expression has been found in the endothelium of endoneurial and epineurial vessels in the presence of proinflammatory cytokines (35). This is believed to play a role in perpetuating local inflammation in peripheral nerves. All of these factors are associated with the overall picture of sepsis. Retrospective (136) and prospective (126) studies have failed to implicate a variety of potential causes of critical illness polyneuropathy, including types of primary illness or injury, Guillain-Barré syndrome, specific nutritional deficiencies, or immune-mediated disease (124).
Increased levels of complement C4b/c were seen in all 27 intensive care unit patients, 13 of whom developed associated weakness. There was no difference between the weak and intact strength groups in maximum, minimum, and mean levels of complement activation products (128). Critical illness myopathy has most commonly been associated with corticosteroid use, most notably in asthmatics or post-transplant patients, and the use of neuromuscular blockade. However, neither is required to induce the syndrome. There is also evidence linking proteolytic and apoptotic systems as mechanisms of muscle fiber atrophy, necrosis, and cell death. Caspase-3-mediated apoptosis has been shown to cause extensive cell death in multiple organ systems during sepsis (54). A large histopathologic review of muscle biopsies in patients with critical illness myopathy showed fiber atrophy associated with myosin filament depolymerization that may be caused by increased expression of lysosomal enzymes and ubiquitin (47). Upregulation of part of the apoptotic system, specifically TGF-beta/MAPK, has been found to be uniquely involved in critical illness myopathy (28). Serum amyloid A1 transiently accumulates within the first few days in the muscle of critical illness myopathy patients and is in vitro induced by interleukin 6 and tumor necrosis factor-alpha (68).
During electrophysiological studies, severely affected muscle has been found to be electrically inexcitable to direct muscle stimulation, which should be preserved in a purely neurogenic process (102). In studies with animal models, this inexcitability has been proposed to be caused by a hyperpolarizing shift in the voltage dependence of fast inactivation of sodium channels (101).
The morphologic features of critical illness polyneuropathy have been demonstrated through the biopsy of peripheral nerve and muscle and a comprehensive autopsy study of both the central and peripheral nervous system in nine patients (136). There is a primary axonal degeneration of peripheral nerve motor and sensory fibers, but no evidence of inflammation, as may be seen in Guillain-Barré syndrome. Muscle shows scattered atrophic fibers in acute denervation and grouped atrophy in chronic denervation. Immunohistochemistry of muscle biopsy specimens demonstrates upregulation of HLA-DR, the proinflammatory cytokine tumor necrosis factor-R75, and the anti-inflammatory cytokine IL-10 when compared to controls (26). The only CNS manifestation is central chromatolysis of anterior horn cells and loss of dorsal root ganglion cells, secondary to the peripheral nerve axonal damage. No changes appear distinctive of critical illness polyneuropathy.
In systemic inflammatory response syndrome, cellular and humoral responses are activated (39).
The chief humoral response, the cytokines, are locally activated mediators and include interleukins-1, -2, and -6, tumor necrosis factor-alpha, arachidonic acid, coagulation factors, free oxygen radicals, and proteases. The cellular response involves lymphocytes, monocytes, and neutrophils. These cellular and humoral factors interact with themselves and with adhesion molecules, which are increased in the blood of septic patients (22). Adhesion molecules adhere to leukocytes, platelets, and endothelial cells; they also induce "rolling neutrophils" and fibrin platelet aggregates that obstruct capillary flow. In a study of 22 patients admitted with sepsis and neuromuscular disorders, superficial peroneal nerve biopsies were studied for expression of adhesion molecules, E-selectin, and TNF-alpha. Eight of these patients had findings of axonal neuropathy consistent with critical illness polyneuropathy. Expression of E-selectin was significantly elevated in the endoneurial and epineurial vessels of patients with suspected critical illness polyneuropathy, supporting the idea that microvascular changes may contribute to the axonopathy of critical illness polyneuropathy (35). Moreover, cytokines that are secreted in sepsis have histamine-like properties that may also increase microvascular permeability (136). Increased capillary permeability induces local tissue edema. Activation of nitric oxide, now known to be the endovascular relaxing factor (95), causes arteriolar dilation, which may further slow capillary flow. Blood vessels supplying the peripheral nerves lack autoregulation, rendering these vessels susceptible to such disturbances (75). Thus, essential nutrients fail to reach the organ parenchyma. Severe energy deficits would result, inducing a primary axonal degeneration, most likely distally, if highly energy-dependent systems involving axonal transport of structural proteins are involved.
The microcirculation is disturbed in sepsis.
Disturbances of the microcirculation to nerve and muscle may also explain the effects of neuromuscular blocking agents and corticosteroids. Through increased capillary permeability induced by sepsis, neuromuscular blocking agents, notably vecuronium or its metabolite, 3-desacetyl-vecuronium, could have a direct toxic effect on peripheral nerve axons (107). These neuromuscular blocking agents may also cause functional denervation through their prolonged neuromuscular blocking action (123). The result would be denervation atrophy of muscle and a relatively pure motor neuropathy.
There has always been a concern that antibiotics, particularly aminoglycosides with their known neural toxicity, might cause critical illness polyneuropathy. These drugs might gain access to the peripheral nerve as a result of increased capillary permeability. However, there has been no statistical proof that antibiotics cause peripheral nerve dysfunction in sepsis (112).
Z'Graggen and colleagues compared the nerve excitability of 10 patients with critical illness polyneuropathy to 20 control patients (133). The authors used the strength-duration time constant, threshold electrotonus, current-threshold relationship, and recovery cycle (refractoriness, superexcitability, and late subexcitability). They found in patients with critical illness polyneuropathy evidence of chronic depolarization of motor nerve membranes due to raised extracellular potassium and to hypoperfusion. Similar findings are also described in critical illness myopathy. Z’Graggen and colleagues investigated velocity recovery cycles recorded by direct muscle stimulation of 10 critical illness myopathy patients and 10 normal controls (132). They demonstrated that, in patients, muscle fibers were depolarized or that sodium inactivation was increased.
Critical illness myopathy often occurs in association with the use of neuromuscular blocking agents and steroids that are used to treat acute severe asthma or to treat patients in the post-transplant state (64). In many reports, it has been concluded that neuromuscular blocking agents and steroids, singly or in combination, induced either a pure axonal motor neuropathy (41; 63; 08; 38) or a primary myopathy (79; 108; 25; 30; 93; 52; 66; 137; 64; 65; 45). A distinctive feature of the primary myopathy is a selective loss of myosin filaments (108; 25), although other neuropathologic findings are also seen. A combination of functional denervation due to neuromuscular blocking agents and the subsequent direct effects of steroids on muscle is one possible mechanism. Protracted immobility and a high stress catabolic state, such as prolonged acute illness, might induce a lesser degree of this process.
It is possible that many of these patients suffer from systemic inflammatory response syndrome because infection is often a precipitating event in acute, severe asthma, and is common in post-transplant patients. Animal experiments by Karpati and colleagues have shown that if the muscle is first denervated by nerve transection and then corticosteroids are given, a thick filament myopathy similar to that myopathy seen in humans can be induced (57). Thus, in the human condition, critical illness polyneuropathy and the additional effects of neuromuscular blocking agents could denervate muscle, and steroids could then induce the typical myopathic changes. Both the rapidly evolving myopathy reported by al-Lozi and colleagues, which is characterized by destruction of thick filaments throughout the muscle fibers (02), and the acute necrotizing myopathy of intensive care may simply represent further stages of this process (99; 137).
Mozaffar and colleagues found that combining glucocorticoid administration and denervation reduces rat skeletal muscle mRNA expression of myosin heavy chain and to a lesser extent actin mRNA (87). The authors observed selective depletion of myosin heavy chain protein with sparing of actin protein levels in severely atrophic muscle.
In another study, muscle inactivity was shown to play a central role in triggering the development of critical illness myopathy (122). Under normal conditions, glucose transporter-4 (GLUT4) is stored in vesicles in perinuclear compartments and is rapidly translocated to the sarcolemma after insulin receptor binding or after muscle contraction. The insulin-signaling cascade involves protein kinase B/Akt, whereas contraction-induced GLUT4 translocation is mediated through phosphorylation of the 5’-adenosine monophosphate-activated protein kinase. Failure of GLUT4 translocation from the perinuclear spaces to the sarcolemmal membrane was demonstrated to be a central mechanism of impaired glucose supply to muscle cells in critical illness myopathy patients. Glucose metabolism could not be stimulated during euglycemic-hyperinsulinergic clamp during critical illness. These investigators not only demonstrated significant insulin resistance in critically ill patients as compared to healthy control subjects, but also demonstrated that insulin resistance was most severe in critical illness myopathy patients. However, electrical muscle stimulation corrected muscle-specific adenosine monophosphate-activated protein kinase failure, restored GLUT4 translocation in the sarcolemmal membrane, and prevented type-2 fiber atrophy in critical illness myopathy patients.
Several protein degradative and apoptotic systems are found in muscle that have been implicated in critical illness myopathy. Minetti and colleagues studied the expression of ubiquitin and myosin isoforms by immunofluorescence in muscle biopsies of two patients who developed severe weakness after receiving high-dose steroids (85). These samples were compared to biopsies from patients with other neuromuscular disorders as well as biopsies from normal subjects. Only in the muscle that had been exposed to high-dose steroids was there increased ubiquitin expression. This suggests that the ubiquitin-ATP-dependent proteolytic system may be initiated by corticosteroid use. In a similar study, several proteolytic systems were evaluated in five patients with flaccid hypotonia. This study again found increased expression of ubiquitin but also found a significant increase in expression of the calpains, which are calcium-activated proteases (110). Helliwell found that multiple proteolytic systems are involved in muscle fiber atrophy, including ubiquitin, cathepsin B, lysosomal enzymes, and heat shock protein 72 (47). In a study of septic patients with multiple organ failure, caspase-3-mediated apoptosis played a role in cell death in multiple organ systems and is thought to involve the nervous system as well (54). The proteolytic pathway TGF-beta/MAPK was also found to be induced in five patients with critical illness myopathy (28).
To investigate the pathophysiologic mechanisms underlying diaphragm weakness, Hooijman and colleagues obtained diaphragm muscle biopsies from 22 critically ill patients who received mechanical ventilation before surgery and compared these with control subjects who received biopsies during thoracic surgery for resection of a suspected early lung malignancy (53). In critically ill patients, both slow-twitch and fast-twitch diaphragmatic fibers had approximately 25% smaller cross-sectional area, with reduced contractile force by 50% or more. Markers of the ubiquitin-proteasome pathway in the diaphragm of critically ill patients were also up-regulated. In a proof-of-concept study in a muscle-specific ring finger protein-1 (MuRF-1) knockout mouse model by the same authors, they went on to evaluate the role of the ubiquitin-proteasome pathway in the development of contractile weakness. MuRF-1 knockout mice were protected against the development of diaphragm contractile weakness during mechanical ventilation.
Patients with critical illness myopathy routinely undergo electrophysiologic testing for diagnostic and prognostic purposes. One challenge in electrodiagnostic studies with critically ill patients is that they are often unable to cooperate with the examination and provide adequate numbers of voluntary activation of motor unit potentials for meaningful analysis. This limitation can make it difficult to distinguish between myopathic and neuropathic processes. Motor nerve conduction studies typically show reduced evoked amplitudes in both conditions. Sensory nerve responses are often spared or mildly reduced, which leads to some descriptions of pure motor neuropathy without additional pathologic confirmation. Reliable sensory nerve conduction studies are also sometimes problematic to accomplish in this setting; some patients may have both conditions. One additional aid in differentiation is direct muscle stimulation studies, which demonstrate reduced or loss of muscle electrical inexcitability in critical illness myopathy (102). One cause of the inexcitability may be a hyperpolarizing shift in the voltage dependence of fast inactivation of sodium channels. This may result from post-translation modification of sodium channels by glycosylation or phosphorylation or from increased expression of the embryonic sodium channel isoform that causes a shift toward more negative potentials (101).
Sepsis occurs in 20% to 50% of patients in major medical or surgical critical care units (115). Seventy percent of patients with sepsis may suffer from critical illness polyneuropathy (126). Critical illness polyneuropathy has also been shown to be associated with increased length of stay in the intensive care unit, with increased serum glucose, and with decreased serum albumin (126).
The occurrence of critical illness myopathy is unknown. However, some studies have indicated that it may affect 33% of patients admitted to the intensive care unit with status asthmaticus (30). In patients undergoing liver transplantation, 7% may develop critical illness myopathy (20). However, all incidence data are clouded by primary difficulties with the diagnosis and classification of these processes; the incidence of both conditions is suspected to be higher.
In a systematic review of 24 published studies (113), critical illness neuromuscular abnormalities affected 46% of adult intensive care unit patients. In one report, this frequency was previously 55% from 1990 to 1995 but has declined to 39% (92). In children, the risk is less well defined as there are no prospective clinical and electrophysiologic studies. In a prospective multicenter clinical and electrophysiologic study of 92 adult intensive care unit patients, 28 patients (30%) developed neuromuscular complications of critical illness (69).
The prediction of which patients will develop ICU-associated weakness has been the subject of a systematic review (135). There are five predictors that emerged as risk factors for developing intensive care unit-associated weakness included duration of mechanical ventilation, age, APACHE II score, blood lactate levels, and duration of stay in the ICU. These models demonstrated strong discriminative abilities.
Critical illness polyneuropathy can be best prevented or mitigated by the prevention or rapid treatment of sepsis and other concurrent abnormalities. Some evidence indicates that strict control of blood glucose with insulin therapy may decrease the risk of developing critical illness polyneuropathy (118).
Critical illness myopathy may be prevented by cautious use of neuromuscular blocking agents and corticosteroids, especially by limiting the total dose of medication as well as reducing the duration of therapy.
In a systematic literature review, some of these concepts were challenged (113). Surprisingly, the authors found no consistent relationship between critical illness neuromuscular complications and the critical illness severity or the use of implicated drugs (glucocorticoids, neuromuscular blockers, aminoglycosides, or midazolam). Hyperglycemia was the main modifiable risk factor for neuromuscular dysfunction acquired in the intensive care unit (113). However, a meta-analysis of 14 prospective studies led to a different set of conclusions (129). In particular, the odds ratio of weakness was associated with critical illness severity (Acute Physiology and Chronic Health Evaluation II score) and drugs such as neuromuscular blocking agents and aminoglycosides. Multivariate analysis confirmed hyperglycemia as a risk factor and added several others, including female gender, multiple organ failure, systemic inflammatory response syndrome, sepsis, electrolyte disturbances, hyperosmolarity, high lactate level, duration of mechanical ventilation, parenteral nutrition, and use of norepinephrine. A systematic review and meta-analysis of randomized controlled trials investigated the effect of neuromuscular blocking agents in acute respiratory distress syndrome (ARDS) (78). Early NMBAs infusion for 48 hours in moderate-to-severe ARDS was associated with reduced intensive care unit mortality and did not increase the risk of intensive care unit-associated weakness.
|
Critical illness polyneuropathy | |
|
Incidence: Common | |
|
Motor neuropathy | |
|
Incidence: Common with neuromuscular blocking agents | |
|
Transient neuromuscular blockade | |
|
Incidence: Common with neuromuscular blocking agents | |
|
Thick filament myopathy | |
|
Incidence: Common with steroids, neuromuscular blocking agents, and asthma | |
|
Disuse myopathy; cachectic myopathy | |
|
Incidence: Common | |
|
Necrotizing myopathy of intensive care | |
|
Incidence: Rare | |
|
| |
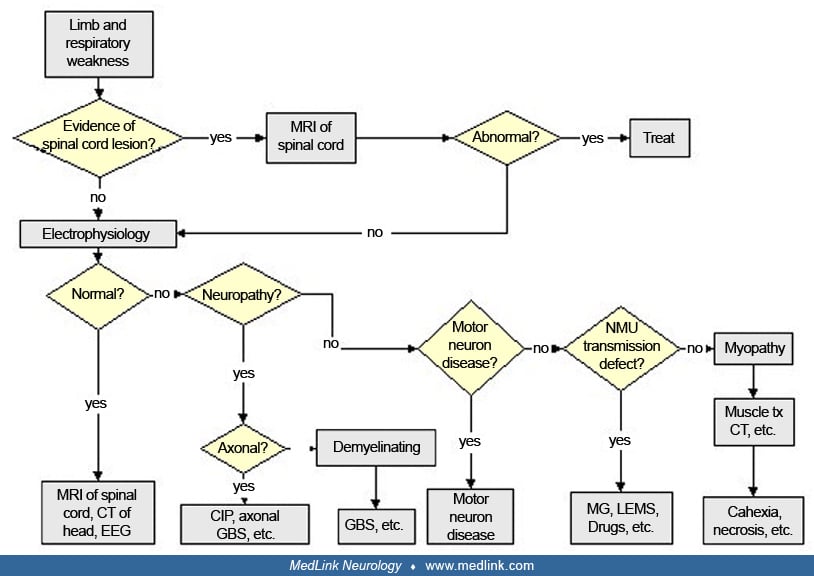
There should be a systematic approach to patients in the critical care unit who have weakness of limb and respiratory muscles.
The problem is considered in two main categories. First are those patients who develop paralysis rapidly before admission to the critical care unit. Because of the acuteness of the situation, there is not sufficient time for investigation of the underlying cause until stabilization has been achieved in the critical care unit. Conditions to be considered are high cervical spinal cord dysfunction due to trauma, neoplasm, or infection; motor neuron disease in which the respiratory muscles are affected before the other muscles; Guillain-Barré syndrome and other acute polyneuropathies such as porphyria; and the acute axonal forms of Guillain-Barré syndrome, including the pure motor variety particularly common in Northern China (83). Mild, chronic polyneuropathies such as diabetic polyneuropathy may affect the nerves of respiration predominantly. After admission to the critical care unit, sepsis may worsen a preexisting polyneuropathy. Occasionally, defects in neuromuscular transmission, myasthenia gravis, and Lambert-Eaton myasthenic syndrome present with primary respiratory failure.
The second category is patients who are admitted to a critical care unit for unrelated primary illnesses or trauma and later develop severe weakness. Diagnostic considerations in this group include anoxic myelopathy, affecting mainly anterior horn cells, which may result from cardiac arrest, atherosclerosis, surgery of the aorta, or severe pulmonary disease (06); Guillain-Barré syndrome related to infections; or critical illness myopathy or polyneuropathy.
A variety of infections predispose severely ill patients to develop critical illness myopathy and polyneuropathy. In a retrospective single-center study of 949 patients with COVID-19 admitted to the ICU, 63% were able to get out of bed, and 21% were able to walk greater than or equal to 30 meters at ICU discharge (89). In severe COVID-19 illness, retrospective studies have described weakness in up to 46% of patients (98). Risk factors are similar to other critically ill cases and include premorbid health status, sepsis, multiple organ failure, mechanical ventilation, immobilization, neuromuscular blockade, corticosteroid use, and glycemic control. Similar to non-COVID cases, prolonged mechanical ventilation and long hospital stays render critical illness weakness more likely to occur with COVID-19 infection.
The earliest electrophysiologic sign of critical illness polyneuropathy is a reduction of compound muscle action potential amplitudes, with a minor change in latency. These changes are typical of axonal damage but are also associated with severe myopathy, including critical illness myopathy. Fibrillation potentials and positive sharp waves may not appear in the muscle until 3 weeks after onset. Motor unit potentials, if they can be voluntarily activated by the patient, will often appear polyphasic and normal or somewhat low in amplitude, suggesting an associated primary involvement of muscle. These electrophysiologic changes, however, are not specific and are also produced by a primary myopathy. Despite electrodiagnostic technical difficulty in critically ill patients, it is important to demonstrate a reduction of sensory nerve action potential amplitudes before a firm electrophysiologic diagnosis of polyneuropathy can be made; this finding is not present in many instances. There is some evidence that some patients with critical illness myopathy demonstrate increased compound motor unit potential duration (96). In support of that, in vitro single muscle fiber excitability studies by Allen and colleagues have demonstrated a reduction in muscle fiber conduction velocity that is inversely proportional to the degree of muscle weakness in critical illness myopathy (03). The CMAP duration prolongation was also proportional to the degree of weakness and phase of illness. A study indicates that this finding can be detectable in cases of critical illness neuropathy or myopathy, as evidenced by CMAP duration greater than 8.0 msec (even longer for peroneal at the tibialis anterior) elicited by proximal or distal stimulation sites but not in controls (62). If present in greater than one nerve, the specificity of prolonged CMAP duration approached 100%.
In a prospective multicenter study of 92 intensive care unit patients, peroneal CMAP performed daily showed reduced amplitudes by more than two standard deviations in all 28 patients (30%) with neuromuscular complications of critical illness, with a 67% specificity (69). All 28 patients developed the electrophysiological change within 13 days of intensive care unit admission, with a median time of 6 days. This change in CMAP amplitude was abrupt over 1 day in most cases but took place over several days in others. Others similarly were able to predict the development of acquired neuromuscular dysfunction based on nerve conduction studies done at day 7 of intensive care unit admission (59).
Repetitive nerve stimulation studies to demonstrate a defect in neuromuscular transmission should also be performed in suspicious cases. This defect does not occur in sepsis, but will be present if neuromuscular blocking agents have been used. Their effects may persist beyond several hours to a number of days if the patient is in renal failure or liver failure (107).
Phrenic nerve conduction studies and, in some cases, needle electromyography of chest wall muscles and the diaphragm are safe and can establish respiratory muscle involvement in patients who are difficult to wean from the ventilator (11). Electrophysiologic features of critical illness myopathy include reduction of compound motor action potential amplitudes (less than 80%) and perhaps, in some cases, increased duration in two or more nerves with normal or near normal sensory nerve action potential amplitudes (more than 80%). There should be an absence of decrement on repetitive nerve stimulation. Myopathic changes should be seen on needle examination, with or without fibrillation potentials (67).
Continuing debate regarding whether unexplained weakness in critically ill patients is due to primary nerve or muscle pathology led to the use of direct muscle stimulation, particularly in those patients who have inexcitable nerves on routine nerve conduction studies (102; 100). The ratio of nerve-stimulated compound motor action potential amplitude to muscle-stimulated compound motor action potential amplitude should be more than 0.5 in critical illness myopathy (and in normal controls) and less than 0.5 in critical illness polyneuropathy. Trojaborg and colleagues applied those techniques in addition to motor unit number estimates and quantitative electromyography to classify 22 consecutive patients with critical illness-related weakness as having primarily neurogenic or myopathic features. These authors found that electrophysiologic abnormalities supported a myopathy in all cases (116).
Creatine kinase levels vary in critical illness myopathy but are usually normal or mildly elevated, except in acute necrotizing myopathy, where creatine kinase levels are high. The signature pathological finding is a distinctive loss of thick myosin filaments. However, there can be various degrees of type 1 or type 2 muscle fiber atrophy as well as necrosis (46). In an observational study of the first week of critical illness myopathy, Bierbrauer and colleagues described histological evidence in the vastus lateralis muscle of selective type 2 fiber atrophy coupled with depletion of myosin heavy chain IIa isoform messenger RNA in patients with inexcitable peroneal muscle membrane as defined by distal CMAP amplitudes below 3.0 mV (10). There has been debate regarding the need for a muscle biopsy in patients with possible critical illness myopathy (67; 86). The controversy surrounds whether the biopsy results alter treatment decisions. Although Lacomis and colleagues have listed histopathologic findings of myosin loss as one of their diagnostic criteria, they do not advocate muscle biopsy in cases without apparent diagnostic uncertainty. Muscle ultrasound data from healthy controls were compared to that derived from septic patients in the first 2 weeks; most septic cases have elevated mean muscle echotexture and fasciculations in more arm and leg muscle groups (43). There was limited accuracy of muscle thickness, echo intensity, and homogeneity, as well as nerve cross-sectional area, thickness, and vascularization for the diagnosis of ICU-associated weakness relatively early (7 to 9 days) in the disease course (127). In the study conducted by Latronico and colleagues, Giuseppe Lauria’s group described a reduction in intraepidermal nerve density on skin biopsy of critical illness patients (71).
Another emerging use of muscle ultrasound that may be of prognostic value is evaluating the thickness of the diaphragm muscle (40). Development of increased thickness predicted prolonged ventilation, perhaps due to excessive ventilatory effort. Decreased diaphragm muscle thickness was associated with a lower probability of liberation from ventilation, due to abnormally low inspiratory effort. Tibial high-resolution nerve ultrasound showed increased cross-sectional area, suggesting inflammatory edema as well as hypoechoic nerves as a possible sign of inflammation in critical illness polyneuromyopathy (36).
The management of all of the above neuromuscular disorders involves the close collaboration of neurologists, neurophysiologists, and intensivists. Treatment of critical illness polyneuropathy involves treatment of systemic inflammatory response syndrome (sepsis) and includes antibiotics to counteract infection, surgical drainage of an infected focus, and the use of inotropic drugs and fluid replacement to control hypotension. Attempts to intervene at an early stage of the systemic inflammatory response syndrome have shown variable results. These attempts at intervention have used agents such as monoclonal and polyclonal antibodies directed against bacterial endotoxin, monoclonal antibodies to tumor necrosis factor-alpha, fusion protein constructs of soluble tumor necrosis factor receptors, interleukin-1 receptor antagonists (76), and the platelet-activating factor receptor antagonist, BN5202 (27). N-acetylcysteine, a drug that acts as an oxygen radical scavenger, has also been used (111). On the supposition that critical illness polyneuropathy might have an immune-mediated basis, Wijdicks and Fulgham treated three patients with high intravenous immunoglobulins, but there was no beneficial effect (124). A prospective, randomized controlled trial of IgM-enriched intravenous immunoglobulin in critical illness polyneuropathy or myopathy did not demonstrate a benefit in reducing disease severity at 14 days (19).
In one review, avoiding hyperglycemia and postponing parenteral nutrition beyond the first week in the intensive care unit reduces the burden of ICU-acquired weakness (119). Also, early mobilization, which requires minimizing sedation, may be promising, but several studies are ongoing. A systematic review also identified low-quality evidence suggesting that pioglitazone administration would increase skeletal muscle insulin sensitivity and might decrease intramuscular inflammatory mediators such as TNF-α (82). In another systematic review and meta-analysis of 29 studies, high protein nutrition of more than 1.2 g/kg in critically ill patients may improve nitrogen balance and muscle mass on the short-term and possibly 60-day mortality (120).
Though intensive glucose control was earlier thought to be helpful in reducing the incidence and severity of critical illness polyneuropathy (118), a 2012 study argues against tight glucose control given the higher mortality risk associated with increased moderate to severe hypoglycemia (91). In a prospective randomized controlled trial, the effect of intensive insulin therapy was compared to conventional therapy on the morbidity and mortality in critically ill patients (51). Patients remaining in the intensive care unit for at least 7 days had a reduced incidence of critical illness polyneuropathy and myopathy (39%) with tighter glucose control when compared to conventional therapy (51%). There was an associated reduction in the rate of prolonged mechanical ventilation (35% and 48%, respectively) with intensive insulin control. Therefore, an earlier Cochrane review supported the beneficial effects of intensive insulin therapy in reducing the incidence of critical illness myopathy and neuropathy (49). Tight glucose control also reduced the duration of mechanical ventilation, the duration of ICU stay, and the 180-day mortality. However, it has become more difficult to support this recommendation because evidence associates tight glycemic control with an increased mortality risk within 90 days after randomization (91). In this study, 6026 critically ill patients were randomly assigned to either intensive blood glucose control, with a target blood glucose range of 81 to 108 mg per deciliter, or conventional glucose control, with a target of less than or equal to 180 mg per deciliter (10.0 mmol per liter). This group of investigators found that intensive glucose control led to increased risk of moderate (blood glucose 41-70 mg per deciliter) and severe (blood glucose ≤ 40 mg per deciliter) hypoglycemia, both of which were associated with an increased risk of death. The American Diabetes Association recommends a target blood glucose of 144 to 180 mg per deciliter to reduce the risk of hypoglycemia in critically ill patients (04). Therefore, according to a 2014 Cochrane update, intensive insulin therapy reduces critical illness neuropathy and myopathy (risk ratio 0.65), and it reduces duration of mechanical ventilation, intensive care unit stay, and 180-day mortality. Because hypoglycemia is a risk, Cochrane reviewers recommend that the consequences and prevention of hypoglycemia need further study (50).
Preliminary data seem to indicate that early and daily electrical muscle stimulation of the bilateral vastus lateralis, vastus medialis, and peroneus longus muscles may reduce the incidence of critical illness weakness (104). Patients with an MRC sum score of less than 48 of 60 were diagnosed with critical illness weakness without EMG confirmation of a neuropathy or myopathy. In this randomized parallel intervention trial with a near 66% drop-out rate (mainly due to death or impaired cognition), three of 24 evaluable electrical muscle stimulation cases developed critical illness weakness as opposed to 11 of 28 in the control group. There was also an inexplicable benefit in the electrical muscle stimulation group towards earlier weaning from the ventilator (day 0 in the electrical muscle stimulation group vs. day 10 in the placebo group), which suggests an imbalance in the baseline properties of both groups. Given this concern, the lack of EMG data, and the small sample size of this trial, these results are preliminary and have to be taken with caution until further studies are done. In another study, 80 adults admitted to the intensive care unit and supported by mechanical ventilation for 96 or more hours received for 10 days on open-label basis an intervention (61). This consisted of either physical therapy alone or physical therapy with transcutaneous electrical neuromuscular stimulation. Although 40% of 68 completers had myopathy on day 14, this study failed to demonstrate a benefit of transcutaneous electrical neuromuscular stimulation in critically ill patients. In a meta-analysis, electrical muscle stimulation combined with the usual standard of care was compared with usual care alone in critically ill patients (134). This review of six controlled studies did not reveal any significant differences in overall muscle strength, ICU mortality, duration of mechanical ventilation, or ICU length of stay. However, a systematic review of randomized controlled trials identified 182 adults in six trials assessing the impact of physical exercise or neuromuscular electrical stimulation on ICU-acquired weakness (37). The authors reported that these interventions prevented excessive muscle loss in ICU patients and increased muscle strength in those with ICU-acquired weakness.
In patients who are recovering, physiotherapy and rehabilitation tailored to myopathy or polyneuropathy should be instituted. After a mean inpatient neuro-rehabilitation stay of 76 days, some critical illness neuropathy or myopathy patients remained to have a modified Rankin score of 5 (severe disability) (56). However, as a group, the median modified ranking score of 42 cases improved from 5 on admission (all severe) to 3 on discharge (moderate disability, range 0 to 5), with further reduction to 1 on long-term outpatient follow-up (no significant disability despite mild symptoms, range 0 to 5). Although early endurance and resistance training within 48 hours of critical care admission and mechanical ventilation makes intuitive sense, this intervention did not improve functional capacity or independence at hospital discharge in comparison to early standard physiotherapy (31). Another study compared ergometer training, resistance training, and standard physiotherapy in post-hospital rehabilitation of patients with intensive care-acquired weakness. Both ergometer training and resistance training regimens were better than standard care physiotherapy in improving lower limb muscle strength, walking ability, and cardiorespiratory fitness (121).
The mean daily dose of corticosteroids may predict physical outcome at 1 year (90). Physical function at 1 and 2 years is negatively impacted by longer duration of intensive care unit stay (32). A Cochrane review concluded there is no effect of corticosteroids on critical illness neuropathy, except for fewer new shock episodes (50). In an animal study, rats were exposed to the intensive care unit condition for 5 days (sedation, immobilization, and mechanical ventilation) (01). The results of this study suggested that exposure to veramalone has less severe negative effects on fast-twitch muscle fibers than in rats exposed to prednisone. However, human studies to support this preliminary finding have yet to be performed.
Noncompetitive neuromuscular blocking agents are contraindicated.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Mazen M Dimachkie MD
Dr. Dimachkie, Director of the Neuromuscular Disease Division and Executive Vice Chairman for Research Programs, Department of Neurology, The University of Kansas Medical Center received consultant honorariums from Abcuro, Advanced Clinica Cabaletta Bio, Amicus, Annexon Inc., ArgenX, Astellas, Cabaletta Bio, Catalyst, Covance/LabCorp/Fortrea, CSL Behring, Dianthus, EMD Serono/Merck, Horizon, Ig Society Inc, Ipsen, Janssen, Octapharma, Sanofi Genzyme, Shire/Takeda, and Treat NMD/TACT. Dr. Dimachikie also received research grants from Astellas, Catalyst, CSL Behring, EMD Serono/Merck, Horizon, Janssen, Nuvig, Octapharma, Sanofi Genzyme, Sarepta Therapeutics, Shire/Takeda, and TMA.
See Profile
Louis H Weimer MD
Dr. Weimer of Columbia University received a consultant honorarium from Roche and Ovid Therapeutics.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
Jun. 11, 2026
Peripheral Neuropathies
May. 12, 2026
Neuromuscular Disorders
Apr. 23, 2026
Peripheral Neuropathies
Apr. 09, 2026
Infectious Disorders
Apr. 08, 2026
Peripheral Neuropathies
Apr. 07, 2026